Role of NF-κB in Platelet Function

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
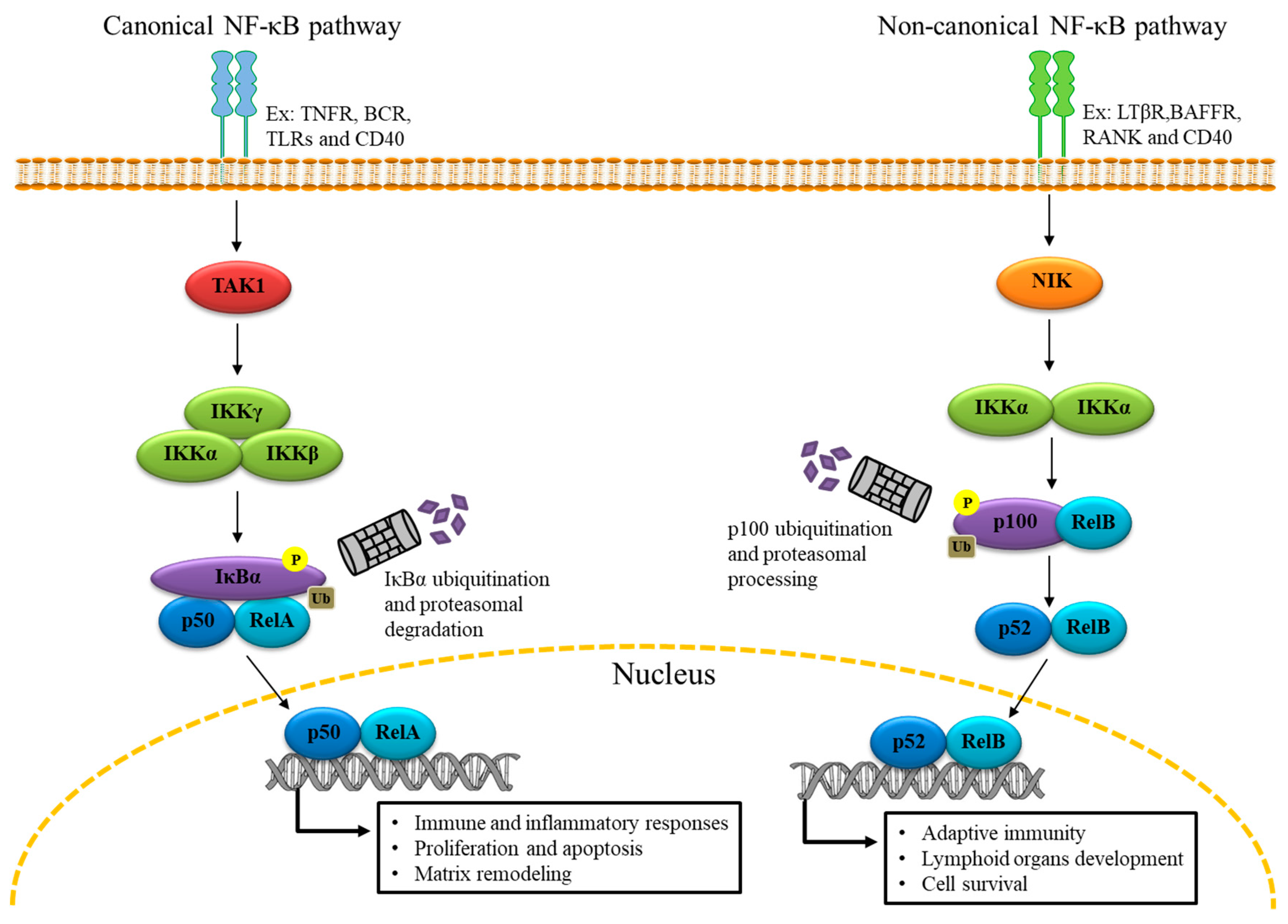
2. The Genomic Role of NF-κB
3. NF-κB Expression in Platelets
4. NF-κB Functions in Platelets
4.1. Platelet Survival and Apoptosis
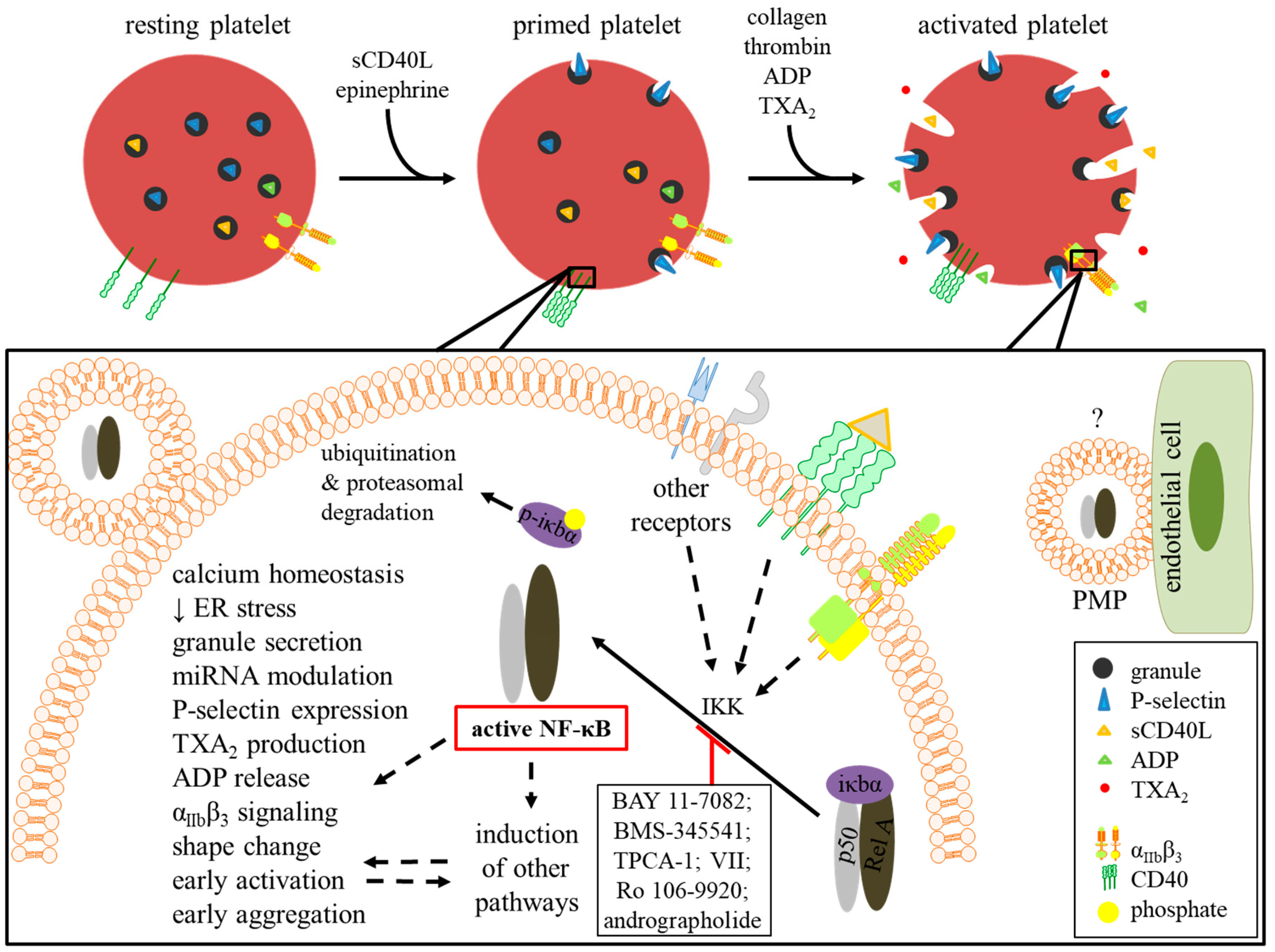
4.2. Platelet Activation and Priming
4.2.1. Thrombin-Activated Platelets
4.2.2. Collagen-Activated Platelets
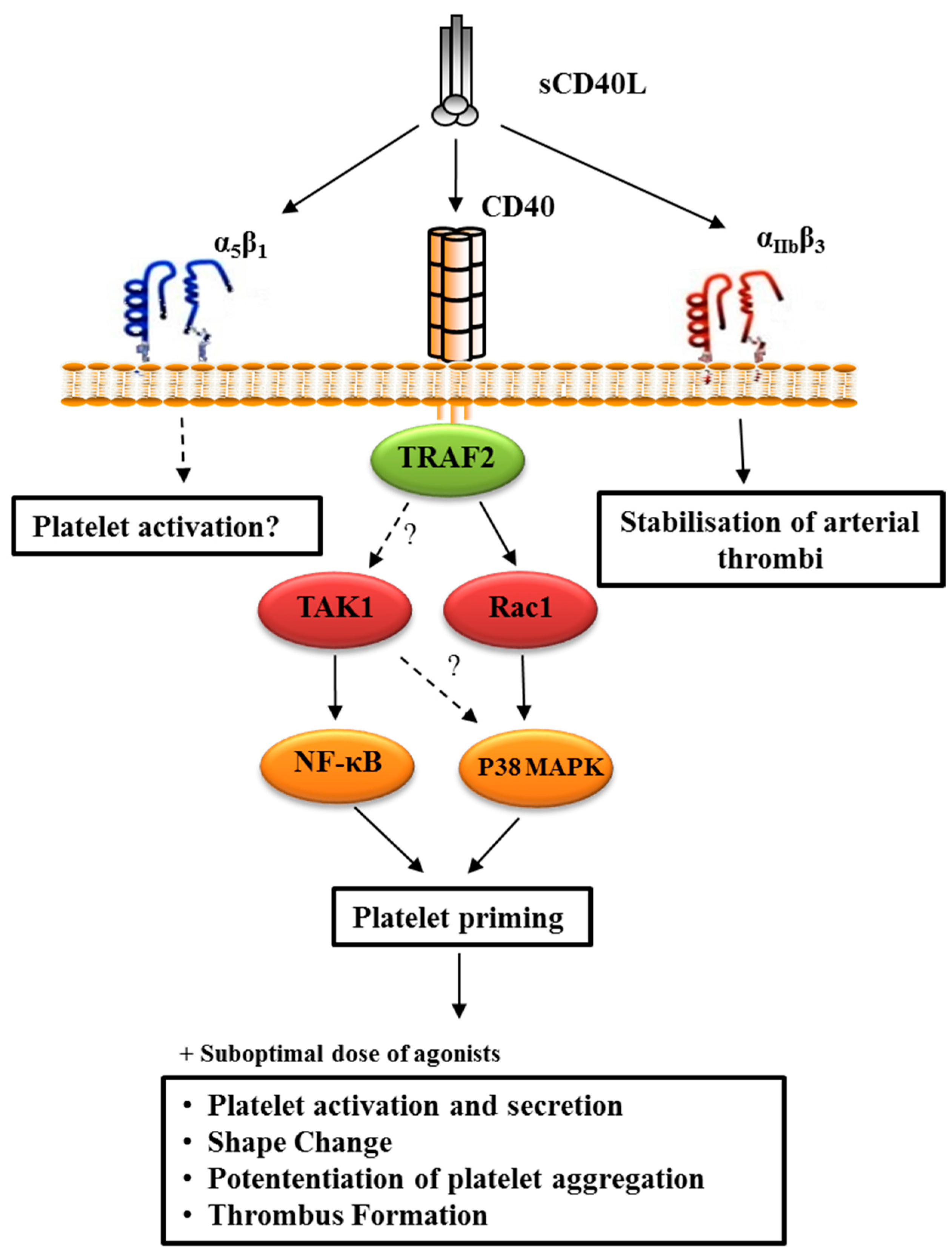
4.2.3. CD40L-Primed Platelets
4.2.4. TLR Ligand-Activated Platelets
4.2.5. AGE-Activated Platelets
4.2.6. Epinephrine-Primed Platelets
4.2.7. ADP-Activated Platelets
4.3. Platelet Aggregation
5. Natural/Pharmacological Compounds and NF-κB
6. The Interplay between mRNA, miRNA, and NF-κB in Platelets
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Balakumar, P.; Maung, U.K.; Jagadeesh, G. Prevalence and prevention of cardiovascular disease and diabetes mellitus. Pharm. Res. 2016, 113, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Gawaz, M. Platelets in the onset of atherosclerosis. Blood Cells Mol. Dis. 2006, 36, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, S.; Kramer, B.; Daub, K.; Stellos, K.; Gawaz, M. Molecular pathways used by platelets to initiate and accelerate atherogenesis. Curr. Opin. Lipidol. 2007, 18, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Duchene, J.; von Hundelshausen, P. Platelet-derived chemokines in atherosclerosis. Hamostaseologie 2015, 35, 137–141. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keating, F.K.; Whitaker, D.A.; Kabbani, S.S.; Ricci, M.A.; Sobel, B.E.; Schneider, D.J. Relation of augmented platelet reactivity to the magnitude of distribution of atherosclerosis. Am. J. Cardiol. 2004, 94, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, X.; Shi, X.; Zhu, M.; Wang, J.; Huang, S.; Huang, X.; Wang, H.; Li, L.; Deng, H.; et al. Platelet integrin alphaIIbbeta3: Signal transduction, regulation, and its therapeutic targeting. J Hematol. Oncol. 2019, 12, 26. [Google Scholar] [CrossRef]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef]
- Prescott, S.M.; McIntyre, T.M.; Zimmerman, G.A.; Stafforini, D.M. Inflammation as an early component of atherosclerosis and vascular damage--a role for P-selectin and platelet-activating factor. Jpn. Circ. J. 1996, 60, 137–141. [Google Scholar] [CrossRef][Green Version]
- Yacoub, D.; Hachem, A.; Theoret, J.F.; Gillis, M.A.; Mourad, W.; Merhi, Y. Enhanced levels of soluble CD40 ligand exacerbate platelet aggregation and thrombus formation through a CD40-dependent tumor necrosis factor receptor-associated factor-2/Rac1/p38 mitogen-activated protein kinase signaling pathway. Arter. Thromb. Vasc. Biol. 2010, 30, 2424–2433. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basilio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-kappaB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef]
- Arenzana-Seisdedos, F.; Turpin, P.; Rodriguez, M.; Thomas, D.; Hay, R.T.; Virelizier, J.L.; Dargemont, C. Nuclear localization of I kappa B alpha promotes active transport of NF-kappa B from the nucleus to the cytoplasm. J. Cell Sci. 1997, 110, 369–378. [Google Scholar]
- Tanaka, M.; Fuentes, M.E.; Yamaguchi, K.; Durnin, M.H.; Dalrymple, S.A.; Hardy, K.L.; Goeddel, D.V. Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity 1999, 10, 421–429. [Google Scholar] [CrossRef]
- Gilmore, T.D. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Baltimore, D. I kappa B: A specific inhibitor of the NF-kappa B transcription factor. Science 1988, 242, 540–546. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Baltimore, D. NF-kappa B: Ten years after. Cell 1996, 87, 13–20. [Google Scholar] [CrossRef]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef]
- Sebastien, G.B. Wired for eating. Med Sci (Paris) 2004, 20, 958–959. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Spinelli, S.L.; Casey, A.E.; Pollock, S.J.; Gertz, J.M.; McMillan, D.H.; Narasipura, S.D.; Mody, N.A.; King, M.R.; Maggirwar, S.B.; Francis, C.W.; et al. Platelets and megakaryocytes contain functional nuclear factor-kappaB. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 591–598. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Toulany, M.; Saki, M.; Rodemann, H.P.; Mrowietz, U.; Lang, F.; Wieder, T. Potential roles of the NFkappaB and glutathione pathways in mature human erythrocytes. Cell. ξ Mol. Biol. Lett. 2012, 17, 11–20. [Google Scholar] [CrossRef]
- Liu, F.; Morris, S.; Epps, J.; Carroll, R. Demonstration of an activation regulated NF-kappaB/I-kappaBalpha complex in human platelets. Thromb. Res. 2002, 106, 199–203. [Google Scholar] [CrossRef]
