Characterization of Tachyplesin Peptides and Their Cyclized Analogues to Improve Antimicrobial and Anticancer Properties

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Results
2.1. Properties of Tachyplesin I–III and Their Cyclic Analogues
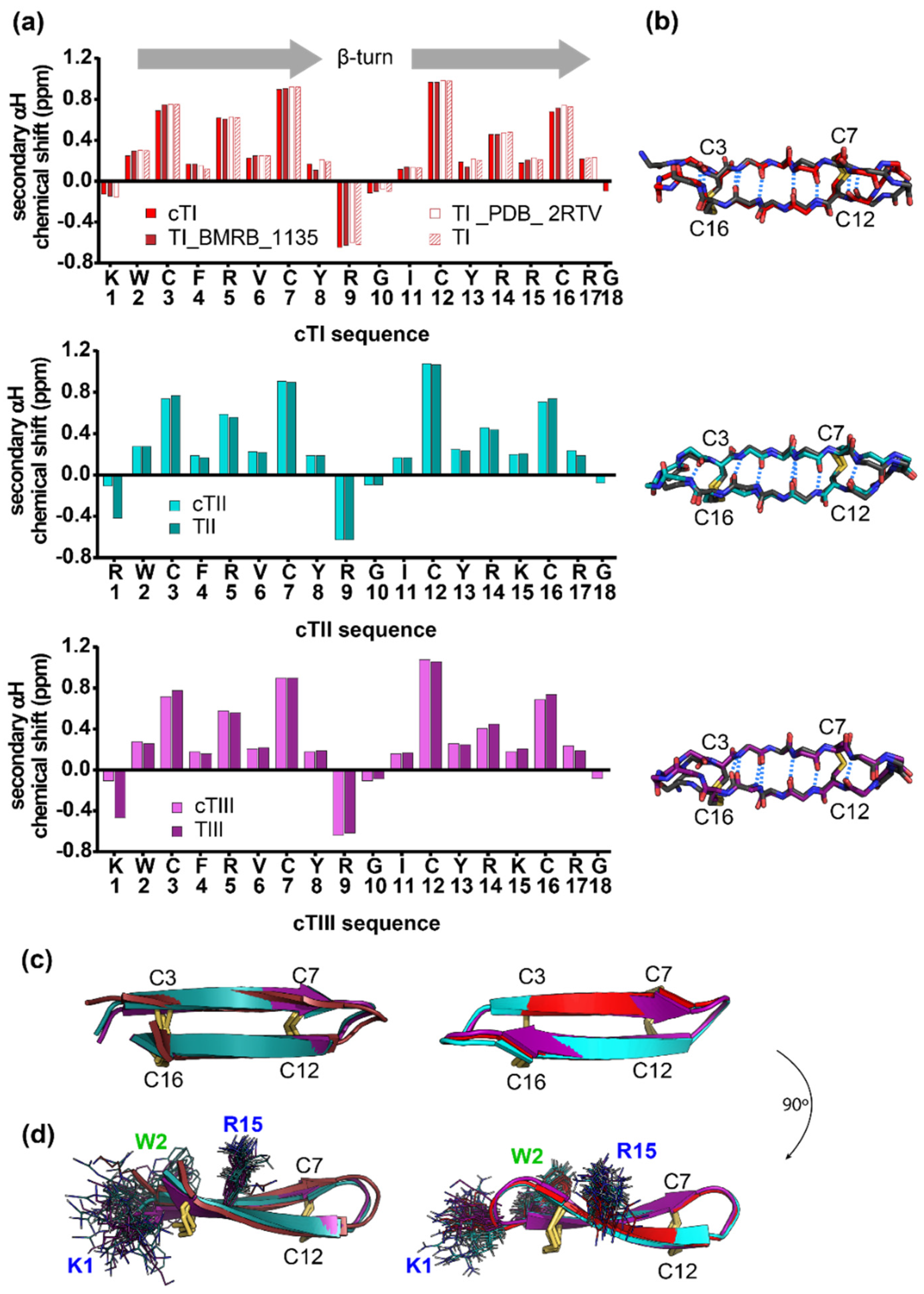
2.2. Structure of Tachyplesins and Backbone-Cyclised Analogues
2.3. Improved Stability and Reduced Hemolytic Activity of Cyclized Tachyplesin Peptides
2.4. Biological Activity of Tachyplesin I–III and of Their Cyclic Analogues
2.4.1. Activity against Bacteria
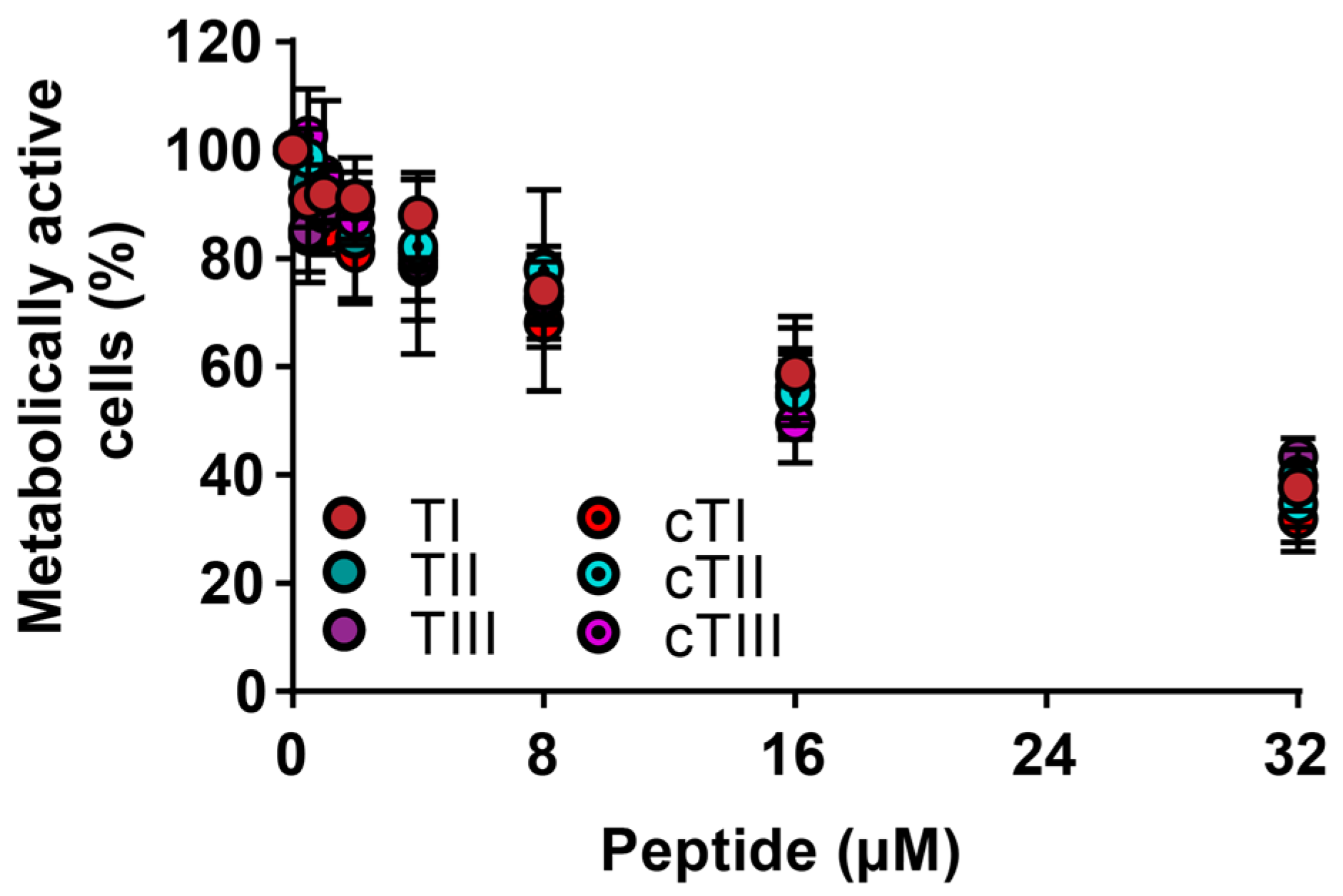
2.4.2. Activity against Cancerous Cells
2.5. Mechanistic Studies
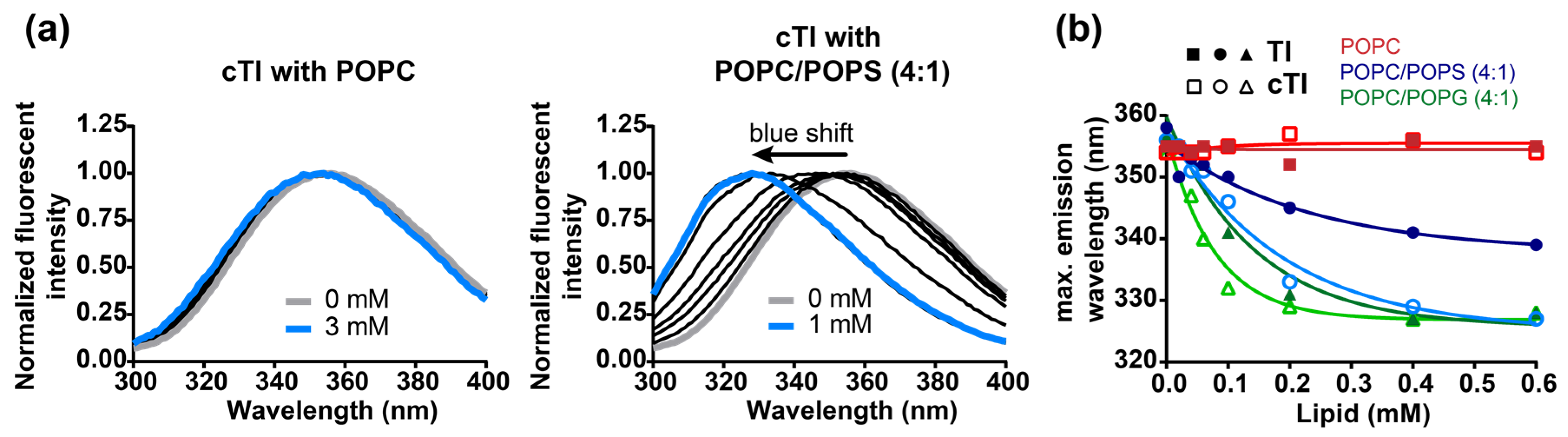
2.5.1. Peptide Binding to Model Membranes
2.5.2. Partitioning of Trp Residue into Model Membranes
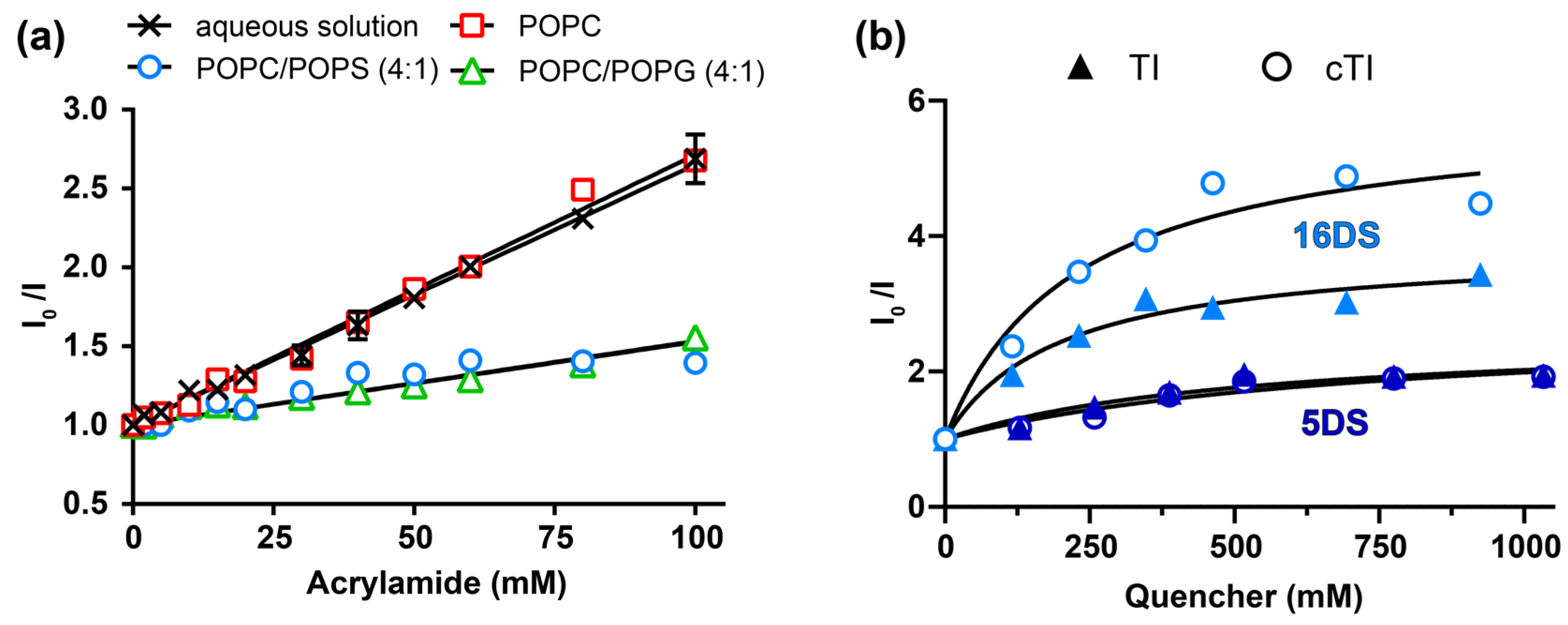
2.5.3. Insertion of Trp Residue into Model Membranes
3. Discussion
4. Material and Methods
4.1. Peptide Synthesis, Folding and Purification
4.2. NMR Spectroscopy
4.3. Serum Stability
4.4. Hemolytic Studies
4.5. Cell Culture
4.6. Cytotoxicity Assays
4.7. Antimicrobial Studies
4.8. Biofilm Studies
4.9. Lipid Vesicle Preparation
4.10. Fluorescence Spectroscopy Assays
4.11. Quenching of Tryptophan Fluorescence
4.12. Surface Plasmon Resonance (SPR)
4.13. Vesicle Leakage Assay
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1D | One-dimensional |
| 2-CTC | 2-chlorotrityl resin |
| 3D | Three-dimensional |
| ACN | Acetonitrile |
| ATI | Activity/toxicity index |
| ATTC | American Type Culture Collection |
| BMRB | Biological magnetic resonance data bank |
| CC | Cytotoxic concentration |
| CF | 5-carboxyfluorescein |
| cTI | Cyclic Tachyplesin I |
| cTII | Cyclic Tachyplesin II |
| cTIII | Cyclic Tachyplesin III |
| cTI–cTIII | Cyclic Tachyplesin I, II and III |
| DS | Doxyl-stearic acid |
| E.COSY | Exclusive correlation spectra |
| ESI-MS | Electrospray ionization mass spectroscopy |
| FBS | Fetal bovine serum |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| HC50 | Hemolytic concentration necessary for 50% lysis of RBCs |
| HDP | Host defense peptide |
| KSV | Stern – Volmer constant |
| LPS | Lipopolysaccharide |
| LUV | Large unilamellar vesicle |
| MCC | Minimal cytotoxic concentration |
| MHC | Minimal hemolytic concentration |
| MIC | Minimal inhibitory concentration |
| NMR | Nuclear magnetic resonance |
| NOESY | Nuclear Overhauser effect spectroscopy |
| PBS | Phosphate buffered saline |
| PDB | Protein data bank |
| PC | Phosphatidylcholine |
| PS | Phosphatidylserine |
| PG | Phosphatidylglycerol |
| P/L | Peptide-to-lipid ratio |
| POPC | 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine |
| POPS | 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine |
| POPG | 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) |
| RBCs | Red blood cells |
| RP-HPLC | Reverse-phase high-performance liquid chromatography |
| RT | Retention time |
| RU | Response unit |
| SEM | Standard error of mean |
| SPPS | Solid phase peptide synthesis |
| SPR | Surface plasmon resonance |
| SUV | Small unilamellar vesicle |
| TI | Tachyplesin I |
| TII | Tachyplesin II |
| TIII | Tachyplesin III |
| TI–TIII | Tachyplesin I, II and III |
| TFA | Trifluoroacetic acid |
| TOCSY | Total correlated spectroscopy |
References
