Immunotherapy-Based Targeting and Elimination of Leukemic Stem Cells in AML and CML

 ,
,  ,
,
Abstract
1. Introduction
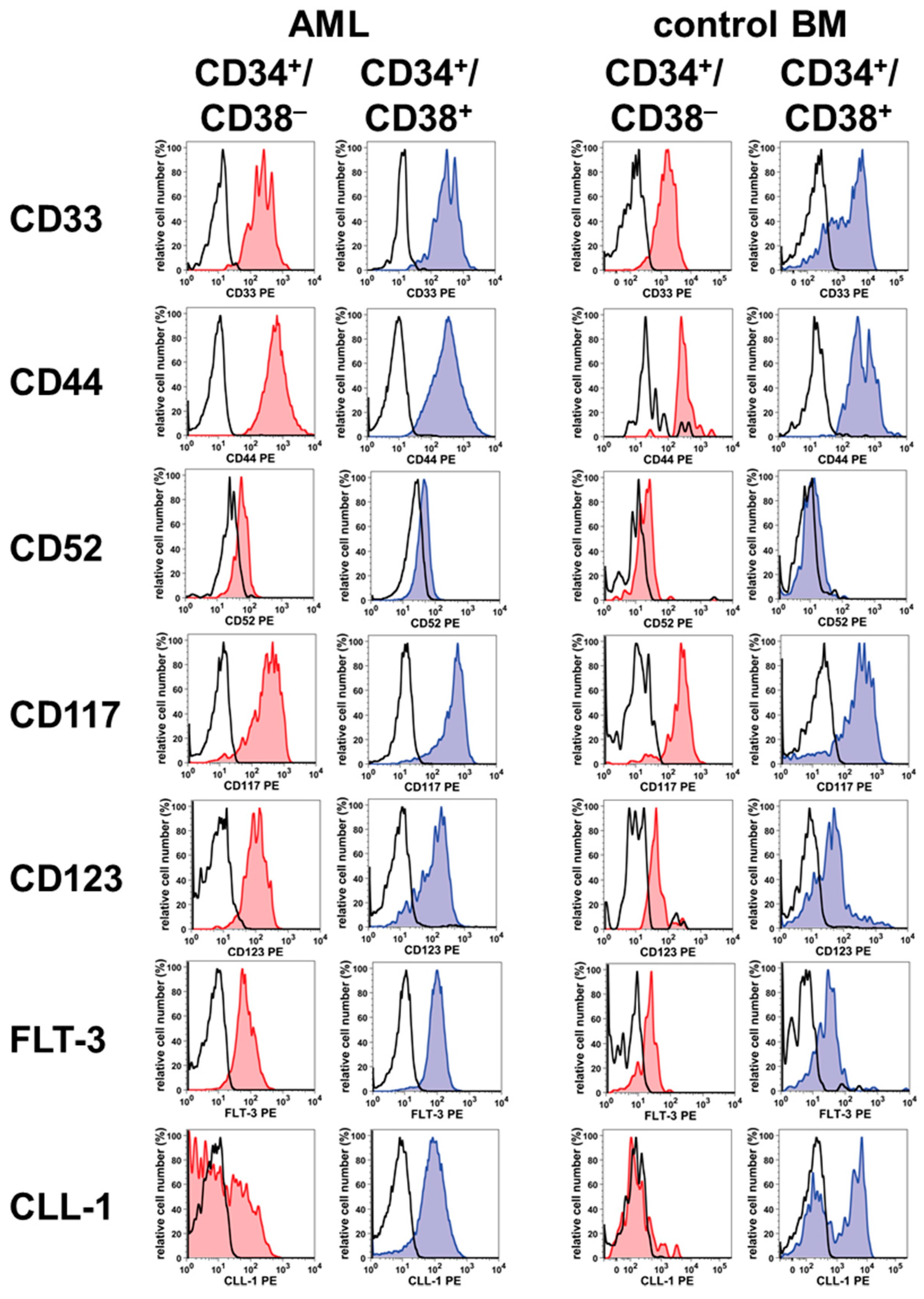
2. Phenotype of LSC in AML and CML
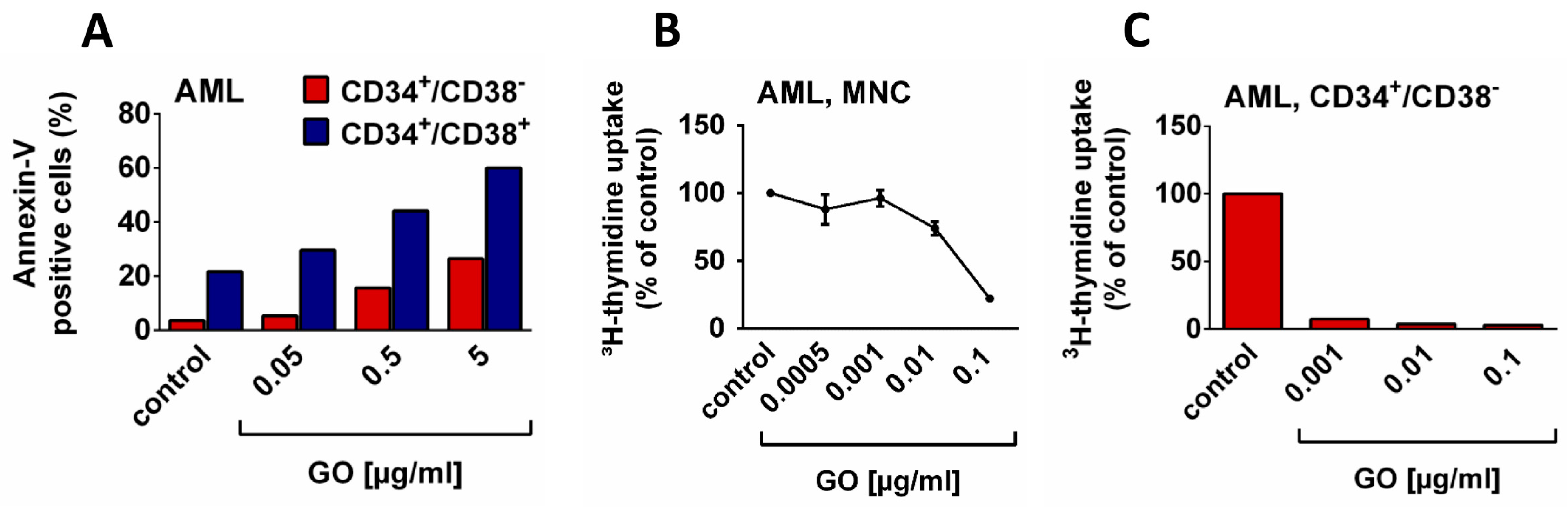
3. Targeting LSC with Antibody-Based Drugs
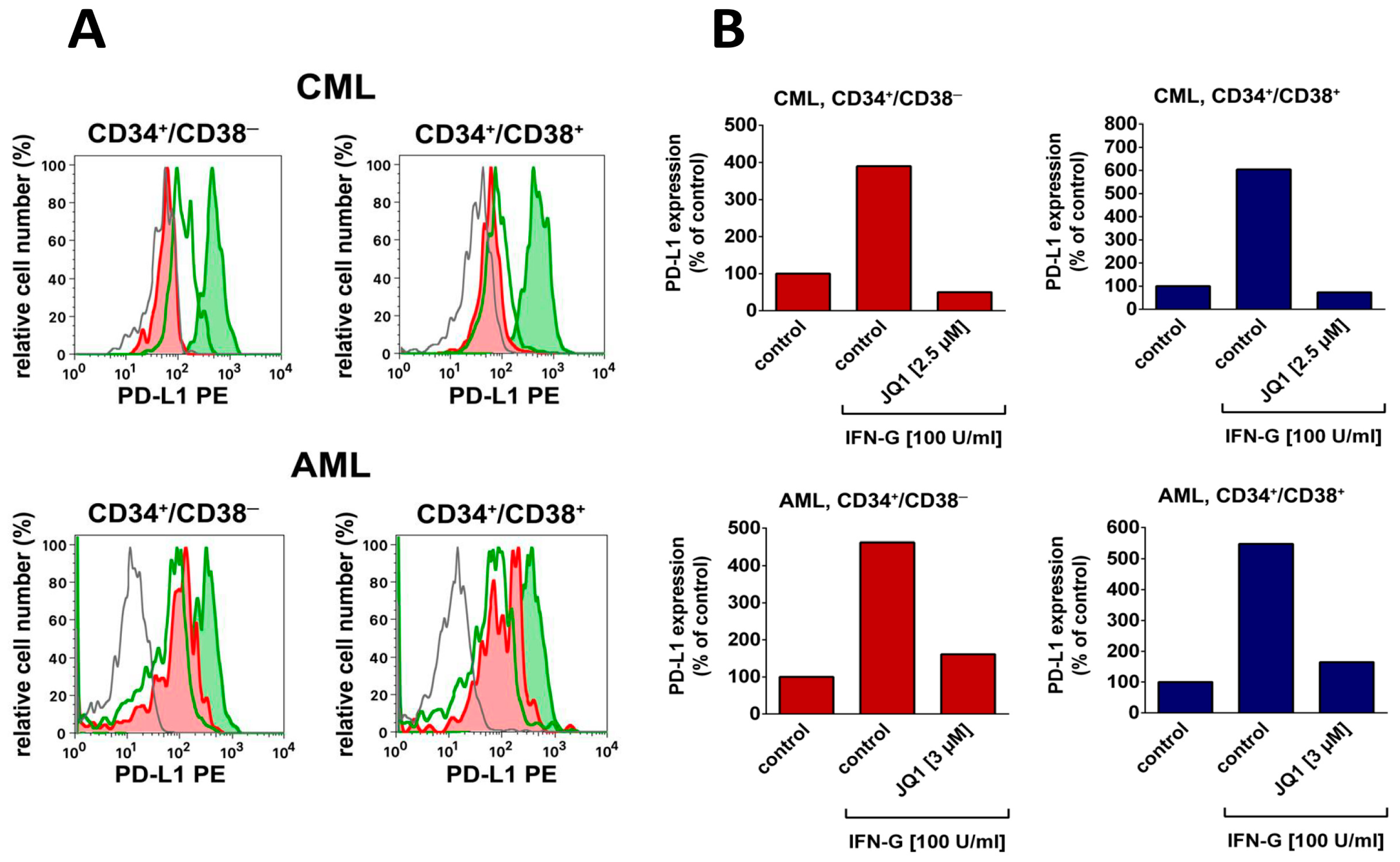
4. Targeting LSC Using Drugs Directed against Immune Checkpoint Molecules
5. Targeting of LSC by Bispecific Antibodies
6. Targeting of LSC by Chimeric Antigen Receptor (CAR) Cell-Based Therapy
7. Targeting LSC by Employing NK Cells and/or T cells
8. Targeting LSC by Suppressing or Promoting LSC Homing
9. Limitations of LSC-Targeting Immunotherapy in AML and CML: LSC Resistance
10. Concluding Remarks and Outlook to the Future
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NBM | normal bone marrow |
| FceRII | Fc-epsilon receptor II |
| IL-2RA | interleukin-2 receptor alpha chain |
| DPPIV | dipeptidyl peptidase IV |
| IAP | integrin associated protein |
| NCAM | neural cell adhesion molecule |
| G-CSFR | granulocyte colony-stimulating factor receptor |
| PROM1 | prominin-1 |
| PD-L1 | programmed cell death-ligand 1 |
| CLL-1 | C-type lectin-like molecule-1 |
| IL-1RAP | interleukin-1 receptor accessory protein |
| n.c. | not yet clustered |
| LSC | leukemic stem cells |
| AML | acute myeloid leukemia |
| CT | chemotherapy |
| HSCT | hematopoietic stem cell transplantation |
| AML | acute myeloid leukemia |
| NCT | national clinical trial identifier |
| BiTE | bispecific T cell engagers |
| TriKE | tri-specific killer engager |
| DART | dual affinity retargeting antibody |
| CAR | chimeric antigen receptor |
| NK | natural killer cell |
| AML | acute myeloid leukemia |
| MDS | myelodysplastic syndrome |
| R/R | refractory/resistant |
| USA | United States of America |
| HSCT | hematopoietic stem cell transplantation |
| DCN | dendritic cell neoplasm |
| NK | killer cells |
| RAEB | refractory anemia with excess of blasts |
| NKG2D | natural killer group 2D antigen |
References
- Estey, E.; Döhner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Goldman, J.M. Advances in CML. Clin. Adv. Hematol. Oncol. 2007, 5, 270–292. [Google Scholar] [PubMed]
- Hehlmann, R.; Hochhaus, A.; Baccarani, M. European LeukemiaNet. Chronic myeloid leukaemia. Lancet 2007, 370, 342–350. [Google Scholar] [CrossRef]
- Smith, M.L.; Hills, R.K.; Grimwade, D. Independent prognostic variables in acute myeloid leukaemia. Blood Rev. 2011, 25, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Haferlach, T.; Döhner, H. Molecular genetics of adult acute myeloid leukemia: Prognostic and therapeutic implications. J. Clin. Oncol. 2011, 29, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Moarii, M.; Papaemmanuil, E. Classification and risk assessment in AML: Integrating cytogenetics and molecular profiling. ASH Educ. Program. Book 2017, 2017, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Visani, G.; Loscocco, F.; Isidori, A.; Piccaluga, P.P. Genetic profiling in acute myeloid leukemia: A path to predicting treatment outcome. Expert Rev. Hematol. 2018, 11, 455–461. [Google Scholar] [CrossRef]
- Kayser, S.; Levis, M.J. Clinical implications of molecular markers in acute myeloid leukemia. Eur. J. Haematol. 2019, 102, 20–35. [Google Scholar] [CrossRef]
- Andreeff, M.; Konopleva, M. Mechanisms of drug resistance in AML. Cancer Treat. Res. 2002, 112, 237–262. [Google Scholar]
- Goldman, J.M. Treatment strategies for CML. Best Pract. Res. Clin. Haematol. 2009, 22, 303–313. [Google Scholar] [CrossRef]
- Burnett, A.; Wetzler, M.; Löwenberg, B. Therapeutic advances in acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Castelli, G.; Pelosi, E.; Testa, U. Targeted therapies in the treatment of adult acute myeloid leukemias: Current status and future perspectives. Int. J. Hematol. Oncol. 2016, 5, 143–164. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743. [Google Scholar] [CrossRef]
- Barnes, D.J.; Melo, J.V. Primitive, quiescent and difficult to kill: The role of non-proliferating stem cells in chronic myeloid leukemia. Cell Cycle 2006, 5, 2862–2866. [Google Scholar] [CrossRef]
- Krause, D.S.; Van Etten, R.A. Right on target: Eradicating leukemic stem cells. Trends Mol. Med. 2007, 13, 470–481. [Google Scholar] [CrossRef]
- Copland, M. Chronic myelogenous leukemia stem cells: What’s new? Curr. Hematol. Malig. Rep. 2009, 4, 66–73. [Google Scholar] [CrossRef]
- Kavalerchik, E.; Goff, D.; Jamieson, C.H. Chronic myeloid leukemia stem cells. J. Clin. Oncol. 2008, 26, 2911–2915. [Google Scholar] [CrossRef]
- Essers, M.A.; Trumpp, A. Targeting leukemic stem cells by breaking their dormancy. Mol. Oncol. 2010, 4, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Valent, P. Targeting of leukemia-initiating cells to develop curative drug therapies: Straightforward but nontrivial concept. Curr. Cancer Drug Targets 2011, 11, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Taussig, D.C.; Miraki-Moud, F.; Anjos-Afonso, F.; Pearce, D.J.; Allen, K.; Ridler, C.; Lillington, D.; Oakervee, H.; Cavenagh, J.; Agrawal, S.G.; et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood 2008, 112, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Taussig, D.C.; Vargaftig, J.; Miraki-Moud, F.; Griessinger, E.; Sharrock, K.; Luke, T.; Lillington, D.; Oakervee, H.; Cavenagh, J.; Agrawal, S.G.; et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(-) fraction. Blood 2010, 115, 1976–1984. [Google Scholar] [CrossRef] [PubMed]
- Lemoli, R.M.; Salvestrini, V.; Bianchi, E.; Bertolini, F.; Fogli, M.; Amabile, M.; Tafuri, A.; Salati, S.; Zini, R.; Testoni, N.; et al. Molecular and functional analysis of the stem cell compartment of chronic myelogenous leukemia reveals the presence of a CD34- cell population with intrinsic resistance to imatinib. Blood 2009, 114, 5191–5200. [Google Scholar] [CrossRef] [PubMed]
- Van Rhenen, A.; van Dongen, G.A.; Kelder, A.; Rombouts, E.J.; Feller, N.; Moshaver, B.; Stigter-van Walsum, M.; Zweegman, S.; Ossenkoppele, G.J.; Jan Schuurhuis, G. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007, 110, 2659–2666. [Google Scholar] [CrossRef] [PubMed]
- Schulenburg, A.; Brämswig, K.; Herrmann, H.; Karlic, H.; Mirkina, I.; Hubmann, R.; Laffer, S.; Marian, B.; Shehata, M.; Krepler, C.; et al. Neoplastic stem cells: current concepts and clinical perspectives. Crit. Rev. Oncol. Hematol. 2010, 76, 79–98. [Google Scholar] [CrossRef]
- Järås, M.; Johnels, P.; Hansen, N.; Agerstam, H.; Tsapogas, P.; Rissler, M.; Lassen, C.; Olofsson, T.; Bjerrum, O.W.; Richter, J.; et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc. Natl. Acad. Sci. USA 2010, 107, 16280–16285. [Google Scholar] [CrossRef]
- Herrmann, H.; Sadovnik, I.; Cerny-Reiterer, S.; Rülicke, T.; Stefanzl, G.; Willmann, M.; Hoermann, G.; Bilban, M.; Blatt, K.; Herndlhofer, S.; et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood 2014, 123, 3951–3962. [Google Scholar] [CrossRef]
- Eisterer, W.; Jiang, X.; Christ, O.; Glimm, H.; Lee, K.H.; Pang, E.; Lambie, K.; Shaw, G.; Holyoake, T.L.; Petzer, A.L.; et al. Different subsets of primary chronic myeloid leukemia stem cells engraft immunodeficient mice and produce a model of the human disease. Leukemia 2005, 19, 435–441. [Google Scholar] [CrossRef]