- Malaver, E.; Romaniuk, M.A.; D’Atri, L.P.; Pozner, R.G.; Negrotto, S.; Benzadon, R.; Schattner, M. NF-kappaB inhibitors impair platelet activation responses. J Thromb. Haemost. 2009, 7, 1333–1343. [Google Scholar] [CrossRef]
- Gambaryan, S.; Kobsar, A.; Rukoyatkina, N.; Herterich, S.; Geiger, J.; Smolenski, A.; Lohmann, S.M.; Walter, U. Thrombin and collagen induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of protein kinase A from an NFkappaB-IkappaB complex. J. Biol. Chem. 2010, 285, 18352–18363. [Google Scholar] [CrossRef]
- Lee, H.S.; Kim, S.D.; Lee, W.M.; Endale, M.; Kamruzzaman, S.M.; Oh, W.J.; Cho, J.Y.; Kim, S.K.; Cho, H.J.; Park, H.J.; et al. A noble function of BAY 11-7082: Inhibition of platelet aggregation mediated by an elevated cAMP-induced VASP, and decreased ERK2/JNK1 phosphorylations. Eur. J. Pharmacol. 2010, 627, 85–91. [Google Scholar] [CrossRef]
- Akbiyik, F.; Ray, D.M.; Gettings, K.F.; Blumberg, N.; Francis, C.W.; Phipps, R.P. Human bone marrow megakaryocytes and platelets express PPARgamma, and PPARgamma agonists blunt platelet release of CD40 ligand and thromboxanes. Blood 2004, 104, 1361–1368. [Google Scholar] [CrossRef]
- Ali, F.Y.; Davidson, S.J.; Moraes, L.A.; Traves, S.L.; Paul-Clark, M.; Bishop-Bailey, D.; Warner, T.D.; Mitchell, J.A. Role of nuclear receptor signaling in platelets: Antithrombotic effects of PPARbeta. FASEB J. 2006, 20, 326–328. [Google Scholar] [CrossRef]
- Chen, T.H.; Shih, C.Y.; Hsu, W.L.; Chou, T.C. Mechanisms of Nifedipine-Downregulated CD40L/sCD40L Signaling in Collagen Stimulated Human Platelets. PLoS ONE 2015, 10, e0127054. [Google Scholar] [CrossRef]
- Shih, C.Y.; Chou, T.C. The antiplatelet activity of magnolol is mediated by PPAR-beta/gamma. Biochem. Pharmacol. 2012, 84, 793–803. [Google Scholar] [CrossRef]
- Moraes, L.A.; Swales, K.E.; Wray, J.A.; Damazo, A.; Gibbins, J.M.; Warner, T.D.; Bishop-Bailey, D. Nongenomic signaling of the retinoid X receptor through binding and inhibiting Gq in human platelets. Blood 2007, 109, 3741–3744. [Google Scholar] [CrossRef]
- Ray, D.M.; Spinelli, S.L.; Pollock, S.J.; Murant, T.I.; O’Brien, J.J.; Blumberg, N.; Francis, C.W.; Taubman, M.B.; Phipps, R.P. Peroxisome proliferator-activated receptor gamma and retinoid X receptor transcription factors are released from activated human platelets and shed in microparticles. Thromb. Haemost. 2008, 99, 86–95. [Google Scholar] [CrossRef]
- Dhawan, L.; Liu, B.; Blaxall, B.C.; Taubman, M.B. A novel role for the glucocorticoid receptor in the regulation of monocyte chemoattractant protein-1 mRNA stability. J. Biol. Chem. 2007, 282, 10146–10152. [Google Scholar] [CrossRef]
- Moraes, L.A.; Paul-Clark, M.J.; Rickman, A.; Flower, R.J.; Goulding, N.J.; Perretti, M. Ligand-specific glucocorticoid receptor activation in human platelets. Blood 2005, 106, 4167–4175. [Google Scholar] [CrossRef]
- Lu, W.J.; Lin, K.C.; Huang, S.Y.; Thomas, P.A.; Wu, Y.H.; Wu, H.C.; Lin, K.H.; Sheu, J.R. Role of a Janus kinase 2-dependent signaling pathway in platelet activation. Thromb. Res. 2014, 133, 1088–1096. [Google Scholar] [CrossRef]
- Gnatenko, D.V.; Dunn, J.J.; McCorkle, S.R.; Weissmann, D.; Perrotta, P.L.; Bahou, W.F. Transcript profiling of human platelets using microarray and serial analysis of gene expression. Blood 2003, 101, 2285–2293. [Google Scholar] [CrossRef]
- Denis, M.M.; Tolley, N.D.; Bunting, M.; Schwertz, H.; Jiang, H.; Lindemann, S.; Yost, C.C.; Rubner, F.J.; Albertine, K.H.; Swoboda, K.J.; et al. Escaping the nuclear confines: Signal-dependent pre-mRNA splicing in anucleate platelets. Cell 2005, 122, 379–391. [Google Scholar] [CrossRef]
- Weyrich, A.S.; Schwertz, H.; Kraiss, L.W.; Zimmerman, G.A. Protein synthesis by platelets: Historical and new perspectives. J. Thromb. Haemost. 2009, 7, 241–246. [Google Scholar] [CrossRef]
- Rowley, J.W.; Oler, A.J.; Tolley, N.D.; Hunter, B.N.; Low, E.N.; Nix, D.A.; Yost, C.C.; Zimmerman, G.A.; Weyrich, A.S. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood 2011, 118, e101–111. [Google Scholar] [CrossRef]
- Zimmerman, G.A.; Weyrich, A.S. Signal-dependent protein synthesis by activated platelets: New pathways to altered phenotype and function. Arterioscler. Thromb. avascular Biol. 2008, 28, s17–24. [Google Scholar] [CrossRef]
- Rivadeneyra, L.; Carestia, A.; Etulain, J.; Pozner, R.G.; Fondevila, C.; Negrotto, S.; Schattner, M. Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB. Thromb. Res. 2014, 133, 235–243. [Google Scholar] [CrossRef]
- Lu, W.J.; Lin, K.H.; Hsu, M.J.; Chou, D.S.; Hsiao, G.; Sheu, J.R. Suppression of NF-kappaB signaling by andrographolide with a novel mechanism in human platelets: Regulatory roles of the p38 MAPK-hydroxyl radical-ERK2 cascade. Biochem. Pharmacol. 2012, 84, 914–924. [Google Scholar] [CrossRef]