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Zuegg, J.; Blaskovich, M.A.T.; Cooper, M.A. Contribution of Amphipathicity and Hydrophobicity to the Antimicrobial Activity and Cytotoxicity of β-Hairpin Peptides. ACS Infect. Dis. 2016, 2, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Tokunaga, F.; Yoneya, T.; Yoshikawa, K.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Antimicrobial peptides, isolated from horseshoe crab hemocytes, tachyplesin II, and polyphemusins I and II: chemical structures and biological activity. J. Biochem. 1989, 106, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Furunaka, H.; Miyata, T.; Tokunaga, F.; Muta, T.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Tachyplesin, a class of antimicrobial peptide from the hemocytes of the horseshoe crab (Tachypleus tridentatus). Isolation and chemical structure. J. Biol. Chem. 1988, 263, 16709–16713. [Google Scholar] [PubMed]
- Ohta, M.; Ito, H.; Masuda, K.; Tanaka, S.; Arakawa, Y.; Wacharotayankun, R.; Kato, N. Mechanisms of antibacterial action of tachyplesins and polyphemusins, a group of antimicrobial peptides isolated from horseshoe crab hemocytes. Antimicrob. Agents Chemother. 1992, 36, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Buri, M.V.; Torquato, H.F.V.; Barros, C.C.; Ide, J.S.; Miranda, A.; Paredes-Gamero, E.J. Comparison of Cytotoxic Activity in Leukemic Lineages Reveals Important Features of beta-Hairpin Antimicrobial Peptides. J. Cell. Biochem. 2017, 118, 1764–1773. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, X.M.; Underhill, C.B.; Yang, S.; Wang, L.; Chen, Y.; Hong, S.; Creswell, K.; Zhang, L. Tachyplesin activates the classic complement pathway to kill tumor cells. Cancer Res. 2005, 65, 4614–4622. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Jin, G.; Zhang, L.; Dai, J.; Dang, J.; Han, Y. Effects of tachyplesin I on human U251 glioma stem cells. Mol. Med. Rep. 2015, 11, 2953–2958. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, D.V.; Emel’yanova, A.A.; Kalashnikova, M.B.; Panteleev, P.V.; Ovchinnikova, T.V. In Vitro Study of Antitumor Effect of Antimicrobial Peptide Tachyplesin I in Combination with Cisplatin. Bull. Exp. Biol. Med. 2018, 165, 220–224. [Google Scholar] [CrossRef]
- Li, Q.F.; Ou Yang, G.L.; Li, C.Y.; Hong, S.G. Effects of tachyplesin on the morphology and ultrastructure of human gastric carcinoma cell line BGC-823. World J. Gastroenterol. 2000, 6, 676–680. [Google Scholar] [CrossRef]
- Li, X.; Dai, J.; Tang, Y.; Li, L.; Jin, G. Quantitative Proteomic Profiling of Tachyplesin I Targets in U251 Gliomaspheres. Mar. Drugs 2017, 15, 20. [Google Scholar] [CrossRef]
- Ouyang, G.L.; Li, Q.F.; Peng, X.X.; Liu, Q.R.; Hong, S.G. Effects of tachyplesin on proliferation and differentiation of human hepatocellular carcinoma SMMC-7721 cells. World J. Gastroenterol. 2002, 8, 1053–1058. [Google Scholar] [CrossRef]
- Paredes-Gamero, E.J.; Martins, M.N.; Cappabianco, F.A.; Ide, J.S.; Miranda, A. Characterization of dual effects induced by antimicrobial peptides: regulated cell death or membrane disruption. Biochim. Biophys. Acta 2012, 1820, 1062–1072. [Google Scholar] [CrossRef]
- Rothan, H.A.; Ambikabothy, J.; Ramasamy, T.S.; Rashid, N.N.; Yusof, R. A Preliminary Study in Search of Potential Peptide Candidates for a Combinational Therapy with Cancer Chemotherapy Drug. Int. J. Pept. Res. Ther. 2017, 25, 115–122. [Google Scholar] [CrossRef]
