Prediction of Potential Drug–Disease Associations through Deep Integration of Diversity and Projections of Various Drug Features

Abstract
1. Introduction
2. Experimental Evaluation and Discussion
2.1. Evaluation Metrics
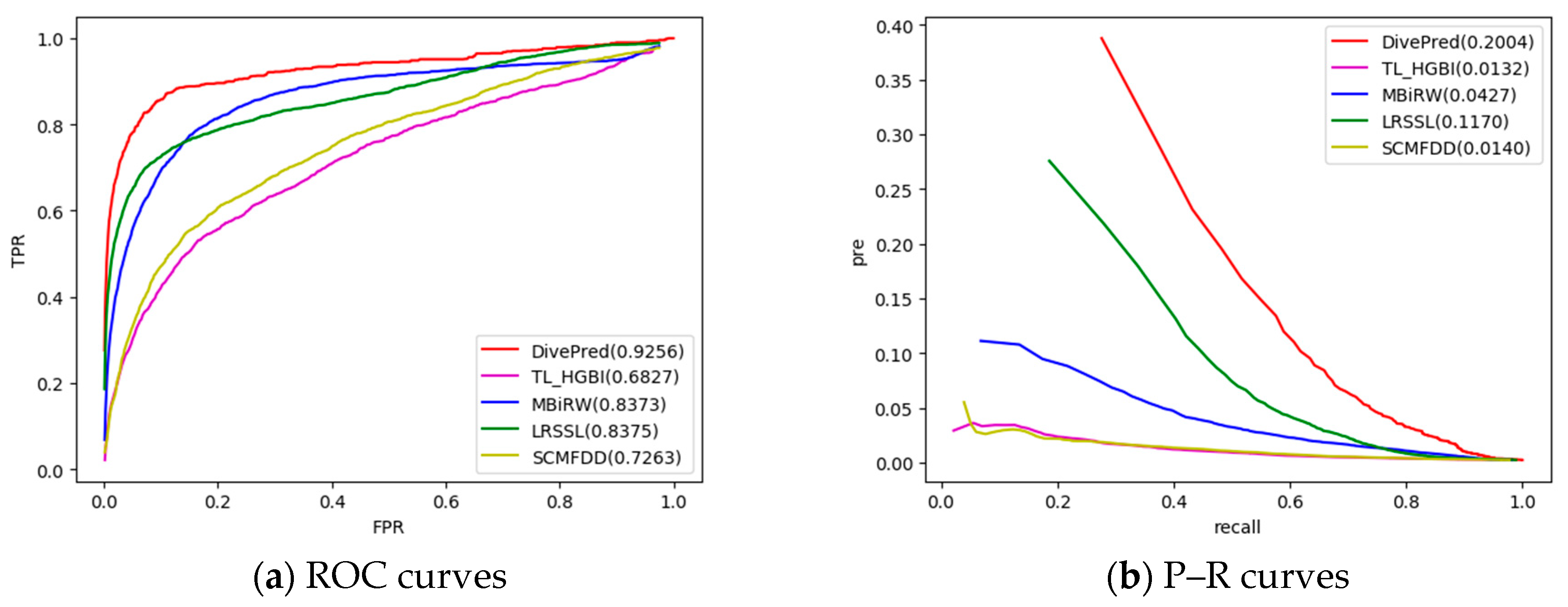
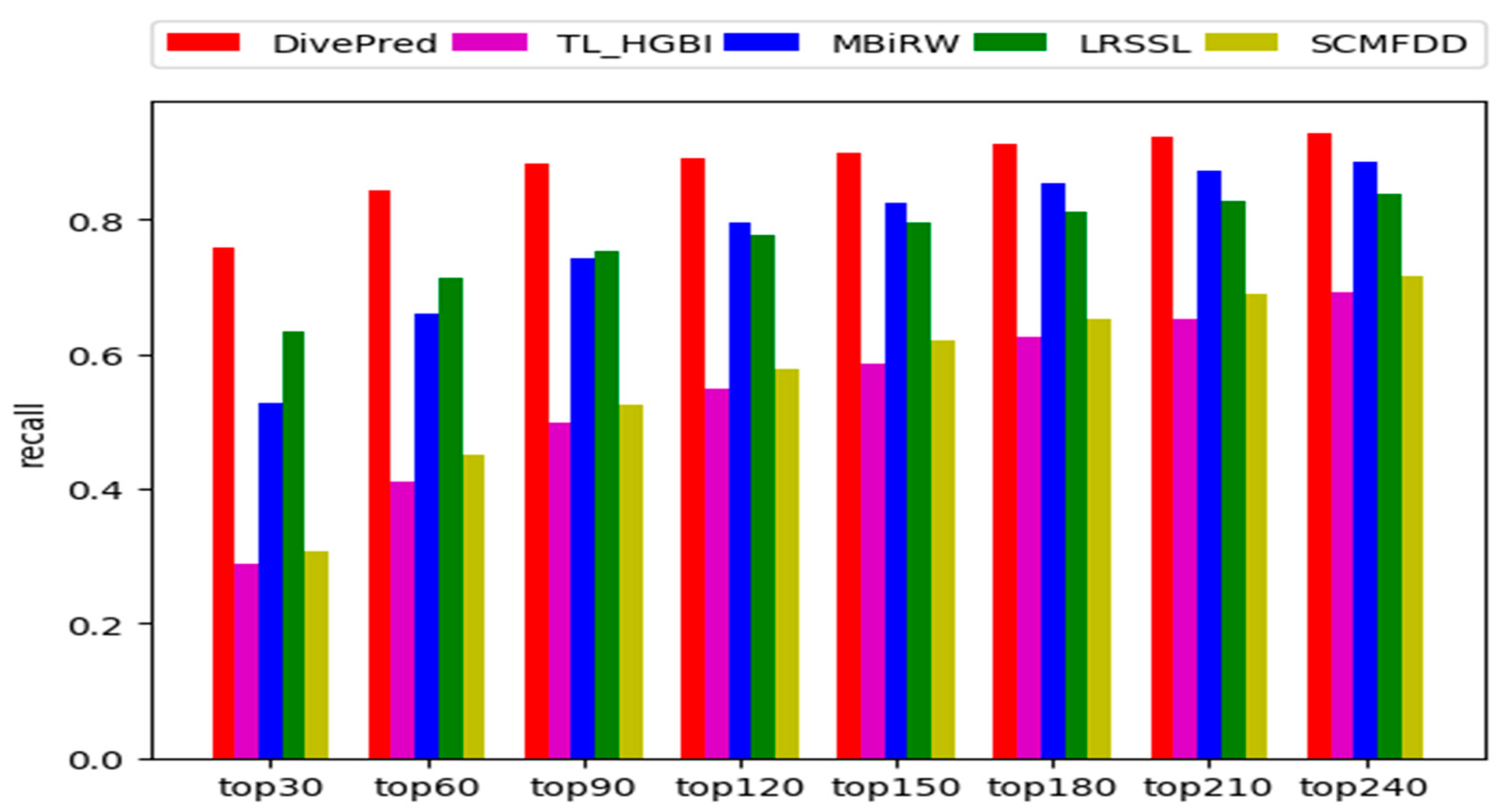
2.2. Comparison with Other Methods
2.3. Case Studies on Five Drugs
2.4. Prediction of Novel Drug–Disease Associations
3. Materials and Methods
3.1. Datasets for Drug–Disease Association Prediction
3.2. Representation of Multi-Source Data
3.3. Drug–Disease Association Prediction Model
3.4. Optimization
| Algorithm 1 DivePred algorithm for predicting the potential drug-disease associations. |
| Input: A drug-disease association matrix and the drugs character matrix. |
| Output: Drug-disease association score matrix , where is the association score for drug and disease . |
|
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, J.; Zheng, S.; Chen, B.; Butte, A.J.; Swamidass, S.J.; Lu, Z. A survey of current trends in computational drug repositioning. Brief. Bioinform. 2015, 17, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, A.; Oraiopoulos, N.; Drakeman, D.L. Renovation as innovation: Is repurposing the future of drug discovery research? Drug Discov. Today 2019, 24, 1. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.P.; Brantner, V.V. Estimating the cost of new drug development: Is it really $802 million? Health Aff. 2006, 25, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.; Gagnon, J.P. Key factors in the rising cost of new drug discovery and development. Nat. Rev. Drug Discov. 2004, 3, 417. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, N.A.; Ellis, P. Drug development: From concept to marketing! Nephron Clin. Pract. 2009, 113, c125–c131. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve r&d productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203. [Google Scholar]
- Sultana, J.; Calabró, M.; Garcia-Serna, R.; Ferrajolo, C.; Crisafulli, C.; Mestres, J. Biological substantiation of antipsychotic-associated pneumonia: Systematic literature review and computational analyses. PloS ONE 2017, 12, e0187034. [Google Scholar] [CrossRef]
- Grabowski, H. Are the economics of pharmaceutical research and development changing? Pharmacoeconomics 2004, 22, 15–24. [Google Scholar] [CrossRef]
- Kinch, M.S.; Griesenauer, R.H. 2017 in review: Fda approvals of new molecular entities. Drug Discov. Today 2018, 23, 1469–1473. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673. [Google Scholar] [CrossRef] [PubMed]
- Nosengo, N. Can you teach old drugs new tricks? Nat. News 2016, 534, 314. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.L.E.; O’Mara, T.A.; Glubb, D.M. Enhancing the promise of drug repositioning through genetics. Front. Pharmacol. 2017, 8, 896. [Google Scholar] [CrossRef] [PubMed]
- Haupt, V.J.; Schroeder, M. Old friends in new guise: Repositioning of known drugs with structural bioinformatics. Brief. Bioinform. 2011, 12, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Lotfi Shahreza, M.; Ghadiri, N.; Mousavi, S.R.; Varshosaz, J.; Green, J.R. A review of network-based approaches to drug repositioning. Brief. Bioinform. 2017, 19, 878–892. [Google Scholar] [CrossRef]
- Sirota, M.; Dudley, J.T.; Kim, J.; Chiang, A.P.; Morgan, A.A.; Sweet-Cordero, A.; Sage, J.; Butte, A.J. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci. Translat. Med. 2011, 3, 96ra77. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Hu, Q.; Li, S. Systematic analysis of new drug indications by drug-gene-disease coherent subnetworks. CPT: Pharmacomet. Syst. Pharmacol. 2014, 3, 1–9. [Google Scholar] [CrossRef]
- Yu, L.; Huang, J.; Ma, Z.; Zhang, J.; Zou, Y.; Gao, L. Inferring drug-disease associations based on known protein complexes. BMC Med. Genomic. 2015, 8, S2. [Google Scholar] [CrossRef]
- Peyvandipour, A.; Saberian, N.; Shafi, A.; Donato, M.; Draghici, S. A novel computational approach for drug repurposing using systems biology. Bioinformatics 2018, 34, 2817–2825. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, S.; Deng, N.; Wang, Y. Drug repositioning by kernel-based integration of molecular structure, molecular activity, and phenotype data. PloS ONE 2013, 8, e78518. [Google Scholar]
- Wang, W.; Yang, S.; Zhang, X.; Li, J. Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics 2014, 30, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Wang, J.; Li, M.; Luo, J.; Peng, X.; Wu, F.X.; Pan, Y. Drug repositioning based on comprehensive similarity measures and bi-random walk algorithm. Bioinformatics 2016, 32, 2664–2671. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Song, Y.; Guan, J.; Luo, L.; Zhuang, Z. Inferring new indications for approved drugs via random walk on drug-disease heterogenous networks. BMC Bioinformatics 2016, 17, 539. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Li, M.; Wang, S.; Liu, Q.; Li, Y.; Wang, J. Computational drug repositioning using low-rank matrix approximation and randomized algorithms. Bioinformatics 2018, 34, 1904–1912. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.; Stein, G.Y.; Ruppin, E.; Sharan, R. Predict: A method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol. 2011, 7, 496. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Sawada, R.; Mizutani, S.; Yamanishi, Y. Systematic drug repositioning for a wide range of diseases with integrative analyses of phenotypic and molecular data. J. Chem. Inf. Modeling 2015, 55, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Zhang, P.; Yan, L.; Fu, Y.; Peng, F.; Qu, L.; Shao, M.; Chen, Y.; Chen, Z. Lrssl: Predict and interpret drug–disease associations based on data integration using sparse subspace learning. Bioinformatics 2017, 33, 1187–1196. [Google Scholar] [CrossRef]
- Zhang, W.; Yue, X.; Lin, W.; Wu, W.; Liu, R.; Huang, F.; Liu, F. Predicting drug-disease associations by using similarity constrained matrix factorization. BMC Bioinformatics 2018, 19, 233. [Google Scholar] [CrossRef]
- Xuan, P.; Cao, Y.; Zhang, T.; Wang, X.; Pan, S.; Shen, T. Drug repositioning through integration of prior knowledge and projections of drugs and diseases. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Bradley, J.D.; Brandt, K.D.; Katz, B.P.; Kalasinski, L.A.; Ryan, S.I. Comparison of an antiinflammatory dose of ibuprofen, an analgesic dose of ibuprofen, and acetaminophen in the treatment of patients with osteoarthritis of the knee. N. Engl. J. Med. 1991, 325, 87–91. [Google Scholar] [CrossRef]
- Stolman, L.P. Hyperhidrosis: Medical and surgical treatment. Eplasty 2008, 8, e22. [Google Scholar] [PubMed]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. Pubchem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microrna functional similarity and functional network based on microrna-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, N.; Dhillon, I.S. Inductive matrix completion for predicting gene–disease associations. Bioinformatics 2014, 30, i60–i68. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, L.; Qu, J.; Guan, N.N.; Li, J.Q. Predicting mirna–disease association based on inductive matrix completion. Bioinformatics 2018, 34, 4256–4265. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, X.; Yin, J. A novel computational method for the identification of potential mirna-disease association based on symmetric non-negative matrix factorization and kronecker regularized least square. Front. Genet. 2018, 9. [Google Scholar] [CrossRef]
- Wang, J.; Tian, F.; Yu, H.; Liu, C.H.; Zhan, K.; Wang, X. Diverse non-negative matrix factorization for multiview data representation. IEEE Trans. Cybern. 2017, 48, 2620–2632. [Google Scholar] [CrossRef]
- Tan, V.Y.; Févotte, C. Automatic relevance determination in nonnegative matrix factorization with the/spl beta/-divergence. IEEE Trans. Pattern Anal. Mach. Intell. 2012, 35, 1592–1605. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug Name | AUC DivePred | TL_HGBI | MBiRW | LRSSL | SCMFDD |
|---|---|---|---|---|---|
| ampicillin | 0.944 | 0.751 | 0.932 | 0.962 | 0.895 |
| cefepime | 0.976 | 0.910 | 0.970 | 0.971 | 0.914 |
| cefotaxime | 0.992 | 0.917 | 0.929 | 0.950 | 0.953 |
| cefotetan | 0.996 | 0.808 | 0.918 | 0.948 | 0.848 |
| cefoxitin | 0.979 | 0.890 | 0.912 | 0.979 | 0.894 |
| ceftazidime | 0.985 | 0.845 | 0.931 | 0.936 | 0.922 |
| ceftizoxime | 0.797 | 0.960 | 0.961 | 0.923 | 0.962 |
| ceftriaxone | 0.907 | 0.945 | 0.898 | 0.955 | 0.811 |
| ciprofloxacin | 0.957 | 0.811 | 0.813 | 0.928 | 0.820 |
| doxorubicin | 0.949 | 0.487 | 0.921 | 0.727 | 0.460 |
| erythromycin | 0.962 | 0.827 | 0.887 | 0.918 | 0.764 |
| itraconazole | 0.952 | 0.445 | 0.877 | 0.845 | 0.730 |
| levofloxacin | 0.975 | 0.943 | 0.975 | 0.964 | 0.872 |
| moxifloxacin | 0.794 | 0.812 | 0.948 | 0.957 | 0.932 |
| ofloxacin | 0.958 | 0.902 | 0.943 | 0.904 | 0.774 |
| Average AUC | 0.926 | 0.683 | 0.837 | 0.838 | 0.726 |
| Drug Name | AUPR DivePred | TL_HGBI | MBIRW | LRSSL | SCMFDD |
|---|---|---|---|---|---|
| ampicillin | 0.189 | 0.032 | 0.023 | 0.285 | 0.068 |
| cefepime | 0.744 | 0.163 | 0.315 | 0.625 | 0.054 |
| cefotaxime | 0.770 | 0.071 | 0.292 | 0.283 | 0.105 |
| cefotetan | 0.486 | 0.054 | 0.197 | 0.512 | 0.059 |
| cefoxitin | 0.580 | 0.151 | 0.394 | 0.286 | 0.065 |
| ceftazidime | 0.675 | 0.032 | 0.201 | 0.488 | 0.694 |
| ceftizoxime | 0.647 | 0.212 | 0.244 | 0.455 | 0.096 |
| ceftriaxone | 0.409 | 0.056 | 0.223 | 0.673 | 0.077 |
| ciprofloxacin | 0.425 | 0.082 | 0.118 | 0.280 | 0.064 |
| doxorubicin | 0.164 | 0.005 | 0.051 | 0.180 | 0.004 |
| erythromycin | 0.425 | 0.023 | 0.038 | 0.144 | 0.022 |
| itraconazole | 0.188 | 0.006 | 0.253 | 0.042 | 0.008 |
| levofloxacin | 0.504 | 0.136 | 0.071 | 0.539 | 0.098 |
| moxifloxacin | 0.565 | 0.049 | 0.065 | 0.384 | 0.088 |
| ofloxacin | 0.378 | 0.091 | 0.130 | 0.201 | 0.078 |
| Average AUC | 0.200 | 0.013 | 0.043 | 0.117 | 0.014 |
| p-Value Between DivePred and Another Method | TL_HGBI | MBiRW | LRSSL | SCMFDD |
|---|---|---|---|---|
| p-value of ROC curve | 5.631 × 10−42 | 7.181 × 10−156 | 3.735 × 10−78 | 6.596 × 10−73 |
| p-value of PR curve | 1.332 × 10−21 | 2.635 × 10−32 | 1.562 × 10−16 | 8.452 × 10−29 |
| Drug Name | Rank | Disease Name | Description | Rank | Disease Name | Description |
|---|---|---|---|---|---|---|
| Acetaminophen | 1 | Osteoarthritis | CTD | 9 | Arthritis | DrugBank |
| 2 | Arthritis, Rheumatoid | CTD | 10 | Pain, Postoperative | CTD | |
| 3 | Inflammation | CTD | 11 | Rheumatic Fever | PubChem | |
| 4 | Dysmenorrhea | inferred candidate by 1 literature | 12 | Arthritis, Gouty | CTD | |
| 5 | Arthritis, Juvenile Rheumatoid | DrugBank | 13 | Premenstrual Syndrome | DrugBank | |
| 6 | Gout | DrugBank | 14 | Menorrhagia | unconfirmed | |
| 7 | Spondylitis, Ankylosing | Clinicaltrials | 15 | Rheumatic Diseases | Clinicaltrials | |
| 8 | Bursitis | literature [30] | ||||
| Ciprofloxacin | 1 | Salmonella Infections | CTD | 9 | Pyelonephritis | CTD |
| 2 | Streptococcal Infections | DrugBank | 10 | Bacterial Infections | CTD | |
| 3 | Bronchitis | CTD | 11 | Serratia Infections | DrugBank | |
| 4 | Pneumonia, Bacterial | CTD | 12 | Tuberculosis, Pulmonary | CTD | |
| 5 | Chlamydia Infections | CTD | 13 | Plague | CTD | |
| 6 | Gram-Negative Bacterial Infections | CTD | 14 | Brucellosis | PubChem | |
| 7 | Enterobacteriaceae Infections | CTD | 15 | Chlamydiaceae Infections | PubChem | |
| 8 | Soft Tissue Infections | CTD | ||||
| Doxorubicin | 1 | Leukemia, Myeloid, Acute | CTD | 9 | Rhabdomyosarcoma | CTD |
| 2 | Precursor Cell Lymphoblastic Leukemia-Lymphoma | CTD | 10 | Histiocytosis | Clinicaltrials | |
| 3 | Carcinoma, Non-Small-Cell Lung | PubChem | 11 | Trophoblastic Neoplasms | DrugBank | |
| 4 | Mycosis Fungoides | PubChem | 12 | Stomach Neoplasms | CTD | |
| 5 | Leukemia, Lymphocytic, Chronic, B-Cell | inferred candidate by 14 literatures | 13 | Hodgkin Disease | CTD | |
| 6 | Head and Neck Neoplasms | CTD | 14 | Melanoma | CTD | |
| 7 | Sarcoma, Kaposi | CTD | 15 | Leukemia, Myelogenous, Chronic, BCR-ABL Positive | DrugBank | |
| 8 | Leukemia, Lymphoid | CTD | ||||
| Hydrocortisone | 1 | Asthma | CTD | 9 | Shock, Septic | CTD |
| 2 | Rhinitis, Allergic, Perennial | DrugBank | 10 | Acne Vulgaris | unconfirmed | |
| 3 | Dermatitis | PubChem | 11 | Rosacea | CTD | |
| 4 | Skin Diseases | CTD | 12 | Addison Disease | CTD | |
| 5 | Pruritus | PubChem | 13 | Hyperhidrosis | literature [31] | |
| 6 | Keratosis | inferred candidate by 1 literature | 14 | Hematologic Diseases | inferred candidate by 1 literature | |
| 7 | Hypersensitivity | inferred candidate by 7 literatures | 15 | Pityriasis Rosea | unconfirmed | |
| 8 | Psoriasis | PubChem | ||||
| Ampicillin | 1 | Proteus Infections | CTD | 9 | Osteomyelitis | Clinicaltrials |
| 2 | Streptococcal Infections | CTD | 10 | Impetigo | unconfirmed | |
| 3 | Septicemia | DrugBank | 11 | Serratia Infections | CTD | |
| 4 | Pneumonia, Bacterial | CTD | 12 | Peritonitis | CTD | |
| 5 | Bone Diseases, Infectious | PubChem | 13 | Bacterial Infections | CTD | |
| 6 | Staphylococcal Skin Infections | DrugBank | 14 | Enterobacteriaceae Infections | DrugBank | |
| 7 | Wound Infection | CTD | 15 | Cellulitis | CTD | |
| 8 | Pseudomonas Infections | PubChem |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xuan, P.; Song, Y.; Zhang, T.; Jia, L. Prediction of Potential Drug–Disease Associations through Deep Integration of Diversity and Projections of Various Drug Features. Int. J. Mol. Sci. 2019, 20, 4102. https://doi.org/10.3390/ijms20174102
Xuan P, Song Y, Zhang T, Jia L. Prediction of Potential Drug–Disease Associations through Deep Integration of Diversity and Projections of Various Drug Features. International Journal of Molecular Sciences. 2019; 20(17):4102. https://doi.org/10.3390/ijms20174102
Chicago/Turabian StyleXuan, Ping, Yingying Song, Tiangang Zhang, and Lan Jia. 2019. "Prediction of Potential Drug–Disease Associations through Deep Integration of Diversity and Projections of Various Drug Features" International Journal of Molecular Sciences 20, no. 17: 4102. https://doi.org/10.3390/ijms20174102
APA StyleXuan, P., Song, Y., Zhang, T., & Jia, L. (2019). Prediction of Potential Drug–Disease Associations through Deep Integration of Diversity and Projections of Various Drug Features. International Journal of Molecular Sciences, 20(17), 4102. https://doi.org/10.3390/ijms20174102