- Lebois, M.; Josefsson, E.C. Regulation of platelet lifespan by apoptosis. Platelets 2016, 27, 497–504. [Google Scholar] [CrossRef]
- Dowling, M.R.; Josefsson, E.C.; Henley, K.J.; Hodgkin, P.D.; Kile, B.T. Platelet senescence is regulated by an internal timer, not damage inflicted by hits. Blood 2010, 116, 1776–1778. [Google Scholar] [CrossRef]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef]
- Nayak, M.K.; Kulkarni, P.P.; Dash, D. Regulatory role of proteasome in determination of platelet life span. J. Biol. Chem. 2013, 288, 6826–6834. [Google Scholar] [CrossRef]
- Brown, S.B.; Clarke, M.C.; Magowan, L.; Sanderson, H.; Savill, J. Constitutive death of platelets leading to scavenger receptor-mediated phagocytosis. A caspase-independent cell clearance program. J. Biol. Chem. 2000, 275, 5987–5996. [Google Scholar] [CrossRef]
- Guttridge, D.C.; Albanese, C.; Reuther, J.Y.; Pestell, R.G.; Baldwin, A.S., Jr. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol. 1999, 19, 5785–5799. [Google Scholar] [CrossRef]
- Ghosh, S.; Hayden, M.S. New regulators of NF-kappaB in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef]
- Piva, R.; Belardo, G.; Santoro, M.G. NF-kappaB: A stress-regulated switch for cell survival. Antioxid. Redox Signal. 2006, 8, 478–486. [Google Scholar] [CrossRef]
- Paul, M.; Kemparaju, K.; Girish, K.S. Inhibition of constitutive NF-kappaB activity induces platelet apoptosis via ER stress. Biochem. Biophys. Res. Commun. 2017, 493, 1471–1477. [Google Scholar] [CrossRef]
- Quach, M.E.; Chen, W.; Li, R. Mechanisms of platelet clearance and translation to improve platelet storage. Blood 2018, 131, 1512–1521. [Google Scholar] [CrossRef]
- Chen, W.; Liang, X.; Syed, A.K.; Jessup, P.; Church, W.R.; Ware, J.; Josephson, C.D.; Li, R. Inhibiting GPIbalpha Shedding Preserves Post-Transfusion Recovery and Hemostatic Function of Platelets After Prolonged Storage. Arter. Thromb. Vasc. Biol. 2016, 36, 1821–1828. [Google Scholar] [CrossRef]
- Chen, W.; Druzak, S.A.; Wang, Y.; Josephson, C.D.; Hoffmeister, K.M.; Ware, J.; Li, R. Refrigeration-Induced Binding of von Willebrand Factor Facilitates Fast Clearance of Refrigerated Platelets. Arter. Thromb. Vasc. Biol. 2017, 37, 2271–2279. [Google Scholar] [CrossRef]
- Bergmeier, W.; Piffath, C.L.; Cheng, G.; Dole, V.S.; Zhang, Y.; von Andrian, U.H.; Wagner, D.D. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates GPIbalpha shedding from platelets in vitro and in vivo. Circ. Res. 2004, 95, 677–683. [Google Scholar] [CrossRef]
- Deng, W.; Xu, Y.; Chen, W.; Paul, D.S.; Syed, A.K.; Dragovich, M.A.; Liang, X.; Zakas, P.; Berndt, M.C.; Di Paola, J.; et al. Platelet clearance via shear-induced unfolding of a membrane mechanoreceptor. Nat. Commun. 2016, 7, 12863. [Google Scholar] [CrossRef]
- Quach, M.E.; Dragovich, M.A.; Chen, W.; Syed, A.K.; Cao, W.; Liang, X.; Deng, W.; De Meyer, S.F.; Zhu, G.; Peng, J.; et al. Fc-independent immune thrombocytopenia via mechanomolecular signaling in platelets. Blood 2018, 131, 787–796. [Google Scholar] [CrossRef]
- Wei, S.; Wang, H.; Zhang, G.; Lu, Y.; An, X.; Ren, S.; Wang, Y.; Chen, Y.; White, J.G.; Zhang, C.; et al. Platelet IkappaB kinase-beta deficiency increases mouse arterial neointima formation via delayed glycoprotein Ibalpha shedding. Arter. Thromb. Vasc. Biol. 2013, 33, 241–248. [Google Scholar] [CrossRef]
- Malerba, M.; Clini, E.; Malagola, M.; Avanzi, G.C. Platelet activation as a novel mechanism of atherothrombotic risk in chronic obstructive pulmonary disease. Expert. Rev. Hematol. 2013, 6, 475–483. [Google Scholar] [CrossRef]
- Zucker, M.B.; Nachmias, V.T. Platelet activation. Arteriosclerosis 1985, 5, 2–18. [Google Scholar] [CrossRef]
- Nurden, A.T. Platelets, inflammation and tissue regeneration. Thromb. Haemost. 2011, 105, S13–33. [Google Scholar] [CrossRef]
- Flevaris, P.; Li, Z.; Zhang, G.; Zheng, Y.; Liu, J.; Du, X. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood 2009, 113, 893–901. [Google Scholar] [CrossRef]
- Karim, Z.A.; Zhang, J.; Banerjee, M.; Chicka, M.C.; Al Hawas, R.; Hamilton, T.R.; Roche, P.A.; Whiteheart, S.W. IkappaB kinase phosphorylation of SNAP-23 controls platelet secretion. Blood 2013, 121, 4567–4574. [Google Scholar] [CrossRef]
- Burke, J.R.; Pattoli, M.A.; Gregor, K.R.; Brassil, P.J.; MacMaster, J.F.; McIntyre, K.W.; Yang, X.; Iotzova, V.S.; Clarke, W.; Strnad, J.; et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J. Biol. Chem. 2003, 278, 1450–1456. [Google Scholar] [CrossRef]