- Shi, S.L.; Wang, Y.Y.; Liang, Y.; Li, Q.F. Effects of tachyplesin and n-sodium butyrate on proliferation and gene expression of human gastric adenocarcinoma cell line BGC-823. World J. Gastroenterol. 2006, 12, 1694–1698. [Google Scholar] [CrossRef]
- Zhang, H.T.; Wu, J.; Zhang, H.F.; Zhu, Q.F. Efflux of potassium ion is an important reason of HL-60 cells apoptosis induced by tachyplesin. Acta Pharmacol. Sin. 2006, 27, 1367–1374. [Google Scholar] [CrossRef]
- Muta, T.; Fujimoto, T.; Nakajima, H.; Iwanaga, S. Tachyplesins Isolated from Hemocytes of Southeast Asian Horseshoe Crabs (Carcinoscorpius rotundicauda and Tachypleus gigas): Identification of a New Tachyplesin, Tachyplesin III, and a Processing Intermediate of Its Precursor1. J. Biochem. 1990, 108, 261–266. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Teixeira, V.; Feio, M.J.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef]
- Utsugi, T.; Schroit, A.J.; Connor, J.; Bucana, C.D.; Fidler, I.J. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991, 51, 3062–3066. [Google Scholar]
- Ran, S.; Downes, A.; Thorpe, P.E. Increased Exposure of Anionic Phospholipids on the Surface of Tumor Blood Vessels. Cancer Res. 2002, 62, 6132. [Google Scholar]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef]
- Riedl, S.; Rinner, B.; Asslaber, M.; Schaider, H.; Walzer, S.; Novak, A.; Lohner, K.; Zweytick, D. In search of a novel target - phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim. Biophys. Acta 2011, 1808, 2638–2645. [Google Scholar] [CrossRef]
- Iwasaki, T.; Ishibashi, J.; Tanaka, H.; Sato, M.; Asaoka, A.; Taylor, D.; Yamakawa, M. Selective cancer cell cytotoxicity of enantiomeric 9-mer peptides derived from beetle defensins depends on negatively charged phosphatidylserine on the cell surface. Peptides 2009, 30, 660–668. [Google Scholar] [CrossRef]
- Leite Natália, B.; Aufderhorst-Roberts, A.; Palma Mario, S.; Connell Simon, D.; Neto João, R.; Beales Paul, A. PE and PS Lipids Synergistically Enhance Membrane Poration by a Peptide with Anticancer Properties. Biophys. J. 2015, 109, 936–947. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Sugishita, K.; Fujii, N.; Miyajima, K. Molecular Basis for Membrane Selectivity of an Antimicrobial Peptide, Magainin 2. Biochemistry 1995, 34, 3423–3429. [Google Scholar] [CrossRef]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta 2008, 1778, 357–375. [Google Scholar] [CrossRef]
- Harris, F.; Dennison, S.R.; Singh, J.; Phoenix, D.A. On the selectivity and efficacy of defense peptides with respect to cancer cells. Med. Res. Rev. 2013, 33, 190–234. [Google Scholar] [CrossRef]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef]
- Matsuzaki, K. Why and how are peptide–lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta 1999, 1462, 1–10. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Fukui, M.; Fujii, N.; Miyajima, K. Interactions of an antimicrobial peptide, tachyplesin I, with lipid membranes. Biochim. Biophys. Acta 1991, 1070, 259–264. [Google Scholar] [CrossRef]
- Iwanaga, S.; Muta, T.; Shigenaga, T.; Seki, N.; Kawano, K.; Katsu, T.; Kawabata, S. Structure-function relationships of tachyplesins and their analogues. Ciba Found. Symp. 1994, 186, 160–174. [Google Scholar]
