Anti-Diabetic Countermeasures Against Tobacco Smoke-Dependent Cerebrovascular Toxicity: Use and Effect of Rosiglitazone



Abstract
1. Introduction
2. Result
2.1. Decreased Harmful Effect of TS on Body Weight by RSG
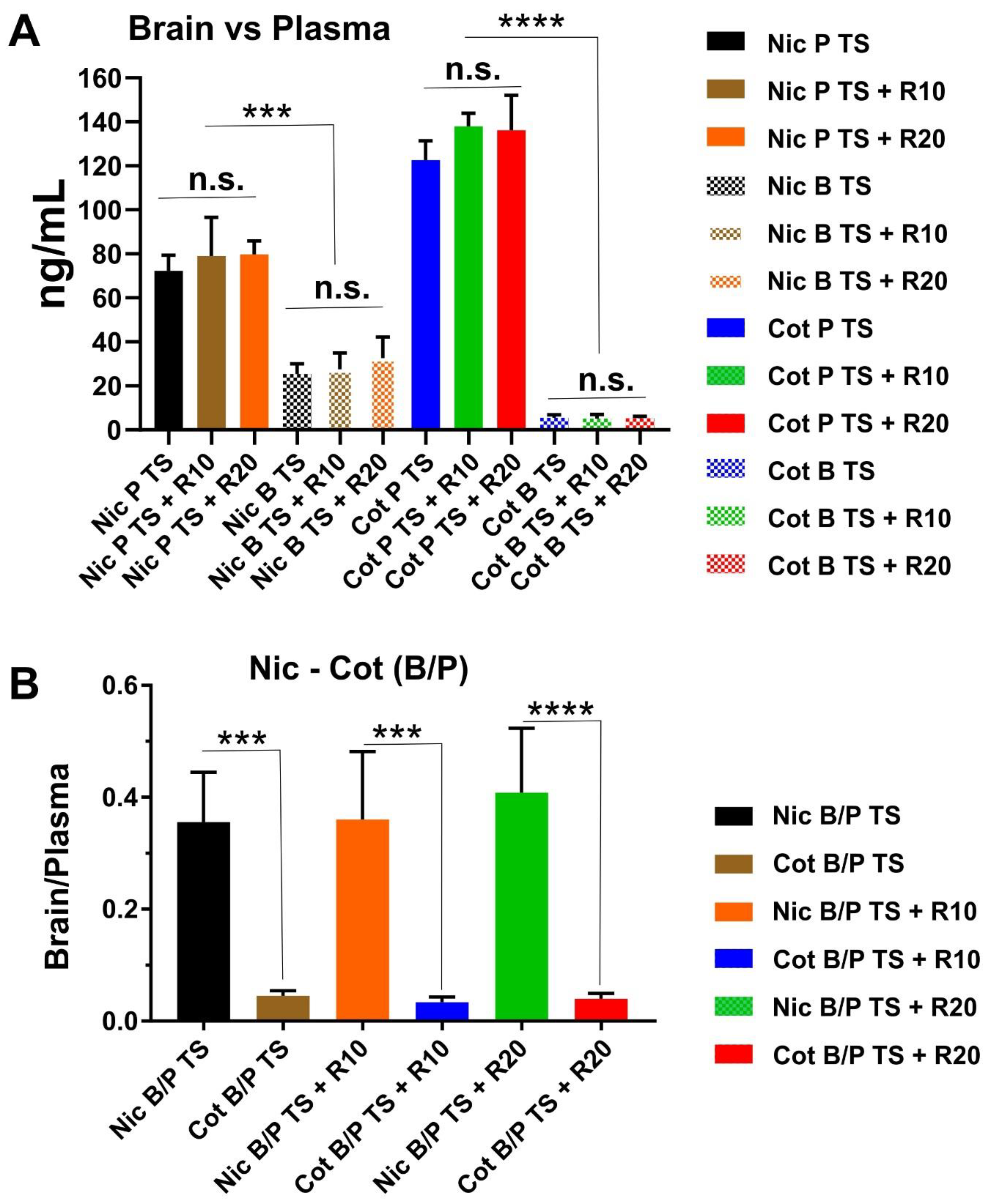
2.2. Result for Nicotine and Cotinine Measurements
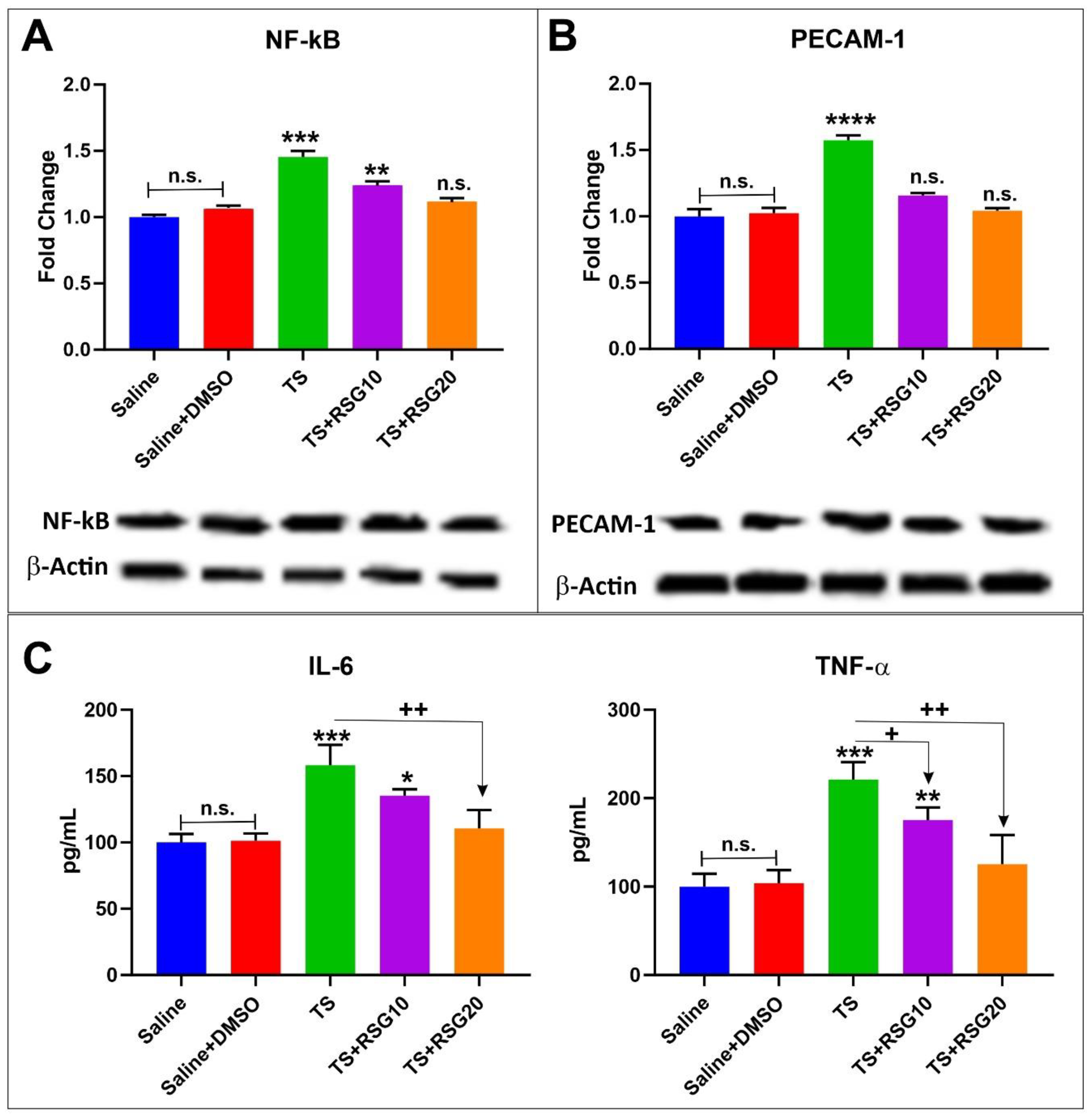
2.3. Upregulation of PPARγ, NRF2—and Its Downstream Effectors NQO-1 and HO-1’s Expressions in a Dose-Dependent Manner
2.4. RSG Decreases TS-Induced Loss of Blood–Brain Barrier Integrity
2.5. Decreased Pro-Inflammatory Effect of TS Exposure by RSG
3. Discussion
4. Materials and Methods
4.1. In Vivo Experimental Design
4.2. Materials and Reagents
4.3. Drug Administration
4.4. Tissue Preparation
4.5. Preparation of Protein Extracts and Western Blotting
4.6. ELISA
4.7. Nicotine and Cotinine Measurements in Brain and Plasma.
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ARE | Antioxidant Response Element |
| BBB | Blood–brain barrier |
| CNS | Central nervous system |
| CS | Cigarette Smoke |
| 2DM | Type 2 Diabetes Mellitus |
| EC | Electronic Cigarette |
| FTC | Federal Trade Control |
| HG | Hyperglycemia |
| HO-1 | Heme Oxygenase 1 |
| ISO | International Organization for Standardization |
| Keap1 | Kelch-like ECH-associated protein 1 |
| NF-ĸB | Nuclear factor kappa-light chain-enhancer of activated B cells |
| NQO-1 | NAD(P)H: Quinone reductase I |
| Nrf2 | Nuclear factor erythroid 2-related factor |
| OS | Oxidative stress |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PECAM-1 | Platelet Endothelial Cell Adhesion Molecule-1 |
| ROS | Reactive Oxygen Species |
| RSG | Rosiglitazone |
| SCSM | Single Cigarette Smoking Machine |
| TJ | Tight Junction |
| TS | Tobacco smoke |
| TZD | Thiazolidinediones |
| ZO-1 | Zonula occludens-1 |
References
- Mazzone, P.; Tierney, W.; Hossain, M.; Puvenna, V.; Janigro, D.; Cucullo, L. Pathophysiological Impact of Cigarette Smoke Exposure on the Cerebrovascular System with a Focus on the Blood-brain Barrier: Expanding the Awareness of Smoking Toxicity in an Underappreciated Area. Int. J. Environ. Res. Public Health 2010, 7, 4111–4126. [Google Scholar] [CrossRef]