- Jamieson, C.H.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A.; et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N. Engl. J. Med. 2004, 351, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Blatt, K.; Menzl, I.; Eisenwort, G.; Cerny-Reiterer, S.; Herrmann, H.; Herndlhofer, S.; Stefanzl, G.; Sadovnik, I.; Berger, D.; Keller, A.; et al. Phenotyping and Target Expression Profiling of CD34+/CD38- and CD34+/CD38+ Stem- and Progenitor cells in Acute Lymphoblastic Leukemia. Neoplasia 2018, 20, 632–642. [Google Scholar] [CrossRef] [PubMed]
- De Boer, B.; Prick, J.; Pruis, M.G.; Keane, P.; Imperato, M.R.; Jaques, J.; Brouwers-Vos, A.Z.; Hogeling, S.M.; Woolthuis, C.M.; Nijk, M.T.; et al. Prospective Isolation and Characterization of Genetically and Functionally Distinct AML Subclones. Cancer Cell 2018, 34, 674–689.e8. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Upchurch, D.; Szilvassy, S.J.; Guzman, M.L.; Howard, D.S.; Pettigrew, A.L.; Meyerrose, T.; Rossi, R.; Grimes, B.; Rizzieri, D.A.; et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000, 14, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Florian, S.; Sonneck, K.; Hauswirth, A.W.; Krauth, M.T.; Schernthaner, G.H.; Sperr, W.R.; Valent, P. Detection of molecular targets on the surface of CD34+/CD38—Stem cells in various myeloid malignancies. Leuk. Lymphoma 2006, 47, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Hauswirth, A.W.; Florian, S.; Printz, D.; Sotlar, K.; Krauth, M.T.; Fritsch, G.; Schernthaner, G.H.; Wacheck, V.; Selzer, E.; Sperr, W.R.; et al. Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur. J. Clin. Investig. 2007, 37, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Hosen, N.; Park, C.Y.; Tatsumi, N.; Oji, Y.; Sugiyama, H.; Gramatzki, M.; Krensky, A.M.; Weissman, I.L. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2007, 104, 11008–11013. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef]
- Kersten, B.; Valkering, M.; Wouters, R.; van Amerongen, R.; Hanekamp, D.; Kwidama, Z.; Valk, P.; Ossenkoppele, G.; Zeijlemaker, W.; Kaspers, G.; et al. CD45RA, a specific marker for leukaemia stem cell sub-populations in acute myeloid leukaemia. Br. J. Haematol. 2016, 173, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Jia, M.; Chen, Y.; Zhao, H.; Luo, Z.; Tang, Y. Re-evaluation of various molecular targets located on CD34+CD38-Lin- leukemia stem cells and other cell subsets in pediatric acute myeloid leukemia. Oncol. Lett. 2016, 11, 891–897. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sutherland, H.J.; Blair, A.; Zapf, R.W. Characterization of a hierarchy in human acute myeloid leukemia progenitor cells. Blood 1996, 87, 4754–4761. [Google Scholar] [PubMed]
- Saito, Y.; Kitamura, H.; Hijikata, A.; Tomizawa-Murasawa, M.; Tanaka, S.; Takagi, S.; Uchida, N.; Suzuki, N.; Sone, A.; Najima, Y.; et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci. Transl. Med. 2010, 2, 17ra9. [Google Scholar] [CrossRef] [PubMed]
- Sperr, W.R.; Hauswirth, A.W.; Florian, S.; Ohler, L.; Geissler, K.; Valent, P. Human leukaemic stem cells: A novel target of therapy. Eur. J. Clin. Investig. 2004, 34, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Liedtke, M.; Gentles, A.J.; Cleary, M.L. CD93 Marks a Non-Quiescent Human Leukemia Stem Cell Population and Is Required for Development of MLL-Rearranged Acute Myeloid Leukemia. Cell Stem Cell 2015, 17, 412–421. [Google Scholar] [CrossRef]
- Hanekamp, D.; Cloos, J.; Schuurhuis, G.J. Leukemic stem cells: Identification and clinical application. Int. J. Hematol. 2017, 105, 549–557. [Google Scholar] [CrossRef]
- Blatt, K.; Herrmann, H.; Hoermann, G.; Willmann, M.; Cerny-Reiterer, S.; Sadovnik, I.; Herndlhofer, S.; Streubel, B.; Rabitsch, W.; Sperr, W.R.; et al. Identification of Campath-1 (CD52) as novel drug target in neoplastic stem cells in 5q-patients with MDS and AML. Clin. Cancer Res. 2014, 20, 3589–3602. [Google Scholar] [CrossRef]
- Blair, A.; Hogge, D.E.; Ailles, L.E.; Lansdorp, P.M.; Sutherland, H.J. Lack of expression of Thy-1 (CD90) on acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo. Blood 1997, 89, 3104–3112. [Google Scholar]
- Valent, P.; Sadovnik, I.; Ráčil, Z.; Herrmann, H.; Blatt, K.; Cerny-Reiterer, S.; Eisenwort, G.; Lion, T.; Holyoake, T.; Mayer, J. DPPIV (CD26) as a novel stem cell marker in Ph+ chronic myeloid leukaemia. Eur. J. Clin. Investig. 2014, 44, 1239–1245. [Google Scholar] [CrossRef]
- Sadovnik, I.; Hoelbl-Kovacic, A.; Herrmann, H.; Eisenwort, G.; Cerny-Reiterer, S.; Warsch, W.; Hoermann, G.; Greiner, G.; Blatt, K.; Peter, B.; et al. Identification of CD25 as STAT5-Dependent Growth Regulator of Leukemic Stem Cells in Ph+ CML. Clin. Cancer Res. 2016, 22, 2051–2061. [Google Scholar] [CrossRef]
- Warfvinge, R.; Geironson, L.; Sommarin, M.N.E.; Lang, S.; Karlsson, C.; Roschupkina, T.; Stenke, L.; Stentoft, J.; Olsson-Strömberg, U.; Hjorth-Hansen, H.; et al. Single-cell molecular analysis defines therapy response and immunophenotype of stem cell subpopulations in CML. Blood 2017, 129, 2384–2394. [Google Scholar] [CrossRef]
- Culen, M.; Borsky, M.; Nemethova, V.; Razga, F.; Smejkal, J.; Jurcek, T.; Dvorakova, D.; Zackova, D.; Weinbergerova, B.; Semerad, L.; et al. Quantitative assessment of the CD26+ leukemic stem cell compartment in chronic myeloid leukemia: patient-subgroups, prognostic impact, and technical aspects. Oncotarget 2016, 7, 33016–33024. [Google Scholar] [CrossRef] [PubMed]
- Gerber, J.M.; Gucwa, J.L.; Esopi, D.; Gurel, M.; Haffner, M.C.; Vala, M.; Nelson, W.G.; Jones, R.J.; Yegnasubramanian, S. Genome-wide comparison of the transcriptomes of highly enriched normal and chronic myeloid leukemia stem and progenitor cell populations. Oncotarget 2013, 4, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J.J.; Deenik, W.; Smolders, K.G.; van Kuijk, B.J.; Pouwels, W.; Kelder, A.; Cornelissen, J.J.; Schuurhuis, G.J.; Ossenkoppele, G.J. Residual normal stem cells can be detected in newly diagnosed chronic myeloid leukemia patients by a new flow cytometric approach and predict for optimal response to imatinib. Leukemia 2012, 26, 977–984. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Herrmann, H.; Cerny-Reiterer, S.; Gleixner, K.V.; Blatt, K.; Herndlhofer, S.; Rabitsch, W.; Jäger, E.; Mitterbauer-Hohendanner, G.; Streubel, B.; Selzer, E.; et al. CD34+/CD38- stem cells in chronic myeloid leukemia express Siglec-3 (CD33) and are responsive to the CD33-targeting drug gemtuzumab/ozogamicin. Haematologica 2012, 97, 219–226. [Google Scholar] [CrossRef]
- Nievergall, E.; Ramshaw, H.S.; Yong, A.S.; Biondo, M.; Busfield, S.J.; Vairo, G.; Lopez, A.F.; Hughes, T.P.; White, D.L.; Hiwase, D.K. Monoclonal antibody targeting of IL-3 receptor α with CSL362 effectively depletes CML progenitor and stem cells. Blood 2014, 123, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Gerber, J.M.; Qin, L.; Kowalski, J.; Smith, B.D.; Griffin, C.A.; Vala, M.S.; Collector, M.I.; Perkins, B.; Zahurak, M.; Matsui, W.; et al. Characterization of chronic myeloid leukemia stem cells. Am. J. Hematol. 2011, 86, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Riccioni, R.; Militi, S.; Coccia, E.; Stellacci, E.; Samoggia, P.; Latagliata, R.; Mariani, G.; Rossini, A.; Battistini, A.; et al. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood 2002, 100, 2980–2988. [Google Scholar] [CrossRef] [PubMed]