- Imura, Y.; Nishida, M.; Ogawa, Y.; Takakura, Y.; Matsuzaki, K. Action mechanism of tachyplesin I and effects of PEGylation. Biochim. Biophys. Acta 2007, 1768, 1160–1169. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Yoneyama, S.; Fujii, N.; Miyajima, K.; Yamada, K.; Kirino, Y.; Anzai, K. Membrane permeabilization mechanisms of a cyclic antimicrobial peptide, tachyplesin I, and its linear analog. Biochemistry 1997, 36, 9799–9806. [Google Scholar] [CrossRef]
- Kuzmin, D.V.; Emelianova, A.A.; Kalashnikova, M.B.; Panteleev, P.V.; Balandin, S.V.; Serebrovskaya, E.O.; Belogurova-Ovchinnikova, O.Y.; Ovchinnikova, T.V. Comparative in vitro study on cytotoxicity of recombinant beta-hairpin peptides. Chem. Biol. Drug Des. 2017, 91, 294–303. [Google Scholar] [CrossRef]
- Laederach, A.; Andreotti, A.H.; Fulton, D.B. Solution and micelle-bound structures of tachyplesin I and its active aromatic linear derivatives. Biochemistry 2002, 41, 12359–12368. [Google Scholar] [CrossRef]
- Kushibiki, T.; Kamiya, M.; Aizawa, T.; Kumaki, Y.; Kikukawa, T.; Mizuguchi, M.; Demura, M.; Kawabata, S.; Kawano, K. Interaction between tachyplesin I, an antimicrobial peptide derived from horseshoe crab, and lipopolysaccharide. Biochim. Biophys. Acta 2014, 1844, 527–534. [Google Scholar] [CrossRef]
- Kawano, K.; Yoneya, T.; Miyata, T.; Yoshikawa, K.; Tokunaga, F.; Terada, Y.; Iwanaga, S. Antimicrobial peptide, tachyplesin I, isolated from hemocytes of the horseshoe crab (Tachypleus tridentatus). NMR determination of the beta-sheet structure. J. Biol. Chem. 1990, 265, 15365–15367. [Google Scholar]
- Mielke, S.P.; Krishnan, V.V. Characterization of protein secondary structure from NMR chemical shifts. Prog. Nucl. Magn. Reson. Spectrosc. 2009, 54, 141–165. [Google Scholar] [CrossRef]
- Hutchinson, E.G.; Thornton, J.M. PROMOTIF--a program to identify and analyze structural motifs in proteins. Protein Sci. 1996, 5, 212–220. [Google Scholar] [CrossRef]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef]
- Chan, L.Y.; Zhang, V.M.; Huang, Y.H.; Waters, N.C.; Bansal, P.S.; Craik, D.J.; Daly, N.L. Cyclization of the antimicrobial peptide gomesin with native chemical ligation: influences on stability and bioactivity. Chembiochem. 2013, 14, 617–624. [Google Scholar] [CrossRef]
- Troeira Henriques, S.; Lawrence, N.; Chaousis, S.; Ravipati, A.S.; Cheneval, O.; Benfield, A.H.; Elliott, A.G.; Kavanagh, A.M.; Cooper, M.A.; Chan, L.Y.; et al. Redesigned Spider Peptide with Improved Antimicrobial and Anticancer Properties. ACS Chem. Biol. 2017, 12, 2324–2334. [Google Scholar] [CrossRef]
- Kondejewski, L.H.; Farmer, S.W.; Wishart, D.S.; Kay, C.M.; Hancock, R.E.; Hodges, R.S. Modulation of structure and antibacterial and hemolytic activity by ring size in cyclic gramicidin S analogs. J. Biol. Chem. 1996, 271, 25261–25268. [Google Scholar] [CrossRef]
- Tam, J.P.; Lu, Y.A.; Yang, J.L. Marked increase in membranolytic selectivity of novel cyclic tachyplesins constrained with an antiparallel two-beta strand cystine knot framework. Biochem. Biophys. Res. Commun. 2000, 267, 783–790. [Google Scholar] [CrossRef]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Blaskovich, M.A.T.; Cooper, M.A. Structure-Activity and -Toxicity Relationships of the Antimicrobial Peptide Tachyplesin-1. ACS Infect. Dis. 2017, 3, 917–926. [Google Scholar] [CrossRef]