- Cataldi, M.; Shah, N.R.; Felt, S.A.; Grdzelishvili, V.Z. Breaking resistance of pancreatic cancer cells to an attenuated vesicular stomatitis virus through a novel activity of IKK inhibitor TPCA-1. Virology 2015, 485, 340–354. [Google Scholar] [CrossRef]
- Karim, Z.A.; Vemana, H.P.; Khasawneh, F.T. MALT1-ubiquitination triggers non-genomic NF-kappaB/IKK signaling upon platelet activation. PLoS ONE 2015, 10, e0119363. [Google Scholar] [CrossRef]
- Chen, B.C.; Chang, H.M.; Hsu, M.J.; Shih, C.M.; Chiu, Y.H.; Chiu, W.T.; Lin, C.H. Peptidoglycan induces cyclooxygenase-2 expression in macrophages by activating the neutral sphingomyelinase-ceramide pathway. J. Biol. Chem. 2009, 284, 20562–20573. [Google Scholar] [CrossRef]
- Chen, W.F.; Lee, J.J.; Chang, C.C.; Lin, K.H.; Wang, S.H.; Sheu, J.R. Platelet protease-activated receptor (PAR)4, but not PAR1, associated with neutral sphingomyelinase responsible for thrombin-stimulated ceramide-NF-kappaB signaling in human platelets. Haematologica 2013, 98, 793–801. [Google Scholar] [CrossRef]
- Aszodi, A.; Pfeifer, A.; Ahmad, M.; Glauner, M.; Zhou, X.H.; Ny, L.; Andersson, K.E.; Kehrel, B.; Offermanns, S.; Fassler, R. The vasodilator-stimulated phosphoprotein (VASP) is involved in cGMP- and cAMP-mediated inhibition of agonist-induced platelet aggregation, but is dispensable for smooth muscle function. EMBO J. 1999, 18, 37–48. [Google Scholar] [CrossRef]
- Wentworth, J.K.; Pula, G.; Poole, A.W. Vasodilator-stimulated phosphoprotein (VASP) is phosphorylated on Ser157 by protein kinase C-dependent and -independent mechanisms in thrombin-stimulated human platelets. Biochem. J. 2006, 393, 555–564. [Google Scholar] [CrossRef]
- Henn, V.; Steinbach, S.; Buchner, K.; Presek, P.; Kroczek, R.A. The inflammatory action of CD40 ligand (CD154) expressed on activated human platelets is temporally limited by coexpressed CD40. Blood 2001, 98, 1047–1054. [Google Scholar] [CrossRef]
- Viallard, J.F.; Solanilla, A.; Gauthier, B.; Contin, C.; Dechanet, J.; Grosset, C.; Moreau, J.F.; Praloran, V.; Nurden, P.; Pellegrin, J.L.; et al. Increased soluble and platelet-associated CD40 ligand in essential thrombocythemia and reactive thrombocytosis. Blood 2002, 99, 2612–2614. [Google Scholar] [CrossRef]
- Andre, P.; Nannizzi-Alaimo, L.; Prasad, S.K.; Phillips, D.R. Platelet-derived CD40L: The switch-hitting player of cardiovascular disease. Circulation 2002, 106, 896–899. [Google Scholar] [CrossRef]
- Fong, S.W.; Few, L.L.; See Too, W.C.; Khoo, B.Y.; Nik Ibrahim, N.N.; Yahaya, S.A.; Yusof, Z.; Mohd Ali, R.; Abdul Rahman, A.R.; Yvonne-Tee, G.B. Systemic and coronary levels of CRP, MPO, sCD40L and PlGF in patients with coronary artery disease. BMC Res. Notes 2015, 8, 679. [Google Scholar] [CrossRef]
- Heeschen, C.; Dimmeler, S.; Hamm, C.W.; van den Brand, M.J.; Boersma, E.; Zeiher, A.M.; Simoons, M.L.; Investigators, C.S. Soluble CD40 ligand in acute coronary syndromes. N. Engl. J. Med. 2003, 348, 1104–1111. [Google Scholar] [CrossRef]
- Varo, N.; Vicent, D.; Libby, P.; Nuzzo, R.; Calle-Pascual, A.L.; Bernal, M.R.; Fernandez-Cruz, A.; Veves, A.; Jarolim, P.; Varo, J.J.; et al. Elevated plasma levels of the atherogenic mediator soluble CD40 ligand in diabetic patients: A novel target of thiazolidinediones. Circulation 2003, 107, 2664–2669. [Google Scholar] [CrossRef]
- de Lemos, J.A.; Zirlik, A.; Schonbeck, U.; Varo, N.; Murphy, S.A.; Khera, A.; McGuire, D.K.; Stanek, G.; Lo, H.S.; Nuzzo, R.; et al. Associations between soluble CD40 ligand, atherosclerosis risk factors, and subclinical atherosclerosis: Results from the Dallas Heart Study. Arter. Thromb. Vasc. Biol. 2005, 25, 2192–2196. [Google Scholar] [CrossRef]
- Sanguigni, V.; Pignatelli, P.; Lenti, L.; Ferro, D.; Bellia, A.; Carnevale, R.; Tesauro, M.; Sorge, R.; Lauro, R.; Violi, F. Short-term treatment with atorvastatin reduces platelet CD40 ligand and thrombin generation in hypercholesterolemic patients. Circulation 2005, 111, 412–419. [Google Scholar] [CrossRef]
- Varo, N.; Libby, P.; Nuzzo, R.; Italiano, J.; Doria, A.; Schonbeck, U. Elevated release of sCD40L from platelets of diabetic patients by thrombin, glucose and advanced glycation end products. Diab. Vasc. Dis. Res. 2005, 2, 81–87. [Google Scholar] [CrossRef]
- Cipollone, F.; Chiarelli, F.; Davi, G.; Ferri, C.; Desideri, G.; Fazia, M.; Iezzi, A.; Santilli, F.; Pini, B.; Cuccurullo, C.; et al. Enhanced soluble CD40 ligand contributes to endothelial cell dysfunction in vitro and monocyte activation in patients with diabetes mellitus: Effect of improved metabolic control. Diabetologia 2005, 48, 1216–1224. [Google Scholar] [CrossRef]