- Yalcintepe, L.; Frankel, A.E.; Hogge, D.E. Expression of interleukin-3 receptor subunits on defined subpopulations of acute myeloid leukemia blasts predicts the cytotoxicity of diphtheria toxin interleukin-3 fusion protein against malignant progenitors that engraft in immunodeficient mice. Blood 2006, 108, 3530–3537. [Google Scholar] [CrossRef][Green Version]
- Lanza, F.; Castagnari, B.; Rigolin, G.; Moretti, S.; Latorraca, A.; Ferrari, L.; Bardi, A.; Castoldi, G. Flow cytometry measurement of GM-CSF receptors in acute leukemic blasts, and normal hemopoietic cells. Leukemia 1997, 11, 1700–1710. [Google Scholar] [CrossRef]
- Graf, M.; Hecht, K.; Reif, S.; Pelka-Fleischer, R.; Pfister, K.; Schmetzer, H. Expression and prognostic value of hemopoietic cytokine receptors in acute myeloid leukemia (AML): Implications for future therapeutical strategies. Eur. J. Haematol. 2004, 72, 89–106. [Google Scholar] [CrossRef]
- Hogg, S.J.; Vervoort, S.J.; Deswal, S.; Ott, C.J.; Li, J.; Cluse, L.A.; Beavis, P.A.; Darcy, P.K.; Martin, B.P.; Spencer, A.; et al. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. 2017, 18, 2162–2174. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Christiansson, L.; Söderlund, S.; Svensson, E.; Mustjoki, S.; Bengtsson, M.; Simonsson, B.; Olsson-Strömberg, U.; Loskog, A.S. Increased level of myeloid-derived suppressor cells, programmed death receptor ligand 1/programmed death receptor 1, and soluble CD25 in Sokal high risk chronic myeloid leukemia. PLoS ONE 2013, 8, e55818. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene 2011, 30, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Schulenburg, A.; Blatt, K.; Cerny-Reiterer, S.; Sadovnik, I.; Herrmann, H.; Marian, B.; Grunt, T.W.; Zielinski, C.C.; Valent, P. Cancer stem cells in basic science and in translational oncology: can we translate into clinical application? J. Hematol. Oncol. 2015, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Jordan, C.T. Therapeutic targeting of acute myeloid leukemia stem cells. Blood 2017, 129, 1627–1635. [Google Scholar] [CrossRef]
- Bernstein, I.D. Monoclonal antibodies to the myeloid stem cells: Therapeutic implications of CMA-676, a humanized anti-CD33 antibody calicheamicin conjugate. Leukemia 2000, 14, 474–475. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, I.D. CD33 as a target for selective ablation of acute myeloid leukemia. Clin. Lymphoma 2002, 2, S9–S11. [Google Scholar] [CrossRef]
- Candoni, A.; Martinelli, G.; Toffoletti, E.; Chiarvesio, A.; Tiribelli, M.; Malagola, M.; Piccaluga, P.P.; Michelutti, A.; Simeone, E.; Damiani, D.; et al. Gemtuzumab-ozogamicin in combination with fludarabine, cytarabine, idarubicin (FLAI-GO) as induction therapy in CD33-positive AML patients younger than 65 years. Leuk. Res. 2008, 32, 1800–1808. [Google Scholar] [CrossRef]
- Satwani, P.; Bhatia, M.; Garvin, J.H.; George, D.; Dela Cruz, F.; Le Gall, J.; Jin, Z.; Schwartz, J.; Duffy, D.; van de Ven, C.; et al. A Phase I study of gemtuzumab ozogamicin (GO) in combination with busulfan and cyclophosphamide (Bu/Cy) and allogeneic stem cell transplantation in children with poor-risk CD33+ AML: A new targeted immunochemotherapy myeloablative conditioning (MAC) regimen. Biol. Blood Marrow Transplant. 2012, 18, 324–329. [Google Scholar]
- Pelosi, E.; Castelli, G.; Testa, U. Targeting LSCs through membrane antigens selectively or preferentially expressed on these cells. Blood Cells Mol. Dis. 2015, 55, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Appelbaum, F.R.; Estey, E.H.; Bernstein, I.D. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012, 119, 6198–6208. [Google Scholar] [CrossRef] [PubMed]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: Results of the MRC AML15 trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Lichtenegger, F.S.; Krupka, C.; Köhnke, T.; Subklewe, M. Immunotherapy for acute myeloid leukemia. Semin. Hematol. 2015, 52, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukemia: A meta-analysis of individual patient data from randomized controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef]
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: Results of the randomized Phase III EORTC-GIMEMA AML-19 trial. J. Clin. Oncol. 2016, 34, 972–979. [Google Scholar] [CrossRef]
- Pollard, J.A.; Loken, M.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Aplenc, R.; Bernstein, I.D.; Gamis, A.S.; Alonzo, T.A.; Meshinchi, S. CD33 expression and its associated with gemtuzumab ozogamicin response: Results from the randomized Phase III children’s oncology group trial AAML0531. J. Clin. Oncol. 2016, 34, 747–755. [Google Scholar] [CrossRef]
- Jen, E.Y.; Ko, C.W.; Lee, J.E.; Del Valle, P.L.; Aydanian, A.; Jewell, C.; Norsworthy, K.J.; Przepiorka, D.; Nie, L.; Liu, J.; et al. FDA approval: Gemtuzumab ozogamicin for the treatment of adults with newly diagnosed CD33-positive acute myeloid leukemia. Clin. Cancer Res. 2018, 24, 3242–3246. [Google Scholar] [CrossRef]
- Kung Sutherland, M.S.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. SGN–CD33A: A novel targeting antibody–drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef]
- Stein, E.M.; Tallman, M.S. Emerging therapeutic drugs for AML. Blood 2016, 127, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Walter, R.B.; Erba, H.P.; Fathi, A.T.; Advani, A.S.; Lancet, J.E.; Ravandi, F.; Kovacsovics, T.; DeAngelo, D.J.; Bixby, D.; et al. A phase 1 trial of vadastuximab talirine as monotherapy in patients with CD33-positive acute myeloid leukemia. Blood 2018, 131, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B. Investigational CD33-targeted therapeutics for acute myeloid leukemia. Expert Opin. Investig. Drugs 2018, 27, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; McCubrey, J.A.; Miller, M.S.; Delatte, S.; Ramage, J.; Kiser, M.; Kucera, G.L.; Alexander, R.L.; Beran, M.; Tagge, E.P.; et al. Diphteria toxin fused to human interleukin-3 is toxic to blasts from patients with myeloid leukemias. Leukemia 2000, 14, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Riccioni, R.; Biffoni, M.; Diverio, D.; Lo-Coco, F.; Foà, R.; Peschle, C.; Frankel, A.E. Diphtheria toxin fused to variant human interleukin-3 induces cytotoxicity of blasts from patients with acute myeloid leukemia according to the level of interleukin-3 receptor expression. Blood 2005, 106, 2527–2529. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.A.; Liu, T.F.; Cline, J.M.; Wagner, J.D.; Hall, P.D.; Frankel, A.E. Toxicology and pharmacokinetics of DT388IL3, a fusion protein consisting of a truncated diphtheria toxin (DT388) linked to human interleukin 3 (IL3) in cynomolgus monkeys. Leuk. Lymphoma 2004, 45, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Hogge, D.E.; Yalcintepe, L.; Wong, S.H.; Gerhard, B.; Frankel, A.E. Variant diphteria toxin-interleukin-3 fusion proteins with increased receptor affinity have enhanced cytotoxicity against acute myeloid leukemia progenitors. Clin. Cancer Res. 2006, 12, 1284–1291. [Google Scholar] [CrossRef]