- Sivandzade, F.; Cucullo, L. Assessing the protective effect of rosiglitazone against electronic cigarette/tobacco smoke-induced blood–brain barrier impairment. BMC Neurosci. 2019, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Organization, W.H. Who Report on the Global Tobacco Epidemic, 2013: Enforcing Bans on Tobacco Advertising, Promotion and Sponsorship: Executive Summary; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- US Department of Health and Human Services. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General; US Department of Health and Human Services, Centers for Disease: Atlanta, GA, USA, 2014.
- Paulson, J.R.; Yang, T.; Selvaraj, P.K.; Mdzinarishvili, A.; Van der Schyf, C.J.; Klein, J.; Bickel, U.; Abbruscato, T.J. Nicotine exacerbates brain edema during in vitro and in vivo focal ischemic conditions. J. Pharmacol. Exp. Ther. 2010, 332, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.; Fofaria, N.; Prasad, S.; Sajja, R.K.; Weksler, B.; Couraud, P.O.; Romero, I.A.; Cucullo, L. Oxidative and pro-inflammatory impact of regular and denicotinized cigarettes on blood brain barrier endothelial cells: Is smoking reduced or nicotine-free products really safe? BMC Neurosci. 2014, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Iida, M.; Iida, H.; Dohi, S.; Takenaka, M.; Fujiwara, H.; Traystman, R.J. Mechanisms Underlying Cerebrovascular Effects of Cigarette Smoking in Rats in Vivo. Stroke 1998, 29, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Ichiki, T. Collaboration between smokers and tobacco in endothelial dysfunction. Cardiovasc. Res. 2011, 90, 395–396. [Google Scholar] [CrossRef][Green Version]
- Arnson, Y.; Shoenfeld, Y.; Amital, H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J. Autoimmun. 2010, 34, J258–J265. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, I.M.; Cojocaru, M.; Sapira, V.; Ionescu, A. Evaluation of oxidative stress in patients with acute ischemic stroke. Rom. J. Intern. Med. 2013, 51, 97–106. [Google Scholar]
- Cataldo, J.K.; Prochaska, J.J.; Glantz, S.A. Cigarette smoking is a risk factor for Alzheimer’s disease: An analysis controlling for tobacco industry affiliation. J. Alzheimers Dis. 2010, 19, 465–480. [Google Scholar] [CrossRef]
- Kaisar, M.A.; Sivandzade, F.; Bhalerao, A.; Cucullo, L. Conventional and electronic cigarettes dysregulate the expression of iron transporters and detoxifying enzymes at the brain vascular endothelium: In vivo evidence of a gender-specific cellular response to chronic cigarette smoke exposure. Neurosci. Lett. 2018, 682, 1–9. [Google Scholar] [CrossRef]