- Marggraf, M.B.; Panteleev, P.V.; Emelianova, A.A.; Sorokin, M.I.; Bolosov, I.A.; Buzdin, A.A.; Kuzmin, D.V.; Ovchinnikova, T.V. Cytotoxic Potential of the Novel Horseshoe Crab Peptide Polyphemusin III. Mar. Drugs 2018, 16, 466. [Google Scholar] [CrossRef]
- Hilchie, A.L.; Hoskin, D.W.; Power Coombs, M.R. Anticancer Activities of Natural and Synthetic Peptides. In Antimicrobial Peptides: Basics for Clinical Application; Matsuzaki, K., Ed.; Springer Singapore: Singapore, 2019; pp. 131–147. [Google Scholar]
- Kuzmin, D.V.; Emelianova, A.A.; Kalashnikova, M.B.; Panteleev, P.V.; Ovchinnikova, T.V. Effect of N- and C-Terminal Modifications on Cytotoxic Properties of Antimicrobial Peptide Tachyplesin I. Bull. Exp. Biol. Med. 2017, 162, 754–757. [Google Scholar] [CrossRef]
- Rivera, M.; Bertasso, A.; McCaffrey, C.; Georgopapadakou, N.H. Porins and lipopolysaccharide of Escherichia coli ATCC 25922 and isogenic rough mutants. FEMS Microbiol. Lett. 1993, 108, 183–187. [Google Scholar] [CrossRef]
- Treangen, T.J.; Maybank, R.A.; Enke, S.; Friss, M.B.; Diviak, L.F.; Karaolis, D.K.; Koren, S.; Ondov, B.; Phillippy, A.M.; Bergman, N.H.; et al. Complete Genome Sequence of the Quality Control Strain Staphylococcus aureus subsp. aureus ATCC 25923. Genome Announc. 2014, 2, e01110-14. [Google Scholar] [CrossRef]
- Clark, D. Novel antibiotic hypersensitive mutants of Escherichia coli genetic mapping and chemical characterization. FEMS Microbiol. Lett. 1984, 21, 189–195. [Google Scholar] [CrossRef]
- Pinto, S.N.; Dias, S.A.; Cruz, A.F.; Mil-Homens, D.; Fernandes, F.; Valle, J.; Andreu, D.; Prieto, M.; Castanho, M.; Coutinho, A.; et al. The mechanism of action of pepR, a viral-derived peptide, against Staphylococcus aureus biofilms. J. Antimicrob. Chemother. 2019, dkz223. [Google Scholar] [CrossRef]
- Watson, H. Biological membranes. Essays Biochem. 2015, 59, 43–69. [Google Scholar] [CrossRef]
- Ingolfsson, H.I.; Melo, M.N.; van Eerden, F.J.; Arnarez, C.; Lopez, C.A.; Wassenaar, T.A.; Periole, X.; de Vries, A.H.; Tieleman, D.P.; Marrink, S.J. Lipid organization of the plasma membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Eftink, M.R. Fluorescence Quenching Reactions. In Biophysical and Biochemical Aspects of Fluorescence Spectroscopy; Dewey, T.G., Ed.; Springer US: Boston, MA, USA, 1991; pp. 1–41. [Google Scholar]
- Ghisaidoobe, A.B.; Chung, S.J. Intrinsic tryptophan fluorescence in the detection and analysis of proteins: a focus on Forster resonance energy transfer techniques. Int. J. Mol. Sci. 2014, 15, 22518–22538. [Google Scholar] [CrossRef]
- Santos, N.C.; Prieto, M.; Castanho, M.A.R.B. Quantifying molecular partition into model systems of biomembranes: an emphasis on optical spectroscopic methods. Biochim. Biophys. Acta 2003, 1612, 123–135. [Google Scholar] [CrossRef]
- Fernandes, M.X.; García de la Torre, J.; Castanho, M.A.R.B. Joint determination by Brownian dynamics and fluorescence quenching of the in-depth location profile of biomolecules in membranes. Anal. Biochem. 2002, 307, 1–12. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Topics in Fluorescence Spectroscopy Principles; Springer US: Boston, MA, USA, 2002. [Google Scholar]
- Chattopadhyay, A.; London, E. Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry 1987, 26, 39–45. [Google Scholar] [CrossRef]
- Kyrychenko, A.; Ladokhin, A.S. Molecular Dynamics Simulations of Depth Distribution of Spin-Labeled Phospholipids within Lipid Bilayer. J. Phys. Chem. B 2013, 117, 5875–5885. [Google Scholar] [CrossRef]