- Frankel, A.E.; Konopleva, M.; Hogge, D.; Rizzieri, D.; Brooks, C.; Cirrito, T.; Kornblau, S.M.; Borthakur, G.; Bivins, C.; Garcia-Manero, G.; et al. Activity and tolerability of SL-401, a targeted therapy directed to the interleukin-3 receptor on cancer stem cells and tumor bulk, as a single agent in patients with advanced hematologic malignancies. J. Clin. Oncol. 2013, 31, 7029. [Google Scholar]
- Frolova, O.; Benito, J.; Brooks, C.; Wang, R.Y.; Korchin, B.; Rowinsky, E.K.; Cortes, J.; Kantarjian, H.; Andreeff, M.; Frankel, A.E.; et al. SL-401 and SL-501, targeted therapeutics directed at the interleukin-3 receptor, inhibit the growth of leukaemic cells and stem cells in advanced phase chronic myeloid leukaemia. Br. J. Haematol. 2014, 166, 862–874. [Google Scholar] [CrossRef]
- Alkharabsheh, O.; Frankel, A.E. Clinical Activity and Tolerability of SL-401 (Tagraxofusp): Recombinant Diphtheria Toxin and Interleukin-3 in Hematologic Malignancies. Biomedicines 2019, 5, 7. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Lane, A.A.; Sweet, K.L.; Stein, A.S.; Vasu, S.; Blum, W.; Rizzieri, D.A.; Wang, E.S.; Duvic, M.; Sloan, J.M.; et al. Tagraxofusp in Blastic Plasmacytoid Dendritic-Cell Neoplasm. N. Engl. J. Med. 2019, 380, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Tagraxofusp: First global approval. Drugs 2019, 79, 579–583. [Google Scholar] [PubMed]
- Jin, L.; Lee, E.M.; Ramshaw, H.S.; Busfield, S.J.; Peoppl, A.G.; Wilkinson, L.; Guthridge, M.A.; Thomas, D.; Barry, E.F.; Boyd, A.; et al. Monoclonal antibody mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemia stem cells. Cell Stem Cell 2009, 5, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Busfield, S.J.; Biondo, M.; Wong, M.; Ramshaw, H.S.; Lee, E.M.; Ghosh, S.; Braley, H.; Panousis, C.; Roberts, A.W.; He, S.Z.; et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia 2014, 28, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- He, S.Z.; Busfield, S.; Ritchie, D.S.; Hertzberg, M.S.; Durrant, S.; Lewis, I.D.; Marlton, P.; McLachlan, A.J.; Kerridge, I.; Bradstock, K.F.; et al. Phase I study of the safety, pharmacokinetics and anti-leukemic activity of the anti-CD123 monoclonal antibody CSL360 in relapsed, refractory or high-risk acute myeloid leukemia. Leuk. Lymphoma 2015, 56, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J.E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006, 12, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Z.; Ding, X.; Li, X.Y.; Cen, J.N.; Chen, Z.X. In vitro effects of anti-CD44 monoclonal antibody on the adhesion and migration of chronic myeloid leukemia stem cells. Zhonghua Xue Ye Xue Za Zhi 2010, 31, 398–402. [Google Scholar] [PubMed]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-clinical development of a humanized anti-CD47 antibody with anti-cancer therapeutic potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef]
- Vey, N.; Delaunay, J.; Martinelli, G.; Fiedler, W.; Raffoux, E.; Prebet, T.; Gomez-Roca, C.; Papayannidis, C.; Kebenko, M.; Paschka, P.; et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget 2016, 7, 32532–32542. [Google Scholar]
- Krupka, C.; Lichtenegger, F.S.; Köhnke, T.; Bögeholz, J.; Bücklein, V.; Roiss, M.; Altmann, T.; Do, T.U.; Dusek, R.; Wilson, K.; et al. Targeting CD157 in AML using a novel, Fc-engineered antibody construct. Oncotarget 2017, 8, 35707–35717. [Google Scholar] [CrossRef]
- Kong, F.; Gao, F.; Li, H.; Liu, H.; Zhang, Y.; Zheng, R.; Zhang, Y.; Chen, J.; Li, X.; Liu, G.; et al. CD47: a potential immunotherapy target for eliminating cancer cells. Clin. Transl. Oncol. 2016, 18, 1051–1055. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bewersdorf, J.P.; Stahl, M.; Zeidan, A.M. Immunotherapy in acute myeloid leukemia and myelodysplastic syndromes: The dawn of a new era? Blood Rev. 2019, 34, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.C.; Appelbaum, F.R.; Eary, J.F.; Fisher, D.R.; Durack, L.D.; Hui, T.E.; Martin, P.J.; Mitchell, D.; Press, O.W.; Storb, R.; et al. Phase I study of (131) I-anti-CD45 antibody plus cyclophosphamide and total body irradiation for advanced acute leukemia and myelodysplastic syndrome. Blood 1999, 94, 1237–1247. [Google Scholar] [PubMed]
- Bunjes, D.; Buchmann, I.; Duncker, C.; Seitz, U.; Kotzerke, J.; Wiesneth, M.; Dohr, D.; Stefanic, M.; Buck, A.; Harsdorf, S.V.; et al. Rhenium 188-labeled anti-CD66 (a, b, c, e) monoclonal antibody to intensify the conditioning regimen prior to stem cell transplantation for patients with high-risk acute myeloid leukemia or myelodysplastic syndrome: Results of a phase I-II study. Blood 2001, 98, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Pagel, J.M.; Appelbaum, F.R.; Eary, J.F.; Rajendran, J.; Fisher, D.R.; Gooley, T.; Ruffner, K.; Nemecek, E.; Sickle, E.; Durack, L.; et al. 131I-anti-CD45 antibody plus busulfan and cyclophosphamide before allogeneic hematopoietic cell transplantation for treatment of acute myeloid leukemia in first remission. Blood 2006, 107, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Bodet-Milin, C.; Kraeber-Bodéré, F.; Eugène, T.; Guérard, F.; Gaschet, J.; Bailly, C.; Mougin, M.; Bourgeois, M.; Faivre-Chauvet, A.; Chérel, M.; et al. Radioimmunotherapy for Treatment of Acute Leukemia. Semin. Nucl. Med. 2016, 46, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Masarova, L.; Kantarjian, H.; Garcia-Mannero, G.; Ravandi, F.; Sharma, P.; Daver, N. Harnessing the immune system against leukemia: Monoclonal antibodies and checkpoint strategies for AML. Adv. Exp. Med. Biol. 2017, 995, 73–95. [Google Scholar] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef]
- Constantinidou, A.; Alifieris, C.; Trafalis, D.T. Targeting Programmed Cell Death-1 (PD-1) and Ligand (PD-L1): A new era in cancer active immunotherapy. Pharmacol. Ther. 2019, 194, 84–106. [Google Scholar] [CrossRef]
- Haroun, F.; Solola, S.A.; Nassereddine, S.; Tabbara, I. PD-1 signaling and inhibition in AML and MDS. Ann. Hematol. 2017, 96, 1441–1448. [Google Scholar] [CrossRef]
- Boddu, P.; Kantarjian, H.; Garcia-Manero, G.; Allison, J.; Sharma, P.; Daver, N. The emerging role of immune checkpoint based approaches in AML and MDS. Leuk. Lymphoma 2018, 59, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Masarova, L.; Kantarjian, H.; Ravandi, F.; Sharma, P.; Garcia-Manero, G.; Daver, N. Update on immunotherapy in AML and MDS: Monoclonal antibodies and checkpoint inhibitors paving the road for clinical practice. Adv. Exp. Med. Biol. 2018, 995, 97–116. [Google Scholar] [PubMed]
- Giannopoulos, K. Targeting Immune Signaling Checkpoints in Acute Myeloid Leukemia. J. Clin. Med. 2019, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and Biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, Phase II study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Berthon, C.; Driss, V.; Liu, J.; Kuranda, K.; Leleu, X.; Jouy, N.; Hetuin, D.; Quesnel, B. In acute myeloid leukemia, B7-H1 (PD-L1) protection of blasts from cytotoxic T cells is induced by TLR ligands and interferon-gamma and can be reversed using MEK inhibitors. Cancer Immunol. Immunother. 2010, 59, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Krönig, H.; Kremmler, L.; Haller, B.; Englert, C.; Peschel, C.; Andreesen, R.; Blank, C.U. Interferon-induced programmed death-ligand 1 (PD-L1/B7-H1) expression increases on human acute myeloid leukemia blast cells during treatment. Eur. J. Haematol. 2014, 92, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. Tumor-Intrinsic PD-L1 Signaling in Cancer Initiation, Development and Treatment: Beyond Immune Evasion. Front. Oncol. 2018, 8, 386. [Google Scholar] [CrossRef]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; O’Sullivan, D.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic JAK2V617F causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, 429. [Google Scholar] [CrossRef]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef]