- Sajja, R.K.; Rahman, S.; Cucullo, L. Drugs of abuse and blood-brain barrier endothelial dysfunction: A focus on the role of oxidative stress. J. Cereb. Blood Flow Metab. 2016, 36, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Cucullo, L. In-vitro blood–brain barrier modeling: A review of modern and fast-advancing technologies. Br. J. Pharmacol. 2018, 38, 1667–1681. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.R.; Keller, J.N. Oxidative stress and cerebral endothelial cells: Regulation of the blood–brain-barrier and antioxidant based interventions. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2012, 1822, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; McCaffrey, G.; Quigley, C.E.; Finch, J.; Demarco, K.M.; Nametz, N.; Davis, T.P. Oxidative stress increases blood–brain barrier permeability and induces alterations in occludin during hypoxia–reoxygenation. Br. J. Pharmacol. 2010, 30, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Martinez, J.; Miller, B. Cerebral microvascular endothelium and the pathogenesis of neurodegenerative diseases. Expert Rev. Mol. Med. 2011, 13, e19. [Google Scholar] [CrossRef] [PubMed]
- Chrissobolis, S. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front. Biosci. 2011, 16, 1733–1745. [Google Scholar] [CrossRef]
- Sajja, R.K.; Naik, P.; Cucullo, L. Differential Cerebrovascular Toxicity of Various Tobacco Products: A Regulatory Perspective. J. Pharmacovigil. 2015, 3, 1000e130. [Google Scholar]
- Prasad, S.; Sajja, R.K.; Park, J.H.; Naik, P.; Kaisar, M.A.; Cucullo, L. Impact of cigarette smoke extract and hyperglycemic conditions on blood–brain barrier endothelial cells. Fluids Barriers CNS 2015, 12, 18. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Sajja, R.K.; Green, K.N.; Cucullo, L. Altered Nrf2 Signaling Mediates Hypoglycemia-Induced Blood–Brain Barrier Endothelial Dysfunction in Vitro. PLoS ONE 2015, 10, e0122358. [Google Scholar] [CrossRef]
- Malinowski, J.M.; Bolesta, S. Rosiglitazone in the treatment of type 2 diabetes mellitus: A critical review. Clin. Ther. 2000, 22, 1151–1168. [Google Scholar] [CrossRef]
- Ceolotto, G.; Gallo, A.; Papparella, I.; Franco, L.; Murphy, E.; Iori, E.; Pagnin, E.; Fadini, G.P.; Albiero, M.; Semplicini, A.; et al. Rosiglitazone Reduces Glucose-Induced Oxidative Stress Mediated by NAD(P)H Oxidase via AMPK-Dependent Mechanism. Arter. Thromb. Vasc. Biol. 2007, 27, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, R.; Toral, M.; Gómez-Guzmán, M.; Romero, M.; Sánchez, M.; Mahmoud, A.M.; Duarte, J. The Role of Nrf2 Signaling in PPARβ/δ-Mediated Vascular Protection against Hyperglycemia-Induced Oxidative Stress. Oxidative Med. Cell. Longev. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kadam, L.; Gomez-Lopez, N.; Mial, T.N.; Kohan-Ghadr, H.-R.; Drewlo, S. Rosiglitazone Regulates TLR4 and Rescues HO-1 and NRF2 Expression in Myometrial and Decidual Macrophages in Inflammation-Induced Preterm Birth. Reprod. Sci. 2017, 24, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Z.; Liu, J.Z.; Hu, J.X.; Chen, H.L.; Li, W.L.; Hai, C.X. Double antioxidant activities of rosiglitazone against high glucose-induced oxidative stress in hepatocyte. Toxicol. Vitr. 2011, 25, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Kaisar, M.A.; Kallem, R.R.; Sajja, R.K.; Sifat, A.E.; Cucullo, L. A convenient UHPLC-MS/MS method for routine monitoring of plasma and brain levels of nicotine and cotinine as a tool to validate newly developed preclinical smoking model in mouse. BMC Neurosci. 2017, 18, 71. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Sajja, R.K.; Kaisar, M.A.; Park, J.H.; Villalba, H.; Liles, T.; Abbruscato, T.; Cucullo, L. Role of Nrf2 and protective effects of Metformin against tobacco smoke-induced cerebrovascular toxicity. Redox Biol. 2017, 12, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Kaisar, M.A.; Villalba, H.; Prasad, S.; Liles, T.; Sifat, A.E.; Sajja, R.K.; Abbruscato, T.J.; Cucullo, L. Offsetting the impact of smoking and e-cigarette vaping on the cerebrovascular system and stroke injury: Is Metformin a viable countermeasure? Redox Biol. 2017, 13, 353–362. [Google Scholar] [CrossRef]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. Nrf2 and nf-қb interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2018, 21, 101059. [Google Scholar] [CrossRef]
- Ma, Q.; He, X. Molecular Basis of Electrophilic and Oxidative Defense: Promises and Perils of Nrf2. Pharmacol. Rev. 2012, 64, 1055–1081. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H.; Alafuzoff, I. Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2012, 96, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacolgy 2014, 79, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Lin, C.-L.G. Oxidative damage to RNA: Mechanisms, consequences, and diseases. Cell. Mol. Life Sci. 2010, 67, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Cerebrovascular and Neurological Disorders: Protective Role of NRF2. Int. J. Mol. Sci. 2019, 20, 3433. [Google Scholar] [CrossRef] [PubMed]