- Batoni, G.; Maisetta, G.; Esin, S. Antimicrobial peptides and their interaction with biofilms of medically relevant bacteria. Biochim. Biophys. Acta 2016, 1858, 1044–1060. [Google Scholar] [CrossRef]
- Grassi, L.; Maisetta, G.; Esin, S.; Batoni, G. Combination Strategies to Enhance the Efficacy of Antimicrobial Peptides against Bacterial Biofilms. Front. Microbiol. 2017, 8, 2409. [Google Scholar] [CrossRef]
- Minardi, D.; Ghiselli, R.; Cirioni, O.; Giacometti, A.; Kamysz, W.; Orlando, F.; Silvestri, C.; Parri, G.; Kamysz, E.; Scalise, G.; et al. The antimicrobial peptide Tachyplesin III coated alone and in combination with intraperitoneal piperacillin-tazobactam prevents ureteral stent Pseudomonas infection in a rat subcutaneous pouch model. Peptides 2007, 28, 2293–2298. [Google Scholar] [CrossRef]
- Yu, K.; Lo, J.C.; Yan, M.; Yang, X.; Brooks, D.E.; Hancock, R.E.; Lange, D.; Kizhakkedathu, J.N. Anti-adhesive antimicrobial peptide coating prevents catheter associated infection in a mouse urinary infection model. Biomaterials 2017, 116, 69–81. [Google Scholar] [CrossRef]
- Oishi, O.; Yamashita, S.; Nishimoto, E.; Lee, S.; Sugihara, G.; Ohno, M. Conformations and orientations of aromatic amino acid residues of tachyplesin I in phospholipid membranes. Biochemistry 1997, 36, 4352–4359. [Google Scholar] [CrossRef]
- Doherty, T.; Waring, A.J.; Hong, M. Dynamic structure of disulfide-removed linear analogs of tachyplesin-I in the lipid bilayer from solid-state NMR. Biochemistry 2008, 47, 1105–1116. [Google Scholar] [CrossRef]
- Torcato, I.M.; Huang, Y.H.; Franquelim, H.G.; Gaspar, D.; Craik, D.J.; Castanho, M.A.; Troeira Henriques, S. Design and characterization of novel antimicrobial peptides, R-BP100 and RW-BP100, with activity against Gram-negative and Gram-positive bacteria. Biochim. Biophys. Acta 2013, 1828, 944–955. [Google Scholar] [CrossRef]
- Cheneval, O.; Schroeder, C.I.; Durek, T.; Walsh, P.; Huang, Y.H.; Liras, S.; Price, D.A.; Craik, D.J. Fmoc-based synthesis of disulfide-rich cyclic peptides. J. Org. Chem. 2014, 79, 5538–5544. [Google Scholar] [CrossRef]
- Güntert, P. Automated NMR Structure Calculation with CYANA. In Protein NMR Techniques; Downing, A.K., Ed.; Humana Press: Totowa, NJ, USA, 2004; pp. 353–378. [Google Scholar]
- Shen, Y.; Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef]
- Brunger, A.T. Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2007, 2, 2728. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Huang, Y.H.; Colgrave, M.L.; Clark, R.J.; Kotze, A.C.; Craik, D.J. Lysine-scanning mutagenesis reveals an amendable face of the cyclotide kalata B1 for the optimization of nematocidal activity. J. Biol. Chem. 2010, 285, 10797–10805. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, Y.; Zheng, C.; Shen, C. Investigation of Cross-Contamination and Misidentification of 278 Widely Used Tumor Cell Lines. PLoS ONE 2017, 12, e0170384. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. In Assay Guidance Manual; Sittampalam, G.S., Coussens, N.P., Brimacombe, K., Grossman, A., Arkin, M., Auld, D., Austin, C., Baell, J., Bejcek, B., Chung, T.D.Y., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2016. [Google Scholar]
- Mayer, L.D.; Hope, M.J.; Cullis, P.R. Vesicles of variable sizes produced by a rapid extrusion procedure. BBA-Biomembranes 1986, 858, 161–168. [Google Scholar] [CrossRef]