- Herrmann, H.; Blatt, K.; Shi, J.; Gleixner, K.V.; Cerny-Reiterer, S.; Müllauer, L.; Vakoc, C.R.; Sperr, W.R.; Horny, H.P.; Bradner, J.E.; et al. Small-molecule inhibition of BRD4 as a new potent approach to eliminate leukemic stem- and progenitor cells in acute myeloid leukemia AML. Oncotarget 2012, 3, 1588–1599. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.B.; McSweeney, P.; et al. Leukemia and lymphoma society blood cancer research partnership. Ipilimumab for patients with relapse after allogeneic transplantation. N. Engl. J. Med. 2016, 375, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Holderried, T.A.W.; Fraccaroli, A.; Schumacher, M.; Heine, A.; Brossart, P.; Stelljes, M.; Klobuch, S.; Kröger, N.; Apostolova, P.; Finke, J.; et al. The role of checkpoint blockade after allogeneic stem cell transplantation in diseases other than Hodgkin’s Lymphoma. Bone Marrow Transplant. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Miyamoto, T.; Yuda, J.; Jabbarzadeh-Tabrizi, S.; Shima, T.; Takayanagi, S.; Niiro, H.; Yurino, A.; Miyawaki, K.; Takenaka, K.; et al. A TIM-3/Gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem Cell 2015, 17, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Dama, P.; Tang, M.; Fulton, N.; Kline, J.; Liu, H. Gal9/Tim-3 expression level is higher in AML patients who fail chemotherapy. J. Immunother. Cancer 2019, 7, 175. [Google Scholar] [CrossRef] [PubMed]
- Noviello, M.; Manfredi, F.; Ruggiero, E.; Perini, T.; Oliveira, G.; Cortesi, F.; De Simone, P.; Toffalori, C.; Gambacorta, V.; Greco, R.; et al. Bone marrow central memory and memory stem T-cell exhaustion in AML patients relapsing after HSCT. Nat. Commun. 2019, 10, 1065. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Hofmeister, R.; Kufer, P.; Schlereth, B.; Baeuerle, P.A. BiTEs: Bispecific antibody constructs with unique anti-tumor activity. Drug Discov. Today 2005, 10, 1237–1244. [Google Scholar] [CrossRef]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef]
- Schürch, C.M. Therapeutic Antibodies for myeloid neoplasms-current developments and future directions. Front. Oncol. 2018, 8, 152. [Google Scholar] [CrossRef]
- Guy, D.G.; Uy, G.L. Bispecific Antibodies for the treatment of acute myeloid leukemia. Curr. Hematol. Malig. Rep. 2018, 13, 417–425. [Google Scholar] [CrossRef]
- Wilke, A.C.; Gökbuget, N. Clinical applications and safety evaluation of the new CD19 specific T-cell engager antibody construct blinatumomab. Expert Opin. Drug Saf. 2017, 16, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Ribera, J.M. Efficacy and safety of bispecific T-cell engager blinatumomab and the potential to improve leukemia-free survival in B-cell acute lymphoblastic leukemia. Expert Rev. Hematol. 2017, 10, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Burt, R.; Warcel, D.; Fielding, A.K. Blinatumomab, a bispecific B-cell and T-cell engaging antibody, in the treatment of B-cell malignancies. Hum. Vaccin. Immunother. 2019, 15, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Curran, E.; Stock, W. Taking a “BiTE out of ALL”: Blinatumomab approval for MRD-positive ALL. Blood 2019, 133, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Bögeholz, J.; Köhnke, T.; Lichtenegger, F.S.; Schneider, S.; Metzeler, K.H.; Fiegl, M.; et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood 2014, 123, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Henn, A.; Raum, T.; Bajtus, M.; Matthes, K.; Hendrich, L.; Wahl, J.; Hoffmann, P.; Kischel, R.; Kvesic, M.; et al. Preclinical characterization of AMG 330, a CD3/CD33-bispecific T-cell-engaging antibody with potential for treatment of acute myelogenous leukemia. Mol. Cancer Ther. 2014, 13, 1549–1557. [Google Scholar] [CrossRef]
- Laszlo, G.S.; Gudgeon, C.J.; Harrington, K.H.; Dell’Aringa, J.; Newhall, K.J.; Means, G.D.; Sinclair, A.M.; Kischel, R.; Frankel, S.R.; Walter, R.B. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood 2014, 123, 554–561. [Google Scholar] [CrossRef]
- Stamova, S.; Cartellieri, M.; Feldmann, A.; Arndt, C.; Koristka, S.; Bartsch, H.; Bippes, C.C.; Wehner, R.; Schmitz, M.; von Bonin, M.; et al. Unexpected recombinations in single chain bispecific anti-CD3-anti-CD33 antibodies can be avoided by a novel linker module. Mol. Immunol. 2011, 49, 474–482. [Google Scholar] [CrossRef]
- Uy, G.; Godwin, J.; Rettig, M.; Vey, N.; Foster, M.; Arellano, M.; Rizzieri, D.; Topp, M.; Huls, G.; Lowenberg, B.; et al. Preliminary results of a phase 1 study of flotetuzumab, a CD123 × CD3 bispecific Dart® protein, in patients with relapsed/refractory acute myeloid leukemia and myelodysplastic syndrome. Blood 2017, 130, 637. [Google Scholar]
- Gaudet, F.; Nemeth, J.F.; McDaid, R.; Li, Y.; Harman, B.; Millar, H.; Teplyakov, A.; Wheeler, J.; Luo, J.; Tam, S.; et al. Development of a CD123 × CD3 bispecific antibody (JNJ-63709178) for the treatment of acute myeloid leukemia (AML). Blood 2016, 128, 2824. [Google Scholar]
- Chu, S.Y.; Pong, E.; Chen, H.; Phung, S.; Chan, E.W.; Endo, N.A.; Rashid, R.; Bonzon, C.; Leung, I.W.L.; Muchhal, U.S.; et al. Immunotherapy with long-lived anti-CD123 × anti-CD3 bispecific antibodies stimulates potent T cell-mediated killing of human AML cell lines and of CD123+ cells in monkeys: a potential therapy for acute myelogenous leukemia. Blood 2014, 124, 2316. [Google Scholar]
- Van Loo, P.F.; Doornbos, R.; Dolstra, H.; Shamsili, S.; Bakker, L. Preclinical evaluation of MCLA117, a CLEC12AxCD3 bispecific antibody efficiently targeting a novel leukemic stem cell associated antigen in AML. Blood 2015, 126, 325. [Google Scholar]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Lichtenegger, F.S.; Köhnke, T.; Vick, B.; Jeremias, I.; Metzeler, K.H.; Altmann, T.; et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: Reversing a T-cell-induced immune escape mechanism. Leukemia 2016, 30, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, M.; Rettig, M.P.; Ritchey, J.K.; Karpova, D.; Uy, G.L.; Eissenberg, L.G.; Gao, F.; Eades, W.C.; Bonvini, E.; Chichili, G.R.; et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood 2016, 127, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.R.; Sukumaran, S.; Hristopoulos, M.; Totpal, K.; Stainton, S.; Lu, E.; Wong, A.; Tam, L.; Newman, R.; Vuillemenot, B.R.; et al. An anti-CD3/anti-CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood 2017, 129, 609–618. [Google Scholar] [CrossRef]
- Hoseini, S.S.; Cheung, N.K. Acute myeloid leukemia targets for bispecific antibodies. Blood Cancer J. 2017, 7, e522. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, S.S.; Guo, H.; Wu, Z.; Hatano, M.N.; Cheung, N.V. A potent tetravalent T-cell-engaging bispecific antibody against CD33 in acute myeloid leukemia. Blood Adv. 2018, 2, 1250–1258. [Google Scholar] [CrossRef]