- Tufekci, K.U.; Bayin, E.C.; Genc, S.; Genc, K. The Nrf2/ARE Pathway: A Promising Target to Counteract Mitochondrial Dysfunction in Parkinson’s Disease. Park. Dis. 2011, 2011, 314082. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.; Sajja, R.K.; Prasad, S.; Cucullo, L. Effect of full flavor and denicotinized cigarettes exposure on the brain microvascular endothelium: A microarray-based gene expression study using a human immortalized BBB endothelial cell line. BMC Neurosci. 2015, 16, 38. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, A.; Srivastava, S.; Siow, R.C.M.; Modo, M.; Fraser, P.A.; Mann, G.E. Targeting the Nrf2–Keap1 antioxidant defence pathway for neurovascular protection in stroke. J. Physiol. 2011, 589, 4125–4136. [Google Scholar] [CrossRef]
- Tanji, K.; Maruyama, A.; Odagiri, S.; Mori, F.; Itoh, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Keap1 Is Localized in Neuronal and Glial Cytoplasmic Inclusions in Various Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2013, 72, 18–28. [Google Scholar] [CrossRef]
- Heiss, E.H.; Schachner, D.; Zimmermann, K.; Dirsch, V.M. Glucose availability is a decisive factor for Nrf2-mediated gene expression. Redox Biol. 2013, 1, 359–365. [Google Scholar] [CrossRef]
- Aleksunes, L.M.; Reisman, S.A.; Yeager, R.L.; Goedken, M.J.; Klaassen, C.D. Nuclear Factor Erythroid 2-Related Factor 2 Deletion Impairs Glucose Tolerance and Exacerbates Hyperglycemia in Type 1 Diabetic Mice. J. Pharmacol. Exp. Ther. 2010, 333, 140–151. [Google Scholar] [CrossRef]
- Lee, J.E.; Park, J.H.; Jang, S.J.; Koh, H.C. Rosiglitazone inhibits chlorpyrifos-induced apoptosis via modulation of the oxidative stress and inflammatory response in SH-SY5Y cells. Toxicol. Appl. Pharmacol. 2014, 278, 159–171. [Google Scholar] [CrossRef]
- Sayan-Ozacmak, H.; Ozacmak, V.H.; Barut, F.; Jakubowska-Dogru, E. Rosiglitazone treatment reduces hippocampal neuronal damage possibly through alleviating oxidative stress in chronic cerebral hypoperfusion. Neurochem. Int. 2012, 61, 287–290. [Google Scholar] [CrossRef]
- Reddy, R.C.; Standiford, T.J. Nrf2 and PPARγ: Ppartnering against oxidant-induced lung injury. Am. J. Respir. Crit. Care Med. 2010, 182, 134–135. [Google Scholar] [CrossRef]
- Cho, H.Y.; Reddy, S.P.; DeBiase, A.; Yamamoto, M.; Kleeberger, S.R. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic. Biol. Med. 2005, 38, 325–343. [Google Scholar] [CrossRef]
- Cho, H.Y.; Gladwell, W.; Wang, X.; Chorley, B.; Bell, D.; Reddy, S.P.; Kleeberger, S.R. Nrf2-regulated PPARγ Expression Is Critical to Protection against Acute Lung Injury in Mice. Am. J. Respir. Crit. Care Med. 2010, 182, 170–182. [Google Scholar] [CrossRef]