- Garlichs, C.D.; John, S.; Schmeisser, A.; Eskafi, S.; Stumpf, C.; Karl, M.; Goppelt-Struebe, M.; Schmieder, R.; Daniel, W.G. Upregulation of CD40 and CD40 ligand (CD154) in patients with moderate hypercholesterolemia. Circulation 2001, 104, 2395–2400. [Google Scholar] [CrossRef]
- Varo, N.; de Lemos, J.A.; Libby, P.; Morrow, D.A.; Murphy, S.A.; Nuzzo, R.; Gibson, C.M.; Cannon, C.P.; Braunwald, E.; Schonbeck, U. Soluble CD40L: Risk prediction after acute coronary syndromes. Circulation 2003, 108, 1049–1052. [Google Scholar] [CrossRef]
- Andre, P.; Prasad, K.S.; Denis, C.V.; He, M.; Papalia, J.M.; Hynes, R.O.; Phillips, D.R.; Wagner, D.D. CD40L stabilizes arterial thrombi by a beta3 integrin--dependent mechanism. Nat. Med. 2002, 8, 247–252. [Google Scholar] [CrossRef]
- Leveille, C.; Bouillon, M.; Guo, W.; Bolduc, J.; Sharif-Askari, E.; El-Fakhry, Y.; Reyes-Moreno, C.; Lapointe, R.; Merhi, Y.; Wilkins, J.A.; et al. CD40 ligand binds to alpha5beta1 integrin and triggers cell signaling. J. Biol. Chem. 2007, 282, 5143–5151. [Google Scholar] [CrossRef]
- Zirlik, A.; Maier, C.; Gerdes, N.; MacFarlane, L.; Soosairajah, J.; Bavendiek, U.; Ahrens, I.; Ernst, S.; Bassler, N.; Missiou, A.; et al. CD40 ligand mediates inflammation independently of CD40 by interaction with Mac-1. Circulation 2007, 115, 1571–1580. [Google Scholar] [CrossRef]
- Michel, N.A.; Zirlik, A.; Wolf, D. CD40L and Its Receptors in Atherothrombosis-An Update. Front. Cardiovasc. Med. 2017, 4, 40. [Google Scholar] [CrossRef]
- Brass, L.F.; Zhu, L.; Stalker, T.J. Novel therapeutic targets at the platelet vascular interface. Arter. Thromb. Vasc. Biol. 2008, 28, s43–50. [Google Scholar] [CrossRef]
- Portillo, J.A.; Greene, J.A.; Schwartz, I.; Subauste, M.C.; Subauste, C.S. Blockade of CD40-TRAF2,3 or CD40-TRAF6 is sufficient to inhibit pro-inflammatory responses in non-haematopoietic cells. Immunology 2015, 144, 21–33. [Google Scholar] [CrossRef]
- Mukundan, L.; Bishop, G.A.; Head, K.Z.; Zhang, L.; Wahl, L.M.; Suttles, J. TNF receptor-associated factor 6 is an essential mediator of CD40-activated proinflammatory pathways in monocytes and macrophages. J. Immunol. 2005, 174, 1081–1090. [Google Scholar] [CrossRef]
- Kim, C.; Ye, F.; Ginsberg, M.H. Regulation of integrin activation. Annu. Rev. Cell Dev. Biol. 2011, 27, 321–345. [Google Scholar] [CrossRef]
- Hassan, G.S.; Merhi, Y.; Mourad, W. CD40 ligand: A neo-inflammatory molecule in vascular diseases. Immunobiology 2012, 217, 521–532. [Google Scholar] [CrossRef]
- Kojok, K.; Akoum, S.E.; Mohsen, M.; Mourad, W.; Merhi, Y. CD40L Priming of Platelets via NF-kappaB Activation is CD40- and TAK1-Dependent. J Am Heart Assoc 2018, 7, e03677. [Google Scholar] [CrossRef]
- Kuijpers, M.J.; Mattheij, N.J.; Cipolla, L.; van Geffen, J.P.; Lawrence, T.; Donners, M.M.; Boon, L.; Lievens, D.; Torti, M.; Noels, H.; et al. Platelet CD40L Modulates Thrombus Growth Via Phosphatidylinositol 3-Kinase beta, and Not Via CD40 and IkappaB Kinase alpha. Arter. Thromb. Vasc. Biol. 2015, 35, 1374–1381. [Google Scholar] [CrossRef]
- Inwald, D.P.; McDowall, A.; Peters, M.J.; Callard, R.E.; Klein, N.J. CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ. Res. 2003, 92, 1041–1048. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Varghese, S.; Vitseva, O.; Tanriverdi, K.; Freedman, J.E. CD40 ligand influences platelet release of reactive oxygen intermediates. Arter. Thromb. Vasc. Biol. 2005, 25, 2428–2434. [Google Scholar] [CrossRef]
- Danese, S.; de la Motte, C.; Reyes, B.M.; Sans, M.; Levine, A.D.; Fiocchi, C. Cutting edge: T cells trigger CD40-dependent platelet activation and granular RANTES release: A novel pathway for immune response amplification. J. Immunol. 2004, 172, 2011–2015. [Google Scholar] [CrossRef]
- Hachem, A.; Yacoub, D.; Zaid, Y.; Mourad, W.; Merhi, Y. Involvement of nuclear factor kappaB in platelet CD40 signaling. Biochem. Biophys. Res. Commun. 2012, 425, 58–63. [Google Scholar] [CrossRef]
- Beaulieu, L.M.; Freedman, J.E. The role of inflammation in regulating platelet production and function: Toll-like receptors in platelets and megakaryocytes. Thromb. Res. 2010, 125, 205–209. [Google Scholar] [CrossRef]
- Cognasse, F.; Hamzeh, H.; Chavarin, P.; Acquart, S.; Genin, C.; Garraud, O. Evidence of Toll-like receptor molecules on human platelets. Immunol. Cell Biol. 2005, 83, 196–198. [Google Scholar] [CrossRef]
- Stark, R.J.; Aghakasiri, N.; Rumbaut, R.E. Platelet-derived Toll-like receptor 4 (Tlr-4) is sufficient to promote microvascular thrombosis in endotoxemia. PloS ONE 2012, 7, e41254. [Google Scholar] [CrossRef]