- Bartels, L.; de Jong, G.; Gillissen, M.A.; Yasuda, E.; Kattler, V.; Bru, C.; Fatmawati, C.; van Hal-van Veen, S.E.; Cercel, M.G.; Moiset, G.; et al. A chemo-enzymatically linked bispecific antibody retargets T cells to a sialylated epitope on CD43 in acute myeloid leukemia. Cancer Res. 2019, 79, 3372–3382. [Google Scholar] [CrossRef]
- Casucci, M.; Nicolis di Robilant, B.; Falcone, L.; Camisa, B.; Norelli, M.; Genovese, P.; Gentner, B.; Gullotta, F.; Ponzoni, M.; Bernardi, M.; et al. Cd44v6-targeted t cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood 2013, 122, 3461–3472. [Google Scholar] [CrossRef]
- Wang, Q.S.; Wang, Y.; Lv, H.Y.; Han, Q.W.; Fan, H.; Guo, B.; Wang, L.L.; Han, W.D. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol. Ther. 2015, 23, 184–191. [Google Scholar] [CrossRef]
- Kenderian, S.S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Aikawa, V.; Morrissette, J.J.; Scholler, J.; Song, D.; Porter, D.L.; Carroll, M.; et al. CD33-specific chimeric antigen receptor t cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015, 29, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.D.; Sauter, C.T.; Ishii, K.; Nguyen, S.M.; Shen, F.; Tasian, S.K.; Chen, W.; Dimitrov, D.S.; Fry, T.J. Preclinical development of flt3-redirected chimeric antigen receptor t cell immunotherapy for acute myeloid leukemia. Blood 2016, 128, 1072. [Google Scholar]
- Wang, Y.; Xu, Y.; Li, S.; Liu, J.; Xing, Y.; Xing, H.; Tian, Z.; Tang, K.; Rao, Q.; Wang, M.; et al. Targeting flt3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered t cells. J. Hematol. Oncol. 2018, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Laborda, E.; Mazagova, M.; Shao, S.; Wang, X.; Quirino, H.; Woods, A.K.; Hampton, E.N.; Rodgers, D.T.; Kim, C.H.; Schultz, P.G.; et al. Development of a chimeric antigen receptor targeting c-type lectin-like molecule-1 for human acute myeloid leukemia. Int. J. Mol. Sci. 2017, 18, 2259. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, H.; Sauer, T.; Shum, T.; Parikh, K.; Mamonkin, M.; Omer, B.; Rouce, R.H.; Lulla, P.; Rooney, C.M.; Gottschalk, S.; et al. Treatment of acute myeloid leukemia with t cells expressing chimeric antigen receptors directed to c-type lectin-like molecule 1. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, S.; Xiao, W.; Li, W.; Wang, L.; Yang, S.; Wang, W.; Xu, L.; Liao, S.; Liu, W.; et al. CAR-T cells targeting CLL-1 as an approach to treat acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Li, M.; Gao, L.; Geng, S.; Wang, J.; Wang, Y.; Yan, Z.; Yu, L. Chimeric antigen receptors for adoptive T cell therapy in acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Schubert, M.L.; Wang, L.; He, B.; Neuber, B.; Dreger, P.; Müller-Tidow, C.; Schmitt, M. Chimeric Antigen Receptor (CAR) T Cell Therapy in Acute Myeloid Leukemia (AML). J. Clin. Med. 2019, 6, 8. [Google Scholar] [CrossRef]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Hönemann, D.; et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef]
- Warda, W.; Larosa, F.; Neto Da Rocha, M.; Trad, R.; Deconinck, E.; Fajloun, Z.; Faure, C.; Caillot, D.; Moldovan, M.; Valmary-Degano, S.; et al. CML hematopoietic stem cells expressing IL1RAP can be targeted by chimeric antigen receptor-engineered T cells. Cancer Res. 2019, 79, 663–675. [Google Scholar] [CrossRef]
- Kottaridis, P.D.; North, J.; Tsirogianni, M.; Marden, C.; Samuel, E.R.; Jide-Banwo, S.; Grace, S.; Lowdell, M.W. Two-stage priming of allogeneic natural killer cells for the treatment of patients with acute myeloid leukemia: A Phase I trial. PLoS ONE 2015, 10, e0123416. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.A.; Denman, C.J.; Rondon, G.; Woodworth, G.; Chen, J.; Fisher, T.; Kaur, I.; Fernandez-Vina, M.; Cao, K.; Ciurea, S.; et al. Haploidentical natural killer cells infused before allogeneic stem cell transplantation for myeloid malignancies: A Phase I trial. Biol. Blood Marrow Transplant. 2016, 22, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, B.C.; Le Luduec, J.B.; Forlenza, C.; Jakubowski, A.A.; Perales, M.A.; Young, J.W.; Hsu, K.C. Phase II study of haploidentical natural killer cell infusion for treatment of relapsed or persistent myeloid malignancies following allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2016, 22, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Ruggeri, L.; Parisi, S.; Bontadini, A.; Dan, E.; Motta, M.R.; Rizzi, S.; Trabanelli, S.; Ocadlikova, D.; Lecciso, M.; et al. Larger size of donor alloreactive NK cell repertoire correlates with better response to NK cell immunotherapy in elderly acute myeloid leukemia patients. Clin. Cancer Res. 2016, 22, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Björklund, A.T.; Clancy, T.; Goodridge, J.P.; Béziat, V.; Schaffer, M.; Hovig, E.; Ljunggren, H.G.; Ljungman, P.T.; Malmberg, K.J. Naive donor NK cell repertoires associated with less leukemia relapse after allogeneic hematopoietic stem cell transplantation. J. Immunol. 2016, 196, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- Berrien-Elliott, M.M.; Wagner, J.A.; Fehniger, T.A. Human cytokine-induced memory-like natural killer cells. J. Innate Immun. 2015, 7, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef]
- Przespolewski, A.; Szeles, A.; Wang, E.S. Advances in immunotherapy for acute myeloid leukemia. Future Oncol. 2018, 14, 963–978. [Google Scholar] [CrossRef]
- Hansrivijit, P.; Gale, R.P.; Barrett, J.; Ciurea, S.O. Cellular therapy for acute myeloid Leukemia—Current status and future prospects. Blood Rev. 2019, in press. [Google Scholar] [CrossRef]
- Koerner, S.P.; Andre, M.C.; Leibold, J.S.; Kousis, P.C.; Kübler, A.; Pal, M.; Haen, S.P.; Bühring, H.J.; Grosse-Hovest, L.; Jung, G.; et al. An Fc-optimized CD133 antibody for induction of NK cell reactivity against myeloid leukemia. Leukemia 2016, 31, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Urbani, E.; Andre, P.; Mancusi, A.; Tosti, A.; Topini, F.; Bléry, M.; Animobono, L.; Romagné, F.; Wagtmann, N.; et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica 2016, 101, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H. Are natural killer cells superior CAR drivers? Oncoimmunology 2014, 3, e28147. [Google Scholar] [CrossRef] [PubMed]
- Introna, M.; Borleri, G.; Conti, E.; Franceschetti, M.; Barbui, A.M.; Broady, R.; Dander, E.; Gaipa, G.; D’Amico, G.; Biagi, E.; et al. Repeated infusions of donor-derived cytokine-induced killer cells in patients relapsing after allogeneic stem cell transplantation: A Phase I study. Haematologica 2007, 92, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Han, W. Cytokine-induced killer (CIK) cells: From basic research to clinical translation. Chin. J. Cancer 2015, 34, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bo, J.; Dai, H.R.; Lu, X.C.; Lv, H.Y.; Yang, B.; Wang, T.; Han, W.D. CIK cells from recurrent or refractory AML patients can be efficiently expanded in vitro and used for reduction of leukemic blasts in vivo. Exp. Hematol. 2013, 41, 241–252. [Google Scholar] [CrossRef]
- Rettinger, E.; Huenecke, S.; Bonig, H.; Merker, M.; Jarisch, A.; Soerensen, J.; Willasch, A.; Bug, G.; Schulz, A.; Klingebiel, T.; et al. Interleukin-15-activated cytokine-induced killer cells may sustain remission in leukemia patients after allogeneic stem cell transplantation: Feasibility, safety and first insights on efficacy. Haematologica 2016, 101, e153–e156. [Google Scholar] [CrossRef]