- Henriques, S.T.; Pattenden, L.K.; Aguilar, M.-I.; Castanho, M.A.R.B. The Toxicity of Prion Protein Fragment PrP(106−126) is Not Mediated by Membrane Permeabilization as Shown by a M112W Substitution. Biochemistry 2009, 48, 4198–4208. [Google Scholar] [CrossRef]
- Henriques, S.T.; Castanho, M.A.R.B. Environmental factors that enhance the action of the cell penetrating peptide pep-1: A spectroscopic study using lipidic vesicles. Biochim. Biophys. Acta 2005, 1669, 75–86. [Google Scholar] [CrossRef]
- Caputo, G.A.; London, E. Using a novel dual fluorescence quenching assay for measurement of tryptophan depth within lipid bilayers to determine hydrophobic alpha-helix locations within membranes. Biochemistry 2003, 42, 3265–3274. [Google Scholar] [CrossRef]
- Henriques, S.T.; Huang, Y.H.; Castanho, M.A.; Bagatolli, L.A.; Sonza, S.; Tachedjian, G.; Daly, N.L.; Craik, D.J. Phosphatidylethanolamine binding is a conserved feature of cyclotide-membrane interactions. J. Biol. Chem. 2012, 287, 33629–33643. [Google Scholar] [CrossRef]
- Henriques, S.T.; Huang, Y.-H.; Rosengren, K.J.; Franquelim, H.G.; Carvalho, F.A.; Johnson, A.; Sonza, S.; Tachedjian, G.; Castanho, M.A.R.B.; Daly, N.L.; et al. Decoding the membrane activity of the cyclotide kalata B1: the importance of phosphatidylethanolamine phospholipids and lipid organization on hemolytic and anti- HIV activities. J. Biol. Chem. 2011, 286, 24231. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Colgrave, M.L.; Daly, N.L.; Keleshian, A.; Martinac, B.; Craik, D.J. Biological Activity of the Prototypic Cyclotide Kalata B1 Is Modulated by the Formation of Multimeric Pores. J. Biol. Chem. 2009, 284, 20699–20707. [Google Scholar] [CrossRef]
- Stewart, J.C. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 1980, 104, 10–14. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence 1 | Mass (Da) 2 | RT (min) 3 | Charge 4 | |
|---|---|---|---|---|---|
| Calc. | Obs. | ||||
| TI | KWCFRVCYRGICYRRCR * | 2263.8 | 2263.5 | 18.04 | +7 |
| TII | RWCFRVCYRGICYRKCR * | 2263.8 | 2263.5 | 17.79 | +7 |
| TIII | KWCFRVCYRGICYRKCR * | 2235.8 | 2235.6 | 17.68 | +7 |
| cTI | KWCFRVCYRGICYRRCRG | 2303.8 | 2303.7 | 18.69 | +6 |
| cTII | RWCFRVCYRGICYRKCRG | 2303.8 | 2303.7 | 18.53 | +6 |
| cTIII | KWCFRVCYRGICYRKCRG | 2275.8 | 2275.5 | 18.44 | +6 |
| Peptide | HC50 (µM) |
|---|---|
| TI | 34.9 ± 2.8 |
| TII | 55.4 ± 6.6 |
| TIII | 86.4 ± 12.2 |
| cTI | 106.9 ± 21.0 |
| cTII | 64.1 ± 9.4 |
| cTIII | >128 |
| MIC (µM) | ||||
|---|---|---|---|---|
| Peptide | E. coli DC2 CGSC 7139 | E. coli ATCC 25922 | S. aureus ATCC 25923 | S. aureus ATCC 6538 |
| TI | 0.5–1 | 0.0625–0.5 | 1–4 | 4–8 |
| TII | 0.5–1 | 0.125–0.5 | 1–4 | 4–8 |
| TIII | 0.25–0.5 | 0.0625–0.5 | 1–4 | 4–8 |
| cTI | 4 | 1 | 2–8 | 8 |
| cTII | 4–8 | 1–2 | 2–8 | 8 |
| cTIII | 4–8 | 1–2 | 2–8 | 8 |
| Peptide | CC50 (µM) 1 | Metabolically Active Cells (%) 2 |
|---|---|---|
| TI | 21.5 ± 2.2 | 37.7 ± 4.3 |
| TII | 23.1 ± 2.9 | 39.9 ± 2.9 |
| TIII | 24.2 ± 4.1 | 43.3 ± 4.1 |
| cTI | 17.8 ± 3.6 | 31.9 ± 6.0 |
| cTII | 19.4 ± 1.5 | 34.6 ± 7.1 |
| cTIII | 18.0 ± 1.4 | 37.5 ± 7.2 |
| CC50 (µM) 1 | Melanoma Selectivity 5 | |||||
|---|---|---|---|---|---|---|
| Peptide | MM96L 2 | HT144 2 | WM164 2 | HeLa 3 | HaCaT 4 | |
| TI | 1.5 ± 0.1 | 1.7 ± 0.2 | 2.5 ± 0.1 | 13.1 ± 1.2 | 11.6 ± 1.6 | 2–21 |
| TII | 1.6 ± 0.1 | 2.0 ± 0.1 | 1.6 ± 0.1 | 18.0 ± 3.9 | 3.7 ± 0.2 | 3–35 |
| TIII | 1.8 ± 0.1 | 2.0 ± 0.1 | 1.7 ± 0.1 | 21.7 ± 1.1 | 7.3 ± 0.5 | 5–48 |
| cTI | 1.3 ± 0.1 | 1.4 ± 0.1 | 2.7 ± 0.1 | 6.7 ± 0.6 | 7.9 ± 0.5 | 2–76 |
| cTII | 1.1 ± 0.1 | 0.8 ± 0.04 | 2.4 ± 0.3 | 7.2 ± 0.4 | 2.4 ± 0.3 | 3–58 |
| cTIII | 1.7 ± 0.1 | 0.9 ± 0.03 | 1.3 ± 0.1 | 9.3 ± 0.4 | 7.5 ± 0.3 | 3–98 |
| Peptide | Lipid System | P/Lmax (mol/mol) 1 | KD (µM) 1 | koff (x 10−2 s−1) 2 | P/Loff (mol/mol) 2 | LCmax (%) 3 |
|---|---|---|---|---|---|---|
| TI | POPC | 0.26 ± 0.06 | 22.4 ± 10.9 | 1.50 ± 0.11 | 0.046 ± 0.001 | 39.5 ± 4.6 |
| cTI | 0.33 ± 0.07 | 16.7 ± 8.4 | 0.91 ± 0.03 | 0.065 ± 0.001 | 32.9 ± 9.4 | |
| TI | POPC/POPS (4:1) | 0.37 ± 0.04 | 11.8 ± 2.4 | 2.75 ± 0.22 | 0.096 ± 0.001 | 76.0 ± 2.8 |
| cTI | 0.48 ± 0.08 | 9.2 ± 3.4 | 0.70 ± 0.03 | 0.137 ± 0.002 | 58.5 ± 3.6 |
| POPC | POPC/POPS (4:1) | POPC/POPG (4:1) | |||
|---|---|---|---|---|---|
| Peptide | shift (nm) 1 | shift (nm) | 0.5 [L] (mM) 2 | shift (nm) | 0.5 [L] (mM) |
| TI | 2 | 22 | 0.19 | 30 | 0.09 |
| TII | 0 | 19 | 0.30 | 29 | 0.09 |
| TIII | −3 | 18 | 0.30 | 23 | 0.10 |
| cTI | 0 | 28 | 0.11 | 28 | 0.06 |
| cTII | 1 | 27 | 0.08 | 29 | 0.05 |
| cTIII | 3 | 27 | 0.12 | 28 | 0.08 |
| Acrylamide Accessibility (%) 1 | ||||
|---|---|---|---|---|
| 1 mM | 1 mM | 0.1 mM | 0.1 mM | |
| Peptide | POPC | POPC/POPS (4:1) | POPC/POPS (4:1) | POPC/POPG (4:1) |
| TI | 90.5 ± 4.3 | 16.2 ± 21.9 | 72.3 ± 3.9 | 54.2 ± 5.8 |
| TII | 76.2 ± 3.2 | 56.8 ± 6.9 | ||
| TIII | 82.1 ± 4.7 | 75.4 ± 4.6 | ||
| cTI | 103.8 ± 2.3 | 22.5 ± 14.3 | 32.3 ± 8.3 | 32.0 ± 4.1 |
| cTII | 61.4 ± 5.5 | 36.1 ± 7.7 | ||
| cTIII | 48.7 ± 6.5 | 46.2 ± 5.7 | ||
| 5DS | 16DS | ||||
|---|---|---|---|---|---|
| Peptide | KSV (M−1) | fb | KSV (M−1) | fb | Z (Å) 2 |
| TI | 3.7 ± 1.3 | 0.63 ± 0.57 | 19.6 ± 4.3 | 0.74 ± 0.37 | 8.8 |
| cTI | 4.9 ± 1.8 | 0.61 ± 0.54 | 26.3 ± 7.1 | 0.83 ± 0.52 | 5.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vernen, F.; Harvey, P.J.; Dias, S.A.; Veiga, A.S.; Huang, Y.-H.; Craik, D.J.; Lawrence, N.; Troeira Henriques, S. Characterization of Tachyplesin Peptides and Their Cyclized Analogues to Improve Antimicrobial and Anticancer Properties. Int. J. Mol. Sci. 2019, 20, 4184. https://doi.org/10.3390/ijms20174184
Vernen F, Harvey PJ, Dias SA, Veiga AS, Huang Y-H, Craik DJ, Lawrence N, Troeira Henriques S. Characterization of Tachyplesin Peptides and Their Cyclized Analogues to Improve Antimicrobial and Anticancer Properties. International Journal of Molecular Sciences. 2019; 20(17):4184. https://doi.org/10.3390/ijms20174184
Chicago/Turabian StyleVernen, Felicitas, Peta J. Harvey, Susana A. Dias, Ana Salomé Veiga, Yen-Hua Huang, David J. Craik, Nicole Lawrence, and Sónia Troeira Henriques. 2019. "Characterization of Tachyplesin Peptides and Their Cyclized Analogues to Improve Antimicrobial and Anticancer Properties" International Journal of Molecular Sciences 20, no. 17: 4184. https://doi.org/10.3390/ijms20174184
APA StyleVernen, F., Harvey, P. J., Dias, S. A., Veiga, A. S., Huang, Y.-H., Craik, D. J., Lawrence, N., & Troeira Henriques, S. (2019). Characterization of Tachyplesin Peptides and Their Cyclized Analogues to Improve Antimicrobial and Anticancer Properties. International Journal of Molecular Sciences, 20(17), 4184. https://doi.org/10.3390/ijms20174184