Computational Mechanistic Insights on the NO Oxidation Reaction Catalyzed by Non-Heme Biomimetic Cr-N-Tetramethylated Cyclam Complexes





Abstract
1. Introduction
2. Results and Discussion
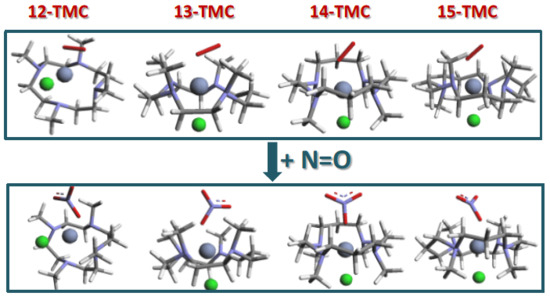
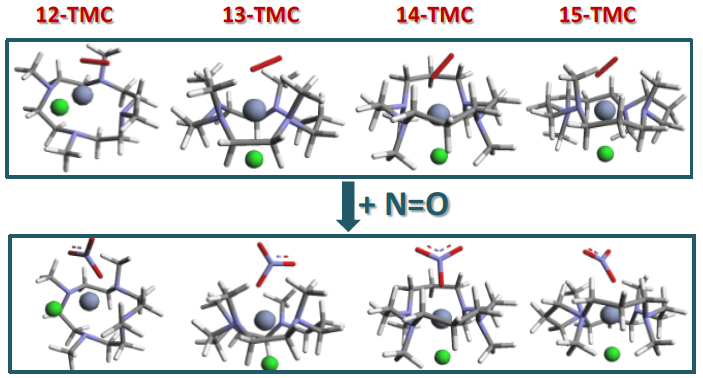
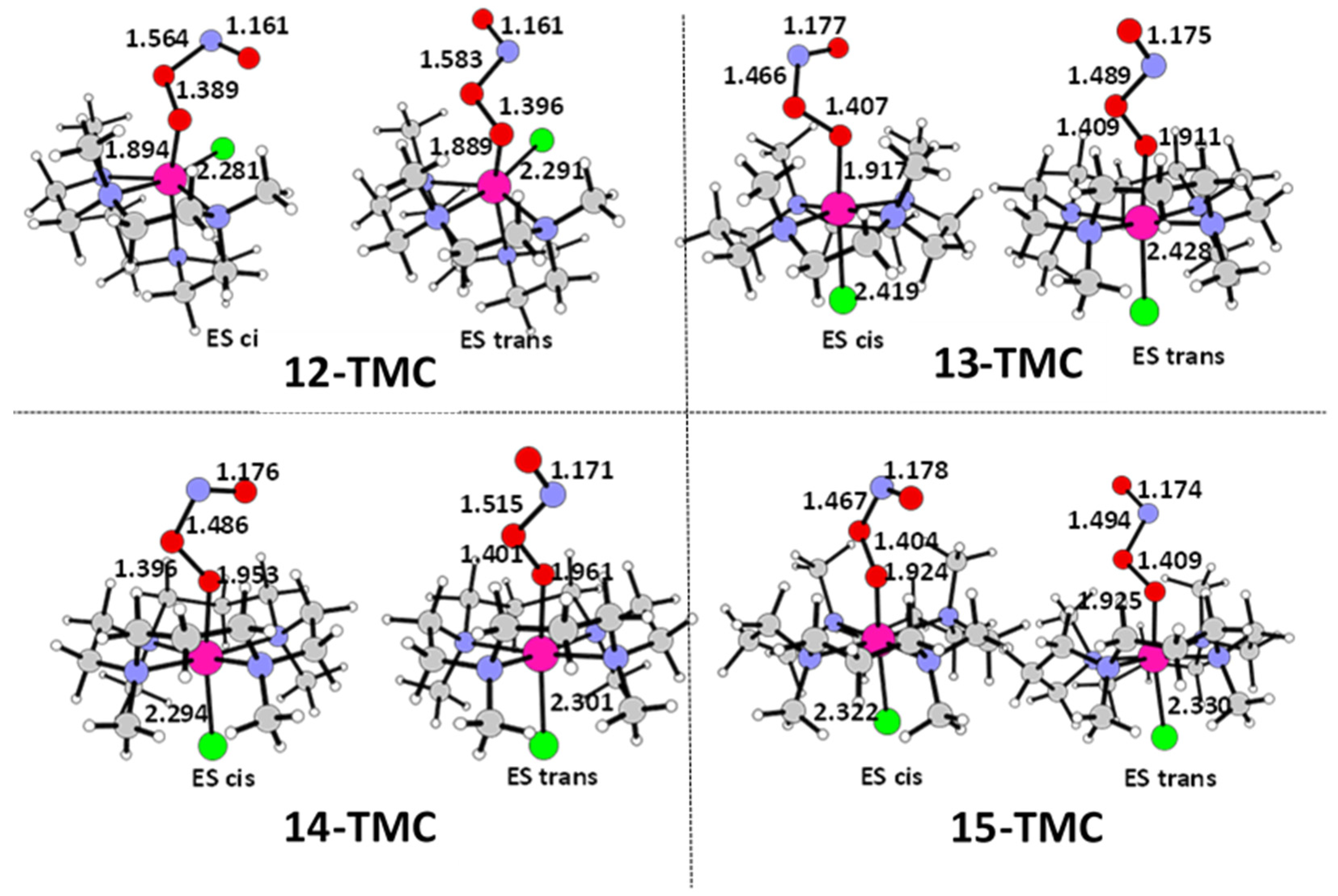
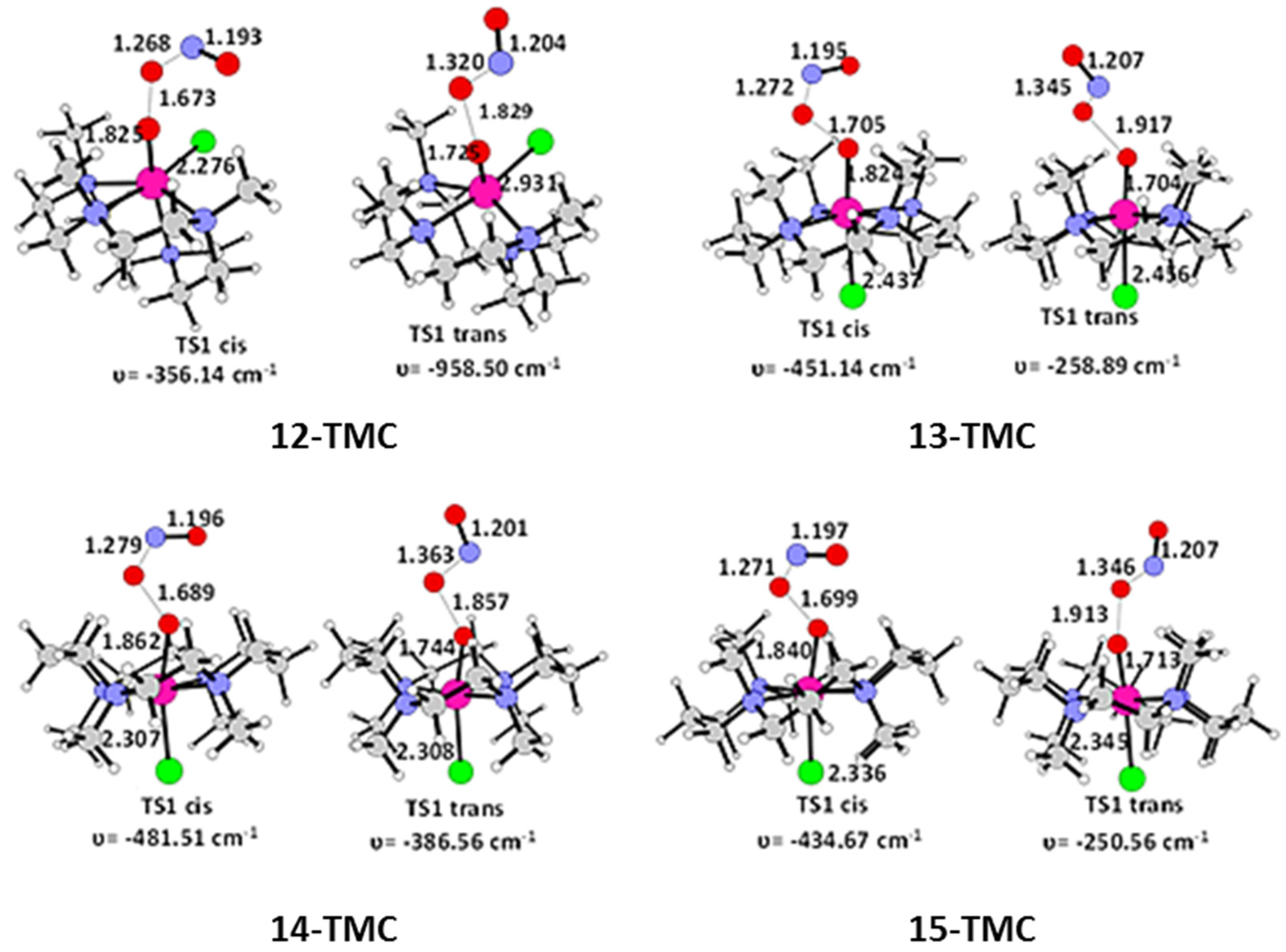