- Tettamanti, S.; Marin, V.; Pizzitola, I.; Magnani, C.F.; Giordano Attianese, G.M.; Cribioli, E.; Maltese, F.; Galimberti, S.; Lopez, A.F.; Biondi, A.; et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br. J. Haematol. 2013, 161, 389–401. [Google Scholar] [CrossRef]
- Mandelli, F.; Vignetti, M.; Tosti, S.; Andrizzi, C.; Foa, R.; Meloni, G. Interleukin 2 treatment in acute myelogenous leukemia. Stem Cells 1993, 11, 263–268. [Google Scholar] [CrossRef]
- Bergmann, L.; Heil, G.; Kolbe, K.; Lengfelder, E.; Puzicha, E.; Martin, H.; Lohmeyer, J.; Mitrou, P.S.; Hoelzer, D. Interleukin-2 bolus infusion as late consolidation therapy in 2nd remission of acute myeloblastic leukemia. Leuk. Lymphoma 1995, 16, 271–279. [Google Scholar] [CrossRef]
- Blaise, D.; Attal, M.; Pico, J.L.; Reiffers, J.; Stoppa, A.M.; Bellanger, C.; Molina, L.; Nedellec, G.; Vernant, J.P.; Legros, M.; et al. The use of a sequential high dose recombinant interleukin 2 regimen after autologous bone marrow transplantation does not improve the disease free survival of patients with acute leukemia transplanted in first complete remission. Leuk. Lymphoma 1997, 25, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Brune, M.; Hansson, M.; Mellqvist, U.H.; Hermodsson, S.; Hellstrand, K. NK cell-mediated killing of AML blasts: Role of histamine, monocytes and reactive oxygen metabolites. Eur J Haematol. 1996, 57, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Hellstrand, K.; Mellqvist, U.H.; Wallhult, E.; Carneskog, J.; Kimby, E.; Celsing, F.; Brune, M. Histamine and interleukin-2 in acute myelogenous leukemia. Leuk. Lymphoma 1997, 27, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Brune, M.; Castaigne, S.; Catalano, J.; Gehlsen, K.; Ho, A.D.; Hofmann, W.K.; Hogge, D.E.; Nilsson, B.; Or, R.; Romero, A.I.; et al. Improved leukemia-free survival after postconsolidation immunotherapy with histamine dihydrochloride and interleukin-2 in acute myeloid leukemia: Results of a randomized phase 3 trial. Blood 2006, 108, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.I.; Thorén, F.B.; Aurelius, J.; Askarieh, G.; Brune, M.; Hellstrand, K. Post-consolidation immunotherapy with histamine dihydrochloride and interleukin-2 in AML. Scand. J. Immunol. 2009, 70, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.S.; Hallner, A.; Brune, M.; Nilsson, S.; Thorén, F.B.; Martner, A.; Hellstrand, K. Immunotherapy with HDC/IL-2 may be clinically efficacious in acute myeloid leukemia of normal karyotype. Hum. Vaccines Immunother. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Cuapio, A.; Post, M.; Cerny-Reiterer, S.; Gleixner, K.V.; Stefanzl, G.; Basilio, J.; Herndlhofer, S.; Sperr, W.R.; Brons, N.H.; Casanova, E.; et al. Maintenance therapy with histamine plus IL-2 induces a striking expansion of two CD56bright NK cell subpopulations in patients with acute myeloid leukemia and supports their activation. Oncotarget 2016, 7, 46466–46481. [Google Scholar] [CrossRef]
- Sander, F.E.; Nilsson, M.; Rydström, A.; Aurelius, J.; Riise, R.E.; Movitz, C.; Bernson, E.; Kiffin, R.; Ståhlberg, A.; Brune, M.; et al. Role of regulatory T cells in acute myeloid leukemia patients undergoing relapse-preventive immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1473–1484. [Google Scholar] [CrossRef]
- Nair, R.R.; Tolentino, J.; Hazlehurst, L.A. The bone marrow microenvironment as a sanctuary for minimal residual disease in CML. Biochem. Pharmacol. 2010, 80, 602–612. [Google Scholar] [CrossRef]
- Shafat, M.S.; Gnaneswaran, B.; Bowles, K.M.; Rushworth, S.A. The bone marrow microenvironment—Home of the leukemic blasts. Blood Rev. 2017, 31, 277–286. [Google Scholar] [CrossRef]
- Wang, A.; Zhong, H. Roles of the bone marrow niche in hematopoiesis, leukemogenesis, and chemotherapy resistance in acute myeloid leukemia. Hematology 2018, 23, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Martín-Henao, G.A.; Quiroga, R.; Sureda, A.; González, J.R.; Moreno, V.; García, J. L-selectin expression is low on CD34+ cells from patients with chronic myeloid leukemia and interferon-a up-regulates this expression. Haematologica 2000, 85, 139–146. [Google Scholar] [PubMed]
- Jongen-Lavrencic, M.; Salesse, S.; Delwel, R.; Verfaillie, C.M. BCR/ABL-mediated downregulation of genes implicated in cell adhesion and motility leads to impaired migration toward CCR7 ligands CCL19 and CCL21 in primary BCR/ABL-positive cells. Leukemia 2005, 19, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, K.; Kohrs, N.; Ptasinska, A.; Assi, S.A.; Herold, T.; Hiddemann, W.; Lausen, J.; Bonifer, C.; Henschler, R.; Wichmann, C. RUNX1/ETO blocks selectin-mediated adhesion via epigenetic silencing of PSGL-1. Oncogenesis 2015, 4, e146. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Rettig, M.P.; Stone, R.M.; Konopleva, M.Y.; Andreeff, M.; McFarland, K.; Shannon, W.; Fletcher, T.R.; Reineck, T.; Eades, W.; et al. A phase 1/2 study of chemosensitization with plerixafor plus G-CSF in relapsed or refractory acute myeloid leukemia. Blood Cancer J. 2017, 7, e542. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cuadrón, D.; Boluda, B.; Martínez, P.; Bergua, J.; Rodríguez-Veiga, R.; Esteve, J.; Vives, S.; Serrano, J.; Vidriales, B.; Salamero, O.; et al. A phase I-II study of plerixafor in combination with fludarabine, idarubicin, cytarabine, and G-CSF (PLERIFLAG regimen) for the treatment of patients with the first early-relapsed or refractory acute myeloid leukemia. Ann. Hematol. 2018, 97, 763–772. [Google Scholar]
- Corces, M.R.; Chang, H.Y.; Majeti, R. Preleukemic Hematopoietic Stem Cells in Human Acute Myeloid Leukemia. Front. Oncol. 2017, 7, 263. [Google Scholar]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Valent, P.; Bonnet, D.; Wöhrer, S.; Andreeff, M.; Copland, M.; Chomienne, C.; Eaves, C. Heterogeneity of neoplastic stem cells: Theoretical, functional, and clinical implications. Cancer Res. 2013, 73, 1037–1045. [Google Scholar] [CrossRef]
- Holyoake, T.L.; Vetrie, D. The chronic myeloid leukemia stem cell: stemming the tide of persistence. Blood 2017, 129, 1595–1606. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, Y.; Smith, C.; Gasparetto, M.; Turhan, A.; Eaves, A.; Eaves, C. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia 2007, 21, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.S.; Carter, B.Z.; Andreeff, M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and development of therapies using chimeric antigen receptor-expressing t cells. Immunol. Rev. 2014, 257, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Wang, M.; Liao, Y.; Li, J.; Niu, T. A review of efficacy and safety of checkpoint inhibitor for the treatment of acute myeloid leukemia. Front. Pharmacol. 2019, 10, 609. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Antigen | CD | Antigen Expression on Stem/Progenitor Cells in ** | |||||
|---|---|---|---|---|---|---|---|
| NBM | AML | CML | |||||
| CD34+/ CD38– | CD34+/ CD38+ | CD34+/ CD38– | CD34+/ CD38+ | CD34+/ CD38– | CD34+/ CD38+ | ||
| B4 | CD19 | – | – | +/– | +/– | +/– | +/– |
| B1 | CD20 | – | – | – | – | – | – |
| FceRII | CD23 | – | – | – | – | – | – |
| IL2RA | CD25 | – | – | +/– | +/– | + | –/+ |
| DPPIV | CD26 | – | – | –/+ | –/+ | + | –/+ |
| Ki-1 | CD30 | +/– | +/– | +/– | +/– | + | + |
| Siglec-3 | CD33 | + | + | + | + | + | + |
| Hermes | CD44 | + | + | + | + | + | + |
| IAP | CD47 | + | + | + | + | + | + |
| Campath1 | CD52 | +/– | +/– | +/– | +/– | + | –/+ |
| NCAM | CD56 | – | – | – | – | + | + |
| G-CSFR | CD114 | +/– | + | + | + | + | + |
| KIT | CD117 | + | + | + | + | + | + |
| IL3RA | CD123 | + | + | + | + | + | + |
| PROM1 | CD133 | + | + | + | + | + | +/– |
| FLT3 | CD135 | +/– | +/– | + | + | +/– | +/– |
| CXCR4 | CD184 | + | + | + | + | + | + |
| PD-L1 | CD274 | +/− | +/− | +/− *** | +/− *** | +/− *** | +/− *** |
| CLL-1 | CD371 | – | + | +/– | + | – | +/– |
| IL-1RAP | n.c. | – | +/– | +/– | + | + | + |
| Target | Name of Agent | Type of Antibody | Development Stage |
|---|---|---|---|
| CD33 | Gemtuzumab ozogamicin (mylotarg) | ADC | Approved for treatment of AML |
| CD33 | SGN-CD33 (lintuzumab) | ADC | Phase III (+CT) completed |
| CD33 | SGN-CD33A (vadastuximab talirine) | ADC | Discontinued (toxicity) |
| CD33 | IMGN779 (CD33-DGN462) | ADC | Phase I completed |
| CD33 | Lintuzumab-90Y | RADA | Phase I completed |
| CD33 | Lintuzumab-213Bi | RADA | Phase I/II completed |
| CD33 | Lintuzumab-225Ac | RADA | Phase I completed |
| CD45 | Various radiolabeled antibodies combined with CT and HSCT | RADA | Phase I, I/II, or III completed/ongoing |
| CD123 | CSL362 | HmAb | Phase I completed |
| CD123 | KHK2823 | HmAb | Phase I, active, not recruiting |
| CD123 | JNJ-56022473 (CSL362) (talacotuzumab) | HmAb * | Discontinued |
| CD123 | SGN-CD123A | ADC | Phase I, terminated |
| CD123 | IMGN632 | ADC | Phase I, recruiting |
| CD123 | SL-401 (tagraxofusp **) | TOX-C | Approved for treatment of plasmacytoid dendritic cell neoplasms |
| CD25 | Denileukin diftitox *** | TOX-C | Marketing discontinued |
| Name of Agent | Type of Agent | Target | Effector * | Phase | NCT |
|---|---|---|---|---|---|
| AMG330 | BiTE | CD33 | CD3 | I | NCT02520427 |
| AMG673 | BiTE | CD33 | CD3 | I | NCT03224819 |
| AMV564 | Tandem diabody | CD33 | CD3 | I | NCT03144245 |
| GEM333 | Single-chain diabody | CD33 | CD3 | I | NCT03516760 |
| 161533 ** | TriKE | CD33 | CD16 | I/II | NCT03214666 |
| MGD006 (flotetuzumab) | DART | CD123 | CD3 | I | NCT02152956 |
| JNJ-63709178 | DuoBody | CD123 | CD3 | I | NCT02715011 |
| XmAb14045 | X-mAb *** | CD123 | CD3 | I | NCT02730312 |
| MCLA-117 | Biclonics **** | CD371 | CD3 | I | NCT03038230 |
| CAR Target | Effector Cell | Phase | Patients/Cells/ Indications | Country | NCT |
|---|---|---|---|---|---|
| Lewis Y | T | I | Myeloma, AML, MDS | Australia | NCT01716364 |
| CD33 | T | I | CD33+ AML | USA | NCT03126864 |
| CD33 | T | I | R/R AML | China | NCT02799680 |
| CD33 | T | I/II | R/R AML | China | NCT01864902 |
| CD33 | NK | I/II | R/R CD33+ AML | China | NCT02944162 |
| CD123 | T | I | CD123+ AML | China | NCT03585517 |
| CD123 | T | I | relapsed AML after HSCT | China | NCT03114670 |
| CD123 | T | I/II | R/R AML | China | NCT03556982 |
| CD123 | T | I | R/R AML | USA | NCT02623582 |
| CD123 | T | I | R/R AML and R/R blastic plasmacytoid DCN | USA | NCT02159495 |
| CD123 | T | I | R/R AML | China | NCT03672851 |
| CD123 | T | I | R/R AML | USA | NCT03766126 |
| UCART 123 | T | I | R/R AML and newly diagnosed high-risk AML | USA | NCT03190278 |
| CD123/CLL-1 | T | II/III | R/R AML | China | NCT03631576 |
| CD33, CD38, CD56, CD117, CD123, CD34, Muc1 | T | I | R/R AML, MDS, ALL | China | NCT03291444 |
| CD33, CD38, CD56, CD117, CD123, CD133, CD34 or Muc1 | T, TT | I | R/R AML | China | NCT03473457 |
| CD33, CD38, CD56, CD123, CLL-1, Muc1 | T | I/II | AML | China | NCT03222674 |
| NKG2D | T | I | AML, MDS-RAEB, Multiple Myeloma | USA | NCT02203825 |
| NKG2D (NKR2) | T | I/II | R/R AML, Myeloma | USA + Belgium | NCT03018405 |
| Therapeutic Approach | Indication/Application |
|---|---|
| Standard therapies: | |
| Allogeneic hematopoietic stem cell transplantation (allo-HSCT) | Refractory or relapsed (R/R) AML and R/R advanced CML |
| Donor lymphocyte infusion (DLI) | Post allo-HSCT R/R AML and R/R CML after successful cytoreduction |
| Injection of IL-2 and histamine | Non-M3 AML-maintenance therapy |
| Experimental therapies *: | |
| Infusion of NK cells and/or T cells | R/R AML ** or AML in MRD |
| Infusion of allogeneic NK cells and/or T cells after HSCT | Post allo-HSCT R/R AML or R/R CML after successful re-induction |
| Infusion of antibody-primed T and/or NK cells | R/R AML ** or AML in MRD |
| Infusion of cytokine-activated T cells and/or NK cells (CIK) | R/R AML ** or AML in MRD |
| Infusion of CAR-T cells | R/R AML ** or AML in MRD |
| Infusion of CAR-NK cells | R/R AML ** or AML in MRD |
| Infusion of CIK CAR cells | R/R AML ** or AML in MRD |
| Mechanism | Possible Strategy to Overcome Resistance |
|---|---|
| Intrinsic resistance | Antibody-based targeting of LSC |
| LSC quiescence | Antibody-based targeting of LSC Priming LSC into the cell cycle |
| Expression of MDR | MDR-targeting drugs or CAR cells |
| Loss of cell surface targets | Mixtures of antibodies or CAR cells directed against two or more surface targets, drug combinations, or combination of drug therapy with HSCT |
| Immune checkpoint-induced LSC resistance | Checkpoint-targeting antibodies Checkpoint-targeting CAR cells BET/MYC-targeting drugs * JAK/STAT-targeting drugs |
| BM niche-related resistance | Niche cell-targeting drugs |
| Osteoblastic niche | BET/MYC-targeting drugs * |
| Vascular niche | Specific anti-angiogenic drugs |
| LSC retention in niche | Mobilizing drugs (plerixafor) |
| LSC hypermobilization | Mobilization blocker (e.g., gliptins) |
| General immunosuppression | Repeated T/NK cell infusion |
| Blocked immune cells | Bispecific antibodies against LSC and immune effector cells |
| Loss of CAR-T cells or CAR-NK cells | Repeated infusions of CAR cells |
| Development of blocking antibodies against CARs | Use of single domain scFvs Humanize the scFvs |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valent, P.; Sadovnik, I.; Eisenwort, G.; Bauer, K.; Herrmann, H.; Gleixner, K.V.; Schulenburg, A.; Rabitsch, W.; Sperr, W.R.; Wolf, D. Immunotherapy-Based Targeting and Elimination of Leukemic Stem Cells in AML and CML. Int. J. Mol. Sci. 2019, 20, 4233. https://doi.org/10.3390/ijms20174233
Valent P, Sadovnik I, Eisenwort G, Bauer K, Herrmann H, Gleixner KV, Schulenburg A, Rabitsch W, Sperr WR, Wolf D. Immunotherapy-Based Targeting and Elimination of Leukemic Stem Cells in AML and CML. International Journal of Molecular Sciences. 2019; 20(17):4233. https://doi.org/10.3390/ijms20174233
Chicago/Turabian StyleValent, Peter, Irina Sadovnik, Gregor Eisenwort, Karin Bauer, Harald Herrmann, Karoline V. Gleixner, Axel Schulenburg, Werner Rabitsch, Wolfgang R. Sperr, and Dominik Wolf. 2019. "Immunotherapy-Based Targeting and Elimination of Leukemic Stem Cells in AML and CML" International Journal of Molecular Sciences 20, no. 17: 4233. https://doi.org/10.3390/ijms20174233
APA StyleValent, P., Sadovnik, I., Eisenwort, G., Bauer, K., Herrmann, H., Gleixner, K. V., Schulenburg, A., Rabitsch, W., Sperr, W. R., & Wolf, D. (2019). Immunotherapy-Based Targeting and Elimination of Leukemic Stem Cells in AML and CML. International Journal of Molecular Sciences, 20(17), 4233. https://doi.org/10.3390/ijms20174233