- Elisia, I.; Nakamura, H.; Lam, V.; Hofs, E.; Cederberg, R.; Cait, J.; Hughes, M.R.; Lee, L.; Jia, W.; Adomat, H.H.; et al. DMSO Represses Inflammatory Cytokine Production from Human Blood Cells and Reduces Autoimmune Arthritis. PLoS ONE 2016, 11, e0152538. [Google Scholar] [CrossRef]
- Liebner, S.; Czupalla, C.J.; Wolburg, H. Current concepts of blood-brain barrier development. Int. J. Dev. Biol. 2011, 55, 467–476. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Carvalho, C.; Moreira, P.I. Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegenerative Events. Front. Physiol. 2018, 9, 806. [Google Scholar] [CrossRef]
- Popescu, B.O. Triggers and Effectors of Oxidative Stress at Blood-Brain Barrier Level: Relevance for Brain Ageing and Neurodegeneration. Oxid Med. Cell Longev. 2013, 2013, 297512. [Google Scholar] [CrossRef]
- De Backer, O.; Elinck, E.; Priem, E.; Leybaert, L.; Lefebvre, R.A. Peroxisome Proliferator-Activated Receptor γ Activation Alleviates Postoperative Ileus in Mice by Inhibition of Egr-1 Expression and Its Downstream Target Genes. J. Pharmacol. Exp. Ther. 2009, 331, 496–503. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group 1 | Group 2 | Group 3 | Group 4 | Group 5 | |
|---|---|---|---|---|---|
| Saline | ✓ | - | - | - | - |
| Saline + DMSO | - | ✓ | ✓ | ✓ | ✓ |
| RSG 10 mg/kg | - | - | - | ✓ | - |
| RSG 20 mg/kg | - | - | - | - | ✓ |
| TS exposure | - | - | ✓ | ✓ | ✓ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivandzade, F.; Cucullo, L. Anti-Diabetic Countermeasures Against Tobacco Smoke-Dependent Cerebrovascular Toxicity: Use and Effect of Rosiglitazone. Int. J. Mol. Sci. 2019, 20, 4225. https://doi.org/10.3390/ijms20174225
Sivandzade F, Cucullo L. Anti-Diabetic Countermeasures Against Tobacco Smoke-Dependent Cerebrovascular Toxicity: Use and Effect of Rosiglitazone. International Journal of Molecular Sciences. 2019; 20(17):4225. https://doi.org/10.3390/ijms20174225
Chicago/Turabian StyleSivandzade, Farzane, and Luca Cucullo. 2019. "Anti-Diabetic Countermeasures Against Tobacco Smoke-Dependent Cerebrovascular Toxicity: Use and Effect of Rosiglitazone" International Journal of Molecular Sciences 20, no. 17: 4225. https://doi.org/10.3390/ijms20174225
APA StyleSivandzade, F., & Cucullo, L. (2019). Anti-Diabetic Countermeasures Against Tobacco Smoke-Dependent Cerebrovascular Toxicity: Use and Effect of Rosiglitazone. International Journal of Molecular Sciences, 20(17), 4225. https://doi.org/10.3390/ijms20174225