A Cell’s Fate: An Overview of the Molecular Biology and Genetics of Apoptosis

 ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
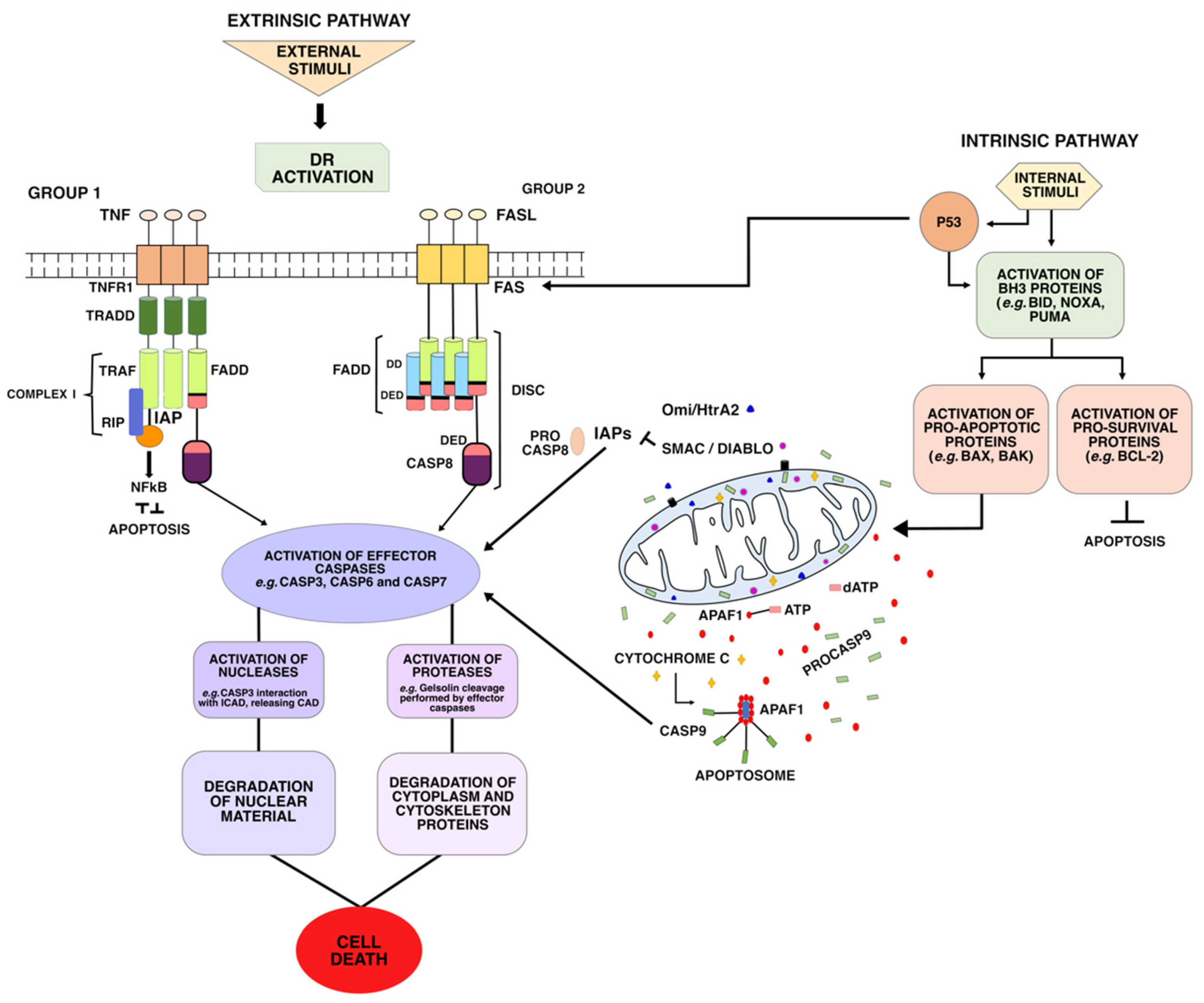
2. Intrinsic or Mitochondrial Pathway
3. Extrinsic or Death Receptor Pathway
- (i)
- The first group includes the receptors Fas (DR2) TRAILR1 (DR4) and TRAILR2 (DR5), which can be activated by Fas ligand (FasL) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), respectively. TRAIL ligands bind to a DR and a TRAIL receptor (TRAIL-R), triggering apoptotic signals and inducing the formation and activation of death-inducing signaling complex (DISC) complex [28]. Once activated, these receptors recruit the death-inducing signaling complex (DISC)—which is composed of FAS-associated via death domain (FADD) and Procaspase-8 (ProC8)—for the transduction of apoptotic signals;
- (ii)
- The second group includes the receptors TNFR1 (DR1), TRAMP (DR3), DR6, and EDAR. These DRs recruit TNF-associated death domain (TRADD) as adapter protein, and bind to the TNF-2,5 receptor-associated factors (TRAF2,5), the receptor-interacting protein kinase (RIP1 or RIPK1), and to cellular inhibitors of apoptosis proteins (cIAP). This forms a signaling complex, called Complex I, for signal transduction of apoptosis and cell survival.
4. Execution Phase
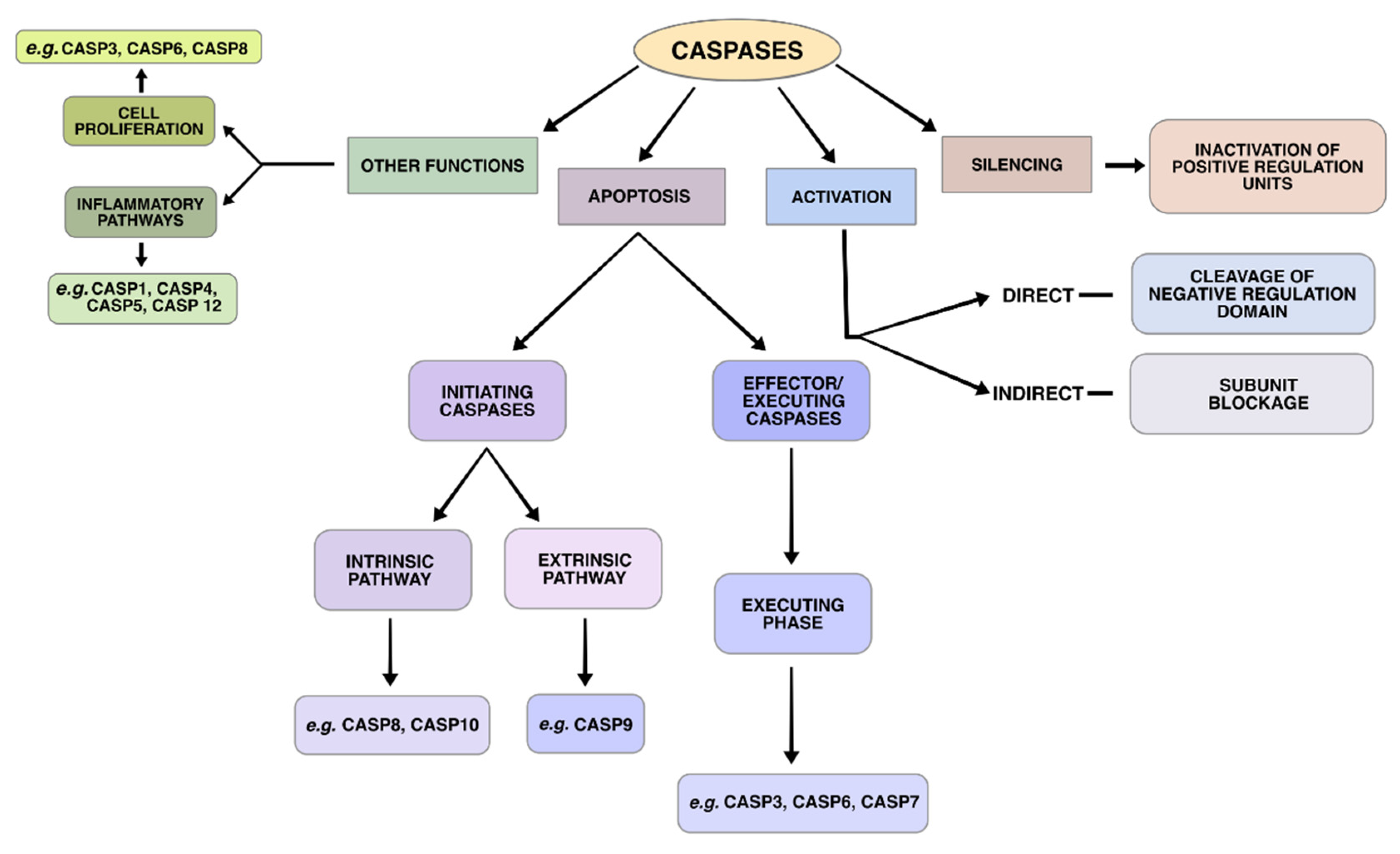
5. Genes Involved in the Apoptotic Pathways
6. Intrinsic Pathway
6.1. APAF1 (Apoptotic Peptidase Activating Factor 1)
6.2. BCL2 (B-Cell Lymphoma Protein 2, Apoptosis Regulator)
6.3. CASP9 (Caspase 9)
7. Extrinsic Pathway
7.1. CASP8 (Caspase 8)
7.2. FADD (FAS-Associated Via Death Domain)
7.3. FAS (FAS Cell Surface Death Receptor)
8. Intrinsic and Extrinsic Pathways
8.1. TP53 (Tumor Protein p53)
8.2. MYC (MYC Proto-Oncogene, bHLH Transcription Factor)
9. Execution Phase
CASP3 (Caspase 3)
10. Mitochondria and Epigenetic Regulation
miRNAs in Apoptosis
11. Conclusions
Funding
Conflicts of Interest
References
- Zhivotovsky, B.; Orrenius, S. Cell death mechanisms: Cross-talk and role in disease. Exp. Cell Res. 2010, 316, 1374–1383. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.A.; Kirby, R. Apoptosis: A review of pro-apoptotic and anti-apoptotic pathways and dysregulation in disease. J. Vet. Emerg. Crit. Care 2008, 18, 572–585. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Lomonosova, E.; Chinnadurai, G. BH3-only proteins in apoptosis and beyond: An overview. Oncogene 2008, 27, S2–S19. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Dewson, G. Mitochondria and apoptosis: Emerging concepts. F1000Prime Rep. 2015, 7, 42. [Google Scholar] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef]
- Cheng, T.C.; Hong, C.; Akey, I.V.; Yuan, S.; Akey, C.W. A near atomic structure of the active human apoptosome. eLife 2016, 5, e17755. [Google Scholar] [CrossRef]
- Yuan, S.; Akey, C.W. Apoptosome structure, assembly, and procaspase activation. Structure 2013, 21, 501–515. [Google Scholar] [CrossRef]
- Gortat, A.; Sancho, M.; Mondragón, L.; Messeguer, À.; Pérez-Payá, E.; Orzáez, M. Apaf1 inhibition promotes cell recovery from apoptosis. Protein Cell 2015, 6, 833–843. [Google Scholar] [CrossRef]
- Bratton, S.B.; Salvesen, G.S. Regulation of the Apaf-1-caspase-9 apoptosome. J. Cell Sci. 2010, 123, 3209–3214. [Google Scholar] [CrossRef]
- Shi, Y. Caspase activation: Revisiting the induced proximity model. Cell 2004, 117, 855–858. [Google Scholar] [CrossRef]
- Yuan, S.; Yu, X.; Asara, J.M.; Heuser, J.E.; Ludtke, S.J.; Akey, C.W. The holo-apoptosome: Activation of procaspase-9 and interactions with caspase-3. Structure 2011, 19, 1084–1096. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Mace, P.D.; Riedl, S.J. Molecular cell death platforms and assemblies. Curr. Opin. Cell Biol. 2010, 22, 828–836. [Google Scholar] [CrossRef]
- Tinel, A.; Tschopp, J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 2004, 304, 843–846. [Google Scholar] [CrossRef]
- Sladky, V.; Schuler, F.; Fava, L.L.; Villunger, A. The resurrection of the PIDDosome - emerging roles in the DNA-damage response and centrosome surveillance. J. Cell Sci. 2017, 130, 3779–3787. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- Elrod, H.A.; Sun, S.-Y. Modulation of death receptors by cancer therapeutic agents. Cancer Biol. Ther. 2008, 7, 163–173. [Google Scholar] [CrossRef]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis—The p53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef]
- Amanullah, A.; Liebermann, D.A.; Hoffman, B. Deregulated c-Myc prematurely recruits both Type I and II CD95/Fas apoptotic pathways associated with terminal myeloid differentiation. Oncogene 2002, 21, 1600–1610. [Google Scholar] [CrossRef]
- Wang, Y.; Engels, I.H.; Knee, D.A.; Nasoff, M.; Deveraux, Q.L.; Quon, K.C. Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway. Cancer Cell 2004, 5, 501–512. [Google Scholar] [CrossRef]
- Mahmood, Z.; Shukla, Y. Death receptors: Targets for cancer therapy. Exp. Cell Res. 2010, 316, 887–899. [Google Scholar] [CrossRef]
- Lee, E.-W.; Seo, J.; Jeong, M.; Lee, S.; Song, J. The roles of FADD in extrinsic apoptosis and necroptosis. BMB Rep. 2012, 45, 496–508. [Google Scholar] [CrossRef]
- Kretz, A.-L.; Trauzold, A.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; von Karstedt, S.; Lemke, J. TRAILblazing Strategies for Cancer Treatment. Cancers 2019, 11, 456. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. Int. J. Mol. Sci. 2015, 16, 30321–30341. [Google Scholar] [CrossRef]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Panayotova-Dimitrova, D.; Leverkus, M. Pick your poison: The Ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle 2012, 11, 460–467. [Google Scholar] [CrossRef]
- Henry, C.M.; Martin, S.J. Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol. Cell 2017, 65, 715–729.e5. [Google Scholar] [CrossRef]
- Mohr, A.; Deedigan, L.; Jencz, S.; Mehrabadi, Y.; Houlden, L.; Albarenque, S.-M.; Zwacka, R.M. Caspase-10: A molecular switch from cell-autonomous apoptosis to communal cell death in response to chemotherapeutic drug treatment. Cell Death Differ. 2018, 25, 340–352. [Google Scholar] [CrossRef]
- Mouasni, S.; Tourneur, L. FADD at the Crossroads between Cancer and Inflammation. Trends Immunol. 2018, 39, 1036–1053. [Google Scholar] [CrossRef]
- Wajant, H. Death receptors. Essays Biochem. 2003, 39, 53–71. [Google Scholar] [CrossRef]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.-Y.; Baldwin, A.S. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- Rytömaa, M.; Lehmann, K.; Downward, J. Matrix detachment induces caspase-dependent cytochrome c release from mitochondria: Inhibition by PKB/Akt but not Raf signalling. Oncogene 2000, 19, 4461–4468. [Google Scholar] [CrossRef][Green Version]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Modur, V.; Nagarajan, R.; Evers, B.M.; Milbrandt, J. FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. J. Biol. Chem. 2002, 277, 47928–47937. [Google Scholar] [CrossRef]
- Stahl, M.; Dijkers, P.F.; Kops, G.J.P.L.; Lens, S.M.A.; Coffer, P.J.; Burgering, B.M.T.; Medema, R.H. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J. Immunol. 2002, 168, 5024–5031. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb. Perspect. Biol. 2013, 5, a008672. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.S. Learning from Deficiency: Gene Targeting of Caspases; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Kuida, K.; Zheng, T.S.; Na, S.; Kuan, C.; Yang, D.; Karasuyama, H.; Rakic, P.; Flavell, R.A. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 1996, 384, 368–372. [Google Scholar] [CrossRef]
- Varfolomeev, E.E.; Schuchmann, M.; Luria, V.; Chiannilkulchai, N.; Beckmann, J.S.; Mett, I.L.; Rebrikov, D.; Brodianski, V.M.; Kemper, O.C.; Kollet, O.; et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 1998, 9, 267–276. [Google Scholar] [CrossRef]
- Kuida, K.; Haydar, T.F.; Kuan, C.Y.; Gu, Y.; Taya, C.; Karasuyama, H.; Su, M.S.; Rakic, P.; Flavell, R.A. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 1998, 94, 325–337. [Google Scholar] [CrossRef]
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef]
- Julien, O.; Zhuang, M.; Wiita, A.P.; O’Donoghue, A.J.; Knudsen, G.M.; Craik, C.S.; Wells, J.A. Quantitative MS-based enzymology of caspases reveals distinct protein substrate specificities, hierarchies, and cellular roles. Proc. Natl. Acad. Sci. USA 2016, 113, E2001–E2010. [Google Scholar] [CrossRef]
- Tucker, M.B.; MacKenzie, S.H.; Maciag, J.J.; Dirscherl Ackerman, H.; Swartz, P.; Yoder, J.A.; Hamilton, P.T.; Clay Clark, A. Phage display and structural studies reveal plasticity in substrate specificity of caspase-3a from zebrafish. Protein Sci. 2016, 25, 2076–2088. [Google Scholar] [CrossRef]
- Wachmann, K.; Pop, C.; van Raam, B.J.; Drag, M.; Mace, P.D.; Snipas, S.J.; Zmasek, C.; Schwarzenbacher, R.; Salvesen, G.S.; Riedl, S.J. Activation and Specificity of human Caspase-10. Biochemistry 2010, 49, 8307–8315. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Orth, J.D.; Loewer, A.; Lahav, G.; Mitchison, T.J. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol. Biol. Cell 2012, 23, 567–576. [Google Scholar] [CrossRef]
- Larsen, B.D.; Sørensen, C.S. The caspase-activated DNase: Apoptosis and beyond. FEBS J. 2017, 284, 1160–1170. [Google Scholar] [CrossRef]
- Kothakota, S.; Azuma, T.; Reinhard, C.; Klippel, A.; Tang, J.; Chu, K.; McGarry, T.J.; Kirschner, M.W.; Koths, K.; Kwiatkowski, D.J.; et al. Caspase-3-generated fragment of gelsolin: Effector of morphological change in apoptosis. Science 1997, 278, 294–298. [Google Scholar] [CrossRef]
- Cecconi, F.; Gruss, P. Human Genome and Diseases: Apaf1 in developmental apoptosis and cancer: How many ways to die? CMLS Cell. Mol. Life Sci. 2001, 58, 1688–1697. [Google Scholar] [CrossRef]
- Karimzadeh, S.; Hosseinkhani, S.; Fathi, A.; Ataei, F.; Baharvand, H. Insufficient Apaf-1 expression in early stages of neural differentiation of human embryonic stem cells might protect them from apoptosis. Eur. J. Cell Biol. 2018, 97, 126–135. [Google Scholar] [CrossRef]
- Spellicy, C.J.; Norris, J.; Bend, R.; Bupp, C.; Mester, P.; Reynolds, T.; Dean, J.; Peng, Y.; Alexov, E.; Schwartz, C.E.; et al. Key apoptotic genes APAF1 and CASP9 implicated in recurrent folate-resistant neural tube defects. Eur. J. Hum. Genet. 2018, 26, 420–427. [Google Scholar] [CrossRef]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008722. [Google Scholar] [CrossRef]
- Chao, D.T.; Korsmeyer, S.J. BCL-2 family: Regulators of cell death. Annu. Rev. Immunol. 1998, 16, 395–419. [Google Scholar] [CrossRef]
- Ke, N.; Godzik, A.; Reed, J.C. Bcl-B, a novel Bcl-2 family member that differentially binds and regulates Bax and Bak. J. Biol. Chem. 2001, 276, 12481–12484. [Google Scholar] [CrossRef]
- Siddiqui, W.A.; Ahad, A.; Ahsan, H. The mystery of BCL2 family: Bcl-2 proteins and apoptosis: An update. Arch. Toxicol. 2015, 89, 289–317. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Morrice, N.; Cohen, P. Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem. J. 2000, 349, 547–557. [Google Scholar] [CrossRef]
- Harada, H.; Becknell, B.; Wilm, M.; Mann, M.; Huang, L.J.; Taylor, S.S.; Scott, J.D.; Korsmeyer, S.J. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol. Cell 1999, 3, 413–422. [Google Scholar] [CrossRef]
- Martinou, J.-C.; Youle, R.J. Mitochondria in Apoptosis: Bcl-2 family Members and Mitochondrial Dynamics. Dev. Cell 2011, 21, 92–101. [Google Scholar] [CrossRef]
- Bergmann, A. Survival signaling goes BAD. Dev. Cell 2002, 3, 607–608. [Google Scholar] [CrossRef]
- Wang, H.G.; Pathan, N.; Ethell, I.M.; Krajewski, S.; Yamaguchi, Y.; Shibasaki, F.; McKeon, F.; Bobo, T.; Franke, T.F.; Reed, J.C. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 1999, 284, 339–343. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Green, D.R. Means to an End: Apoptosis and Other Cell Death Mechanisms; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2011; ISBN 978-0-87969-887-4. [Google Scholar]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Gorham, J.; Cossman, J.; Jaffe, E.; Croce, C.M. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science 1985, 229, 1390–1393. [Google Scholar] [CrossRef]
- Tzifi, F.; Economopoulou, C.; Gourgiotis, D.; Ardavanis, A.; Papageorgiou, S.; Scorilas, A. The Role of BCL2 Family of Apoptosis Regulator Proteins in Acute and Chronic Leukemias. Adv. Hematol. 2012, 2012, 524308. [Google Scholar] [CrossRef]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef]
- Blombery, P.; Anderson, M.A.; Gong, J.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef]
- Ma, S.; Lee, T.K.; Zheng, B.-J.; Chan, K.W.; Guan, X.-Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef]
- Zaanan, A.; Okamoto, K.; Kawakami, H.; Khazaie, K.; Huang, S.; Sinicrope, F.A. The Mutant KRAS Gene Up-regulates BCL-XL Protein via STAT3 to Confer Apoptosis Resistance That Is Reversed by BIM Protein Induction and BCL-XL Antagonism. J. Biol. Chem. 2015, 290, 23838–23849. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. The dark side of Ras: Regulation of apoptosis. Oncogene 2003, 22, 8999–9006. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, M.; Hu, Q.; Bai, X.; Huang, W.; Scheres, S.H.W.; Shi, Y. Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc. Natl. Acad. Sci. USA 2017, 114, 1542–1547. [Google Scholar] [CrossRef]
- Wu, C.-C.; Lee, S.; Malladi, S.; Chen, M.-D.; Mastrandrea, N.J.; Zhang, Z.; Bratton, S.B. The Apaf-1 apoptosome induces formation of caspase-9 homo- and heterodimers with distinct activities. Nature Commun. 2016, 7, 13565. [Google Scholar] [CrossRef]
- Shakeri, R.; Kheirollahi, A.; Davoodi, J. Apaf-1: Regulation and function in cell death. Biochimie 2017, 135, 111–125. [Google Scholar] [CrossRef]
- Guo, R.; Lin, B.; Pan, J.F.; Liong, E.C.; Xu, A.M.; Youdim, M.; Fung, M.L.; So, K.F.; Tipoe, G.L. Inhibition of caspase-9 aggravates acute liver injury through suppression of cytoprotective autophagy. Sci. Rep. 2016, 6, 32447. [Google Scholar] [CrossRef]
- Sommer, M.E.L.; Dalia, R.A.; Nogueira, A.V.B.; Cirelli, J.A.; Vinolo, M.A.R.; Fachi, J.L.; Oliveira, C.A.; Andrade, T.A.M.; Mendonça, F.A.S.; Santamaria, M.; et al. Immune response mediated by Th1 / IL-17 / caspase-9 promotes evolution of periodontal disease. Arch. Oral Biol. 2019, 97, 77–84. [Google Scholar] [CrossRef]
- Ercan, S.; Arinc, S.; Yilmaz, S.G.; Altunok, C.; Yaman, F.; Isbir, T. Investigation of Caspase 9 Gene Polymorphism in Patients With Non-small Cell Lung Cancer. Anticancer Res. 2019, 39, 2437–2441. [Google Scholar] [CrossRef]
- Bellail, A.C.; Tse, M.C.L.; Song, J.H.; Phuphanich, S.; Olson, J.J.; Sun, S.Y.; Hao, C. DR5-mediated DISC controls caspase-8 cleavage and initiation of apoptosis in human glioblastomas. J. Cell. Mol. Med. 2010, 14, 1303–1317. [Google Scholar] [CrossRef]
- Elrod, H.A.; Fan, S.; Muller, S.; Chen, G.Z.; Pan, L.; Tighiouart, M.; Shin, D.M.; Khuri, F.R.; Sun, S.-Y. Analysis of death receptor 5 and caspase-8 expression in primary and metastatic head and neck squamous cell carcinoma and their prognostic impact. PLoS ONE 2010, 5, e12178. [Google Scholar] [CrossRef]
- Camp, N.J.; Parry, M.; Knight, S.; Abo, R.; Elliott, G.; Rigas, S.H.; Balasubramanian, S.P.; Reed, M.W.R.; McBurney, H.; Latif, A.; et al. Fine-mapping CASP8 risk variants in breast cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 176–181. [Google Scholar] [CrossRef]
- Takashina, T.; Nakayama, M. Modifications enhance the apoptosis-inducing activity of FADD. Mol. Cancer Ther. 2007, 6, 1793–1803. [Google Scholar] [CrossRef]
- Svensson, T.; Svensson, A.K.; Kitlinski, M.; Almgren, P.; Engström, G.; Nilsson, J.; Orho-Melander, M.; Nilsson, P.M.; Melander, O. Plasma Concentration of Caspase-8 Is Associated With Short Sleep Duration and the Risk of Incident Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2018, 103, 1592–1600. [Google Scholar] [CrossRef]
- Lehle, A.S.; Farin, H.F.; Marquardt, B.; Michels, B.E.; Magg, T.; Li, Y.; Liu, Y.; Ghalandary, M.; Lammens, K.; Hollizeck, S.; et al. Intestinal Inflammation and Dysregulated Immunity in Patients With Inherited Caspase-8 Deficiency. Gastroenterology 2019, 156, 275–278. [Google Scholar] [CrossRef]
- Jäger, R.; Zwacka, R.M. The enigmatic roles of caspases in tumor development. Cancers 2010, 2, 1952–1979. [Google Scholar] [CrossRef]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef]
- Kaufmann, T.; Strasser, A.; Jost, P.J. Fas death receptor signalling: Roles of Bid and XIAP. Cell Death Differ. 2012, 19, 42–50. [Google Scholar] [CrossRef]
- Fu, Q.; Fu, T.-M.; Cruz, A.C.; Sengupta, P.; Thomas, S.K.; Wang, S.; Siegel, R.M.; Wu, H.; Chou, J.J. Structural Basis and Functional Role of Intramembrane Trimerization of the Fas/CD95 Death Receptor. Mol. Cell 2016, 61, 602–613. [Google Scholar] [CrossRef]
- Tourneur, L.; Chiocchia, G. FADD: A regulator of life and death. Trends Immunol. 2010, 31, 260–269. [Google Scholar] [CrossRef]
- Marín-Rubio, J.L.; de Arriba, M.C.; Cobos-Fernández, M.A.; González-Sánchez, L.; Ors, I.; Sastre, I.; Fernández-Piqueras, J.; Villa-Morales, M. Deregulated FADD expression and phosphorylation in T-cell lymphoblastic lymphoma. Oncotarget 2016, 7, 61485–61499. [Google Scholar]
- Ramos-Miguel, A.; García-Sevilla, J.A.; Barr, A.M.; Bayer, T.A.; Falkai, P.; Leurgans, S.E.; Schneider, J.A.; Bennett, D.A.; Honer, W.G.; García-Fuster, M.J. Decreased cortical FADD protein is associated with clinical dementia and cognitive decline in an elderly community sample. Mol. Neurodegener. 2017, 12, 26. [Google Scholar] [CrossRef]
- Edathara, P.M.; Gorre, M.; Kagita, S.; Vuree, S.; Cingeetham, A.; Nanchari, S.R.; Meka, P.B.; Annamaneni, S.; Digumarthi, R.R.; Satti, V. Association of promoter polymorphisms of Fas-FasL genes with development of Chronic Myeloid Leukemia. Tumor Biol. 2016, 37, 5475–5484. [Google Scholar] [CrossRef]
- Huang, D.; Xiao, J.; Deng, X.; Ma, K.; Liang, H.; Shi, D.; Wu, F.; Shao, Z. Association between Fas/FasL gene polymorphism and musculoskeletal degenerative diseases: A meta-analysis. BMC Musculoskel. Disord. 2018, 19, 137. [Google Scholar] [CrossRef]
- Vallinoto, A.C.R.; Santana, B.B.; dos Santos, E.L.; do Espírito Santo, R.R.; Hermes, R.B.; Sousa, R.C.M.; Cayres-Vallinoto, I.; Machado, L.F.A.; Ishak, M.O.G.; Ishak, R. FAS-670A/G single nucleotide polymorphism may be associated with human T lymphotropic virus-1 infection and clinical evolution to TSP/HAM. Virus Res. 2012, 163, 178–182. [Google Scholar] [CrossRef]
- Vallinoto, A.C.R.; Santana, B.B.; Queiroz, M.A.F.; da Silva, A.N.M.R.; Cayres-Vallinoto, I.M.V.; da Costa, C.A.; de Sousa, M.S.; Ishak, R. Family Aggregation of HTLV-1 Infection Associated with FAS-670A/G Polymorphism: A Case Report. Front. Microbiol. 2018, 8, 2685. [Google Scholar] [CrossRef]
- Menezes, S.M.; Leal, F.E.; Dierckx, T.; Khouri, R.; Decanine, D.; Silva-Santos, G.; Schnitman, S.V.; Kruschewsky, R.; López, G.; Alvarez, C.; et al. A Fashi Lymphoproliferative Phenotype Reveals Non-Apoptotic Fas Signaling in HTLV-1-Associated Neuroinflammation. Front. Immunol. 2017, 8, 97. [Google Scholar] [CrossRef]
- Nguyen, T.-A.T.; Grimm, S.A.; Bushel, P.R.; Li, J.; Li, Y.; Bennett, B.D.; Lavender, C.A.; Ward, J.M.; Fargo, D.C.; Anderson, C.W.; et al. Revealing a human p53 universe. Nucleic Acids Res. 2018, 46, 8153–8167. [Google Scholar] [CrossRef]
- Müller, M.; Wilder, S.; Bannasch, D.; Israeli, D.; Lehlbach, K.; Li-Weber, M.; Friedman, S.L.; Galle, P.R.; Stremmel, W.; Oren, M.; et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 1998, 188, 2033–2045. [Google Scholar] [CrossRef]
- Willms, A.; Schittek, H.; Rahn, S.; Sosna, J.; Mert, U.; Adam, D.; Trauzold, A. Impact of p53 status on TRAIL-mediated apoptotic and non-apoptotic signaling in cancer cells. PLoS ONE 2019, 14, e0214847. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, K.; Yao, Y.; Zhang, K.; Zhou, F.; Zhu, L. Triggering p53 activation is essential in ziyuglycoside I-induced human retinoblastoma WERI-Rb-1 cell apoptosis. J. Biochem. Mol. Toxicol. 2018, 32. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Basu, A.; Haldar, S. The relationship between BcI2, Bax and p53: Consequences for cell cycle progression and cell death. Mol. Hum. Reprod. 1998, 4, 1099–1109. [Google Scholar] [CrossRef]
- Chen, B.-J.; Wu, Y.-L.; Tanaka, Y.; Zhang, W. Small Molecules Targeting c-Myc Oncogene: Promising Anti-Cancer Therapeutics. Int. J. Biol. Sci. 2014, 10, 1084–1096. [Google Scholar] [CrossRef]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef]
- Lüscher, B. Function and regulation of the transcription factors of the Myc/Max/Mad network. Gene 2001, 277, 1–14. [Google Scholar] [CrossRef]
- Murphy, D.J.; Junttila, M.R.; Pouyet, L.; Karnezis, A.; Shchors, K.; Bui, D.A.; Brown-Swigart, L.; Johnson, L.; Evan, G.I. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 2008, 14, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Trudel, M.; Lanoix, J.; Barisoni, L.; Blouin, M.-J.; Desforges, M.; L’Italien, C.; D’Agati, V. C-MYC–induced Apoptosis in Polycystic Kidney Disease Is Bcl-2 and p53 Independent. J. Exp. Med. 1997, 186, 1873–1884. [Google Scholar] [CrossRef] [PubMed]
- Boone, D.N.; Qi, Y.; Li, Z.; Hann, S.R. Egr1 mediates p53-independent c-Myc-induced apoptosis via a noncanonical ARF-dependent transcriptional mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Muthalagu, N.; Junttila, M.R.; Wiese, K.E.; Wolf, E.; Morton, J.; Bauer, B.; Evan, G.I.; Eilers, M.; Murphy, D.J. BIM is the primary mediator of MYC-induced apoptosis in multiple solid tissues. Cell Rep. 2014, 8, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, I.M.; Liu, X.; Zhou, M.; Li, F.; Li, C.-Y. Essential roles of Caspase-3 in facilitating Myc-induced genetic instability and carcinogenesis. Elife 2017, 6, e26371. [Google Scholar] [CrossRef] [PubMed]
- Knoepfler, P.S. Myc goes global: New tricks for an old oncogene. Cancer Res. 2007, 67, 5061–5063. [Google Scholar] [CrossRef]
- Winkle, M.; van den Berg, A.; Tayari, M.; Sietzema, J.; Terpstra, M.; Kortman, G.; de Jong, D.; Visser, L.; Diepstra, A.; Kok, K.; et al. Long noncoding RNAs as a novel component of the Myc transcriptional network. FASEB J. 2015, 29, 2338–2346. [Google Scholar] [CrossRef]
- Walters, J.; Pop, C.; Scott, F.L.; Drag, M.; Swartz, P.; Mattos, C.; Salvesen, G.S.; Clark, A.C. A constitutively active and uninhibitable caspase-3 zymogen efficiently induces apoptosis. Biochem. J. 2009, 424, 335–345. [Google Scholar] [CrossRef]
- Liu, X.; He, Y.; Li, F.; Huang, Q.; Kato, T.A.; Hall, R.P.; Li, C.-Y. Caspase-3 Promotes Genetic Instability and Carcinogenesis. Mol. Cell 2015, 58, 284–296. [Google Scholar] [CrossRef]
- Zhou, M.; Liu, X.; Li, Z.; Huang, Q.; Li, F.; Li, C.-Y. Caspase-3 regulates the migration, invasion and metastasis of colon cancer cells: Metastasis of colon cancer cells. Int. J. Cancer 2018, 143, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Know, L. Life—The Epic Story of Our Mitochondria: How the Original Probiotic Dictates Your Health, Illness, Ageing, and Even Life Itself; FriesenPress: Victoria, BC, Canada, 2014; ISBN 978-1-4602-5181-2. [Google Scholar]
- Karbowski, M.; Lee, Y.-J.; Gaume, B.; Jeong, S.-Y.; Frank, S.; Nechushtan, A.; Santel, A.; Fuller, M.; Smith, C.L.; Youle, R.J. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 2002, 159, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Miyazawa, T.; Kinoshita, K.; Yamada, Y.; Yoshida, T. Functional screening identifies a microRNA, miR-491 that induces apoptosis by targeting Bcl-X(L) in colorectal cancer cells. Int. J. Cancer 2010, 127, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Mullen, T.D.; Obeid, L.M. Ceramide and apoptosis: Exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer Agents Med. Chem. 2012, 12, 340–363. [Google Scholar] [CrossRef]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum–mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Jackson, R.; Harman, C.C.D.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.-Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond—Mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef]
- Kantari, C.; Walczak, H. Caspase-8 and bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta 2011, 1813, 558–563. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, J.; O’Neill, K.L.; Gurumurthy, C.B.; Quadros, R.M.; Tu, Y.; Luo, X. Cleavage by Caspase 8 and Mitochondrial Membrane Association Activate the BH3-only Protein Bid during TRAIL-induced Apoptosis. J. Biol. Chem. 2016, 291, 11843–11851. [Google Scholar] [CrossRef]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef]
- Snustad, D.P.; Simmons, M.J. Fundamentos de Genética; Guanabara Koogan: Rio de Janeiro, Brazil, 2008; Volume 4. [Google Scholar]
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589. [Google Scholar] [CrossRef]
- Picard, M.; Zhang, J.; Hancock, S.; Derbeneva, O.; Golhar, R.; Golik, P.; O’Hearn, S.; Levy, S.; Potluri, P.; Lvova, M.; et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. USA 2014, 111, E4033–E4042. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Castegna, A.; Infantino, V.; Andria, G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 2013, 110, 25–34. [Google Scholar] [CrossRef]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic Regulation of Motor Neuron Cell Death through DNA Methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- D’Aquila, P.; Bellizzi, D.; Passarino, G. Mitochondria in health, aging and diseases: The epigenetic perspective. Biogerontology 2015, 16, 569–585. [Google Scholar] [CrossRef]
- Maia, B.M.; Rocha, R.M.; Calin, G.A. Clinical significance of the interaction between non-coding RNAs and the epigenetics machinery: Challenges and opportunities in oncology. Epigenetics 2014, 9, 75–80. [Google Scholar] [CrossRef]
- Han, B.; Yao, W.; Oh, Y.-T.; Tong, J.-S.; Li, S.; Deng, J.; Yue, P.; Khuri, F.R.; Sun, S.-Y. The novel proteasome inhibitor carfilzomib activates and enhances extrinsic apoptosis involving stabilization of death receptor 5. Oncotarget 2015, 6, 17532–17542. [Google Scholar] [CrossRef]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.-M.J.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef]
- Lambertini, L.; Byun, H.-M. Mitochondrial Epigenetics and Environmental Exposure. Curr. Environ. Health Rep. 2016, 3, 214–224. [Google Scholar] [CrossRef]
- Wang, M.; Huang, T.; Luo, G.; Huang, C.; Xiao, X.-Y.; Wang, L.; Jiang, G.-S.; Zeng, F.-Q. Long non-coding RNA MEG3 induces renal cell carcinoma cells apoptosis by activating the mitochondrial pathway. J. Huazhong Univ. Sci. Technol. Med. Sci. 2015, 35, 541–545. [Google Scholar] [CrossRef]
- Kren, B.T.; Wong, P.Y.-P.; Sarver, A.; Zhang, X.; Zeng, Y.; Steer, C.J. MicroRNAs identified in highly purified liver-derived mitochondria may play a role in apoptosis. RNA Biol. 2009, 6, 65–72. [Google Scholar] [CrossRef]
- Borgna, V.; Villegas, J.; Burzio, V.A.; Belmar, S.; Araya, M.; Jeldes, E.; Lobos-González, L.; Silva, V.; Villota, C.; Oliveira-Cruz, L.; et al. Mitochondrial ASncmtRNA-1 and ASncmtRNA-2 as potent targets to inhibit tumor growth and metastasis in the RenCa murine renal adenocarcinoma model. Oncotarget 2017, 8, 43692–43708. [Google Scholar] [CrossRef]
- Bianchessi, V.; Badi, I.; Bertolotti, M.; Nigro, P.; D’Alessandra, Y.; Capogrossi, M.C.; Zanobini, M.; Pompilio, G.; Raucci, A.; Lauri, A. The mitochondrial lncRNA ASncmtRNA-2 is induced in aging and replicative senescence in Endothelial Cells. J. Mol. Cell. Cardiol. 2015, 81, 62–70. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, L.; Wang, R.R.; Hu, J.-F.; Cui, J. The effects of mitochondria-associated long noncoding RNAs in cancer mitochondria: New players in an old arena. Crit. Rev. Oncol. Hematol. 2018, 131, 76–82. [Google Scholar] [CrossRef]
- Lima, R.T.; Busacca, S.; Almeida, G.M.; Gaudino, G.; Fennell, D.A.; Vasconcelos, M.H. MicroRNA regulation of core apoptosis pathways in cancer. Eur. J. Cancer 2011, 47, 163–174. [Google Scholar] [CrossRef]
- Ji, F.; Zhang, H.; Wang, Y.; Li, M.; Xu, W.; Kang, Y.; Wang, Z.; Wang, Z.; Cheng, P.; Tong, D.; et al. MicroRNA-133a, downregulated in osteosarcoma, suppresses proliferation and promotes apoptosis by targeting Bcl-xL and Mcl-1. Bone 2013, 56, 220–226. [Google Scholar] [CrossRef]
- Zhang, Y.; Schiff, D.; Park, D.; Abounader, R. MicroRNA-608 and microRNA-34a regulate chordoma malignancy by targeting EGFR, Bcl-xL and MET. PLoS ONE 2014, 9, e91546. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A.; Miura, S.; Satoh, K.; Shimosegawa, T. MiR-365 induces gemcitabine resistance in pancreatic cancer cells by targeting the adaptor protein SHC1 and pro-apoptotic regulator BAX. Cell. Signal. 2014, 26, 179–185. [Google Scholar] [CrossRef]
- Zhou, M.; Liu, Z.; Zhao, Y.; Ding, Y.; Liu, H.; Xi, Y.; Xiong, W.; Li, G.; Lu, J.; Fodstad, O.; et al. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J. Biol. Chem. 2010, 285, 21496–21507. [Google Scholar] [CrossRef]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef]
- Koo, K.H.; Kwon, H. MicroRNA miR-4779 suppresses tumor growth by inducing apoptosis and cell cycle arrest through direct targeting of PAK2 and CCND3. Cell Death Dis. 2018, 9, 77. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, J.; Liu, W.; Wang, H.; Zhao, L.; Liu, S.; Li, P.; Zhang, S.; Sun, C.; Wu, Y.; et al. microRNA-378 promotes autophagy and inhibits apoptosis in skeletal muscle. PNAS 2018, 115, E10849–E10858. [Google Scholar] [CrossRef]
- Pernaute, B.; Spruce, T.; Smith, K.M.; Sánchez-Nieto, J.M.; Manzanares, M.; Cobb, B.; Rodríguez, T.A. MicroRNAs control the apoptotic threshold in primed pluripotent stem cells through regulation of BIM. Genes Dev. 2014, 28, 1873–1878. [Google Scholar] [CrossRef]
- Miller, T.E.; Ghoshal, K.; Ramaswamy, B.; Roy, S.; Datta, J.; Shapiro, C.L.; Jacob, S.; Majumder, S. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J. Biol. Chem. 2008, 283, 29897–29903. [Google Scholar] [CrossRef]
- Garofalo, M.; Quintavalle, C.; Di Leva, G.; Zanca, C.; Romano, G.; Taccioli, C.; Liu, C.G.; Croce, C.M.; Condorelli, G. MicroRNA signatures of TRAIL resistance in human non-small cell lung cancer. Oncogene 2008, 27, 3845–3855. [Google Scholar] [CrossRef]
- Wang, P.; Zhuang, L.; Zhang, J.; Fan, J.; Luo, J.; Chen, H.; Wang, K.; Liu, L.; Chen, Z.; Meng, Z. The serum miR-21 level serves as a predictor for the chemosensitivity of advanced pancreatic cancer, and miR-21 expression confers chemoresistance by targeting FasL. Mol. Oncol. 2013, 7, 334–345. [Google Scholar] [CrossRef]
- Shaffiey, F.; Cross, E.; Sathyanarayana, P. Mir-590 Is a Novel STAT5 Regulated Oncogenic miRNA and Targets FasL In Acute Myeloid Leukemia. Blood 2013, 122, 3811. [Google Scholar]
- Huang, G.; Nishimoto, K.; Zhou, Z.; Hughes, D.; Kleinerman, E.S. miR-20a encoded by the miR-17-92 cluster increases the metastatic potential of osteosarcoma cells by regulating Fas expression. Cancer Res. 2012, 72, 908–916. [Google Scholar] [CrossRef]
- Ovcharenko, D.; Kelnar, K.; Johnson, C.; Leng, N.; Brown, D. Genome-scale microRNA and small interfering RNA screens identify small RNA modulators of TRAIL-induced apoptosis pathway. Cancer Res. 2007, 67, 10782–10788. [Google Scholar] [CrossRef]
- Slattery, M.L.; Mullany, L.E.; Sakoda, L.C.; Wolff, R.K.; Samowitz, W.S.; Herrick, J.S. Dysregulated genes and miRNAs in the apoptosis pathway in colorectal cancer patients. Apoptosis 2018, 23, 237–250. [Google Scholar] [CrossRef]
- Abend, J.R.; Uldrick, T.; Ziegelbauer, J.M. Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi’s sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J. Virol. 2010, 84, 12139–12151. [Google Scholar] [CrossRef]
- Yamada, N.; Noguchi, S.; Kumazaki, M.; Shinohara, H.; Miki, K.; Naoe, T.; Akao, Y. Epigenetic regulation of microRNA-128a expression contributes to the apoptosis-resistance of human T-cell leukaemia jurkat cells by modulating expression of fas-associated protein with death domain (FADD). Biochim. Biophys. Acta 2014, 1843, 590–602. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavalcante, G.C.; Schaan, A.P.; Cabral, G.F.; Santana-da-Silva, M.N.; Pinto, P.; Vidal, A.F.; Ribeiro-dos-Santos, Â. A Cell’s Fate: An Overview of the Molecular Biology and Genetics of Apoptosis. Int. J. Mol. Sci. 2019, 20, 4133. https://doi.org/10.3390/ijms20174133
Cavalcante GC, Schaan AP, Cabral GF, Santana-da-Silva MN, Pinto P, Vidal AF, Ribeiro-dos-Santos Â. A Cell’s Fate: An Overview of the Molecular Biology and Genetics of Apoptosis. International Journal of Molecular Sciences. 2019; 20(17):4133. https://doi.org/10.3390/ijms20174133
Chicago/Turabian StyleCavalcante, Giovanna C., Ana Paula Schaan, Gleyce Fonseca Cabral, Mayara Natália Santana-da-Silva, Pablo Pinto, Amanda F. Vidal, and Ândrea Ribeiro-dos-Santos. 2019. "A Cell’s Fate: An Overview of the Molecular Biology and Genetics of Apoptosis" International Journal of Molecular Sciences 20, no. 17: 4133. https://doi.org/10.3390/ijms20174133
APA StyleCavalcante, G. C., Schaan, A. P., Cabral, G. F., Santana-da-Silva, M. N., Pinto, P., Vidal, A. F., & Ribeiro-dos-Santos, Â. (2019). A Cell’s Fate: An Overview of the Molecular Biology and Genetics of Apoptosis. International Journal of Molecular Sciences, 20(17), 4133. https://doi.org/10.3390/ijms20174133