- Zhang, G.; Han, J.; Welch, E.J.; Ye, R.D.; Voyno-Yasenetskaya, T.A.; Malik, A.B.; Du, X.; Li, Z. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J. Immunol. 2009, 182, 7997–8004. [Google Scholar] [CrossRef]
- Liu, S.F.; Malik, A.B. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L622–L645. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Scott, T.; Owens, M.D. Thrombocytes respond to lipopolysaccharide through Toll-like receptor-4, and MAP kinase and NF-kappaB pathways leading to expression of interleukin-6 and cyclooxygenase-2 with production of prostaglandin E2. Mol. Immunol. 2008, 45, 1001–1008. [Google Scholar] [CrossRef]
- Winkler, C.; Ferdous, F.; Dimmick, M.; Scott, T. Lipopolysaccharide induced Interleukin-6 production is mediated through activation of ERK 1/2, p38 MAPK, MEK, and NFkappaB in chicken thrombocytes. Dev. Comp. Immunol. 2017, 73, 124–130. [Google Scholar] [CrossRef]
- Hegab, Z.; Gibbons, S.; Neyses, L.; Mamas, M.A. Role of advanced glycation end products in cardiovascular disease. World J. Cardiol. 2012, 4, 90–102. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar]
- Gawlowski, T.; Stratmann, B.; Ruetter, R.; Buenting, C.E.; Menart, B.; Weiss, J.; Vlassara, H.; Koschinsky, T.; Tschoepe, D. Advanced glycation end products strongly activate platelets. Eur. J. Nutr. 2009, 48, 475–481. [Google Scholar] [CrossRef]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, M.J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003, 370, 1097–1109. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef]
- Yeh, C.H.; Sturgis, L.; Haidacher, J.; Zhang, X.N.; Sherwood, S.J.; Bjercke, R.J.; Juhasz, O.; Crow, M.T.; Tilton, R.G.; Denner, L. Requirement for p38 and p44/p42 mitogen-activated protein kinases in RAGE-mediated nuclear factor-kappaB transcriptional activation and cytokine secretion. Diabetes 2001, 50, 1495–1504. [Google Scholar] [CrossRef]
- Kramer, R.M.; Roberts, E.F.; Strifler, B.A.; Johnstone, E.M. Thrombin induces activation of p38 MAP kinase in human platelets. J. Biol. Chem. 1995, 270, 27395–27398. [Google Scholar] [CrossRef]
- Simons, F.E. First-aid treatment of anaphylaxis to food: Focus on epinephrine. J. Allergy Clin. Immunol. 2004, 113, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.F.; Lockey, R.F.; Simons, F.E.; World Allergy Organization ad hoc Committee on Epinephrine in Anaphylaxis. Epinephrine: The drug of choice for anaphylaxis-a statement of the world allergy organization. World Allergy Organ J 2008, 1, S18–26. [Google Scholar] [CrossRef] [PubMed]
- Lanza, F.; Beretz, A.; Stierle, A.; Hanau, D.; Kubina, M.; Cazenave, J.P. Epinephrine potentiates human platelet activation but is not an aggregating agent. Am. J. Physiol. 1988, 255, H1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Murugappa, S.; Kunapuli, S.P. The role of ADP receptors in platelet function. Front. Biosci. 2006, 11, 1977–1986. [Google Scholar] [CrossRef]
- Puri, R.N.; Colman, R.W. ADP-induced platelet activation. Crit. Rev. Biochem. Mol. Biol. 1997, 32, 437–502. [Google Scholar] [CrossRef]
- Jin, J.; Quinton, T.M.; Zhang, J.; Rittenhouse, S.E.; Kunapuli, S.P. Adenosine diphosphate (ADP)-induced thromboxane A(2) generation in human platelets requires coordinated signaling through integrin alpha(IIb)beta(3) and ADP receptors. Blood 2002, 99, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Lee, J.J.; Chou, D.S.; Jayakumar, T.; Fong, T.H.; Hsiao, G.; Sheu, J.R. A novel role of andrographolide, an NF-kappa B inhibitor, on inhibition of platelet activation: The pivotal mechanisms of endothelial nitric oxide synthase/cyclic GMP. J. Mol. Med. (Berl) 2011, 89, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Schultz, M.; van der Poll, T. Sepsis and thrombosis. Semin. Thromb. Hemost. 2013, 39, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef]
- Hu, L.; Chang, L.; Zhang, Y.; Zhai, L.; Zhang, S.; Qi, Z.; Yan, H.; Yan, Y.; Luo, X.; Zhang, S.; et al. Platelets Express Activated P2Y12 Receptor in Patients With Diabetes Mellitus. Circulation 2017, 136, 817–833. [Google Scholar] [CrossRef]
- Moraes, L.A.; Spyridon, M.; Kaiser, W.J.; Jones, C.I.; Sage, T.; Atherton, R.E.; Gibbins, J.M. Non-genomic effects of PPARgamma ligands: Inhibition of GPVI-stimulated platelet activation. J. Thromb. Haemost. 2010, 8, 577–587. [Google Scholar] [CrossRef][Green Version]
- Schror, K. Aspirin and platelets: The antiplatelet action of aspirin and its role in thrombosis treatment and prophylaxis. Semin. Thromb. Hemost. 1997, 23, 349–356. [Google Scholar] [CrossRef]
- Kopp, E.; Ghosh, S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar] [CrossRef]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef]
- Wang, L.C.; Jiang, R.L.; Zhang, W.; Wei, L.L.; Yang, R.H. Effects of aspirin on the expression of nuclear factor-kappaB in a rat model of acute pulmonary embolism. World J. Emerg. Med. 2014, 5, 229–233. [Google Scholar] [CrossRef]
- Block, R.C.; Abdolahi, A.; Smith, B.; Meednu, N.; Thevenet-Morrison, K.; Cai, X.; Cui, H.; Mousa, S.; Brenna, J.T.; Georas, S. Effects of low-dose aspirin and fish oil on platelet function and NF-kappaB in adults with diabetes mellitus. Prostaglandins Leukot. Essent. Fatty Acids 2013, 89, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Sattler, K.J.; Woodrum, J.E.; Galili, O.; Olson, M.; Samee, S.; Meyer, F.B.; Zhu, X.Y.; Lerman, L.O.; Lerman, A. Concurrent treatment with renin-angiotensin system blockers and acetylsalicylic acid reduces nuclear factor kappaB activation and C-reactive protein expression in human carotid artery plaques. Stroke 2005, 36, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Kubisa, M.J.; Jezewski, M.P.; Gasecka, A.; Siller-Matula, J.M.; Postula, M. Ticagrelor - toward more efficient platelet inhibition and beyond. Ther. Clin. Risk Manag. 2018, 14, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Wu, X.Y.; Chen, J.L.; Chen, G.R.; Xu, J.; Gu, Y.; Song, H.P. Antiplatelet drug ticagrelor delays gastric ulcer healing in rats. Exp. Ther. Med. 2017, 14, 3774–3779. [Google Scholar] [CrossRef] [PubMed]
- Pels, K.; Schwimmbeck, P.L.; Rosenthal, P.; Loddenkemper, C.; Dang-Heine, C.; Rauch, U.; Martens, H.; Schultheiss, H.P.; Dechend, R.; Deiner, C. Long-term clopidogrel administration following severe coronary injury reduces proliferation and inflammation via inhibition of nuclear factor-kappaB and activator protein 1 activation in pigs. Eur. J. Clin. Invest. 2009, 39, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shakur, Y.; Yoshitake, M.; Kambayashi Ji, J. Cilostazol (pletal): A dual inhibitor of cyclic nucleotide phosphodiesterase type 3 and adenosine uptake. Cardiovasc. Drug Rev. 2001, 19, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Ohashi, W.; Tomita, K.; Hattori, K.; Matsuda, N.; Hattori, Y. Anti-inflammatory properties of cilostazol: Its interruption of DNA binding activity of NF-kappaB from the Toll-like receptor signaling pathways. Int. Immunopharmacol. 2018, 62, 120–131. [Google Scholar] [CrossRef]
- da Motta, N.A.; de Brito, F.C. Cilostazol exerts antiplatelet and anti-inflammatory effects through AMPK activation and NF-kB inhibition on hypercholesterolemic rats. Fundam. Clin. Pharmacol. 2016, 30, 327–337. [Google Scholar] [CrossRef]
- Chen, T.H.; Kao, Y.C.; Chen, B.C.; Chen, C.H.; Chan, P.; Lee, H.M. Dipyridamole activation of mitogen-activated protein kinase phosphatase-1 mediates inhibition of lipopolysaccharide-induced cyclooxygenase-2 expression in RAW 264.7 cells. Eur. J. Pharmacol. 2006, 541, 138–146. [Google Scholar] [CrossRef]
- Massaro, M.; Scoditti, E.; Carluccio, M.A.; Pellegrino, M.; Calabriso, N.; Storelli, C.; Martines, G.; De Caterina, R. Dipyridamole decreases inflammatory metalloproteinase-9 expression and release by human monocytes. Thromb. Haemost. 2013, 109, 280–289. [Google Scholar] [CrossRef]
- Gryka, R.J.; Buckley, L.F.; Anderson, S.M. Vorapaxar: The Current Role and Future Directions of a Novel Protease-Activated Receptor Antagonist for Risk Reduction in Atherosclerotic Disease. Drugs R D 2017, 17, 65–72. [Google Scholar] [CrossRef]
- Pang, J.; Hu, P.; Wang, J.; Jiang, J.; Lai, J. Vorapaxar stabilizes permeability of the endothelial barrier under cholesterol stimulation via the AKT/JNK and NFkappaB signaling pathways. Mol. Med. Rep. 2019, 19, 5291–5300. [Google Scholar] [CrossRef]
- Shimada, M.; Imano, H.; Fujiwara, A.; Hashimoto, T.; Kato, R.; Ijiri, Y.; Izumi, Y.; Yoshiyama, M.; Hayashi, T. Direct factor Xa inhibition prevents cardiac remodeling induced by intermittent hypoxia through PAR-1/2 dual signaling pathway. Eur. Heart J. 2017, 38. [Google Scholar] [CrossRef]
- Ma, J.; Li, X.; Wang, Y.; Yang, Z.; Luo, J. Rivaroxaban attenuates thrombosis by targeting the NF-kappaB signaling pathway in a rat model of deep venous thrombus. Int. J. Mol. Med. 2017, 40, 1869–1880. [Google Scholar] [CrossRef][Green Version]
- Hashikata, T.; Yamaoka-Tojo, M.; Namba, S.; Kitasato, L.; Kameda, R.; Murakami, M.; Niwano, H.; Shimohama, T.; Tojo, T.; Ako, J. Rivaroxaban Inhibits Angiotensin II-Induced Activation in Cultured Mouse Cardiac Fibroblasts Through the Modulation of NF-kappaB Pathway. Int. Heart J. 2015, 56, 544–550. [Google Scholar] [CrossRef]
- Shih, C.Y.; Lin, I.H.; Ding, J.C.; Chen, F.C.; Chou, T.C. Antiplatelet activity of nifedipine is mediated by inhibition of NF-kappaB activation caused by enhancement of PPAR-beta/-gamma activity. Br. J. Pharmacol. 2014, 171, 1490–1500. [Google Scholar] [CrossRef]
- Ali, F.Y.; Armstrong, P.C.; Dhanji, A.R.; Tucker, A.T.; Paul-Clark, M.J.; Mitchell, J.A.; Warner, T.D. Antiplatelet actions of statins and fibrates are mediated by PPARs. Arter. Thromb. Vasc. Biol. 2009, 29, 706–711. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef]
- Ishizuka, T.; Itaya, S.; Wada, H.; Ishizawa, M.; Kimura, M.; Kajita, K.; Kanoh, Y.; Miura, A.; Muto, N.; Yasuda, K. Differential effect of the antidiabetic thiazolidinediones troglitazone and pioglitazone on human platelet aggregation mechanism. Diabetes 1998, 47, 1494–1500. [Google Scholar] [CrossRef]
- Rudofsky, G.; Reismann, P.; Groener, J.B.; Djuric, Z.; Fleming, T.; Metzner, C.; Grafe, I.A.; Bierhaus, A.; Nawroth, P.P. Identical LDL-cholesterol lowering but non-identical effects on NF-kappaB activity: High dose simvastatin vs. combination therapy with ezetimibe. Atherosclerosis 2012, 223, 190–196. [Google Scholar] [CrossRef]
- Chang, C.C.; Lu, W.J.; Ong, E.T.; Chiang, C.W.; Lin, S.C.; Huang, S.Y.; Sheu, J.R. A novel role of sesamol in inhibiting NF-kappaB-mediated signaling in platelet activation. J. Biomed. Sci. 2011, 18, 93. [Google Scholar] [CrossRef] [PubMed]
- Abelson, K.S.; Hoglund, A.U. The effects of the alpha2-adrenergic receptor agonists clonidine and rilmenidine, and antagonists yohimbine and efaroxan, on the spinal cholinergic receptor system in the rat. Basic. Clin. Pharmacol. Toxicol. 2004, 94, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Glauert, H.P. Vitamin E and NF-kappaB activation: A review. Vitam. Horm. 2007, 76, 135–153. [Google Scholar] [CrossRef]
- Bugert, P.; Dugrillon, A.; Gunaydin, A.; Eichler, H.; Kluter, H. Messenger RNA profiling of human platelets by microarray hybridization. Thromb. Haemost. 2003, 90, 738–748. [Google Scholar]
- McRedmond, J.P.; Park, S.D.; Reilly, D.F.; Coppinger, J.A.; Maguire, P.B.; Shields, D.C.; Fitzgerald, D.J. Integration of proteomics and genomics in platelets: A profile of platelet proteins and platelet-specific genes. Mol. Cell. Proteomics. 2004, 3, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Landry, P.; Plante, I.; Ouellet, D.L.; Perron, M.P.; Rousseau, G.; Provost, P. Existence of a microRNA pathway in anucleate platelets. Nat. Struct. & Mol. Biol. 2009, 16, 961–966. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Nagalla, S.; Shaw, C.; Kong, X.; Kondkar, A.A.; Edelstein, L.C.; Ma, L.; Chen, J.; McKnight, G.S.; Lopez, J.A.; Yang, L.; et al. Platelet microRNA-mRNA coexpression profiles correlate with platelet reactivity. Blood 2011, 117, 5189–5197. [Google Scholar] [CrossRef]
- Ple, H.; Landry, P.; Benham, A.; Coarfa, C.; Gunaratne, P.H.; Provost, P. The repertoire and features of human platelet microRNAs. PLoS ONE 2012, 7, e50746. [Google Scholar] [CrossRef]
- Osman, A.; Falker, K. Characterization of human platelet microRNA by quantitative PCR coupled with an annotation network for predicted target genes. Platelets 2011, 22, 433–441. [Google Scholar] [CrossRef]
- Ma, X.; Becker Buscaglia, L.E.; Barker, J.R.; Li, Y. MicroRNAs in NF-kappaB signaling. J. Mol. Cell Biol. 2011, 3, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.; Beaulieu, L.M.; Vitseva, O.; Freedman, J.E. Platelets and platelet-like particles mediate intercellular RNA transfer. Blood 2012, 119, 6288–6295. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tan, M.; Xiang, Q.; Zhou, Z.; Yan, H. Thrombin-activated platelet-derived exosomes regulate endothelial cell expression of ICAM-1 via microRNA-223 during the thrombosis-inflammation response. Thromb. Res. 2017, 154, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Barry, O.P.; Pratico, D.; Savani, R.C.; FitzGerald, G.A. Modulation of monocyte-endothelial cell interactions by platelet microparticles. J. Clin. Invest. 1998, 102, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kojok, K.; El-Kadiry, A.E.-H.; Merhi, Y. Role of NF-κB in Platelet Function. Int. J. Mol. Sci. 2019, 20, 4185. https://doi.org/10.3390/ijms20174185
Kojok K, El-Kadiry AE-H, Merhi Y. Role of NF-κB in Platelet Function. International Journal of Molecular Sciences. 2019; 20(17):4185. https://doi.org/10.3390/ijms20174185
Chicago/Turabian StyleKojok, Kevin, Abed El-Hakim El-Kadiry, and Yahye Merhi. 2019. "Role of NF-κB in Platelet Function" International Journal of Molecular Sciences 20, no. 17: 4185. https://doi.org/10.3390/ijms20174185
APA StyleKojok, K., El-Kadiry, A. E.-H., & Merhi, Y. (2019). Role of NF-κB in Platelet Function. International Journal of Molecular Sciences, 20(17), 4185. https://doi.org/10.3390/ijms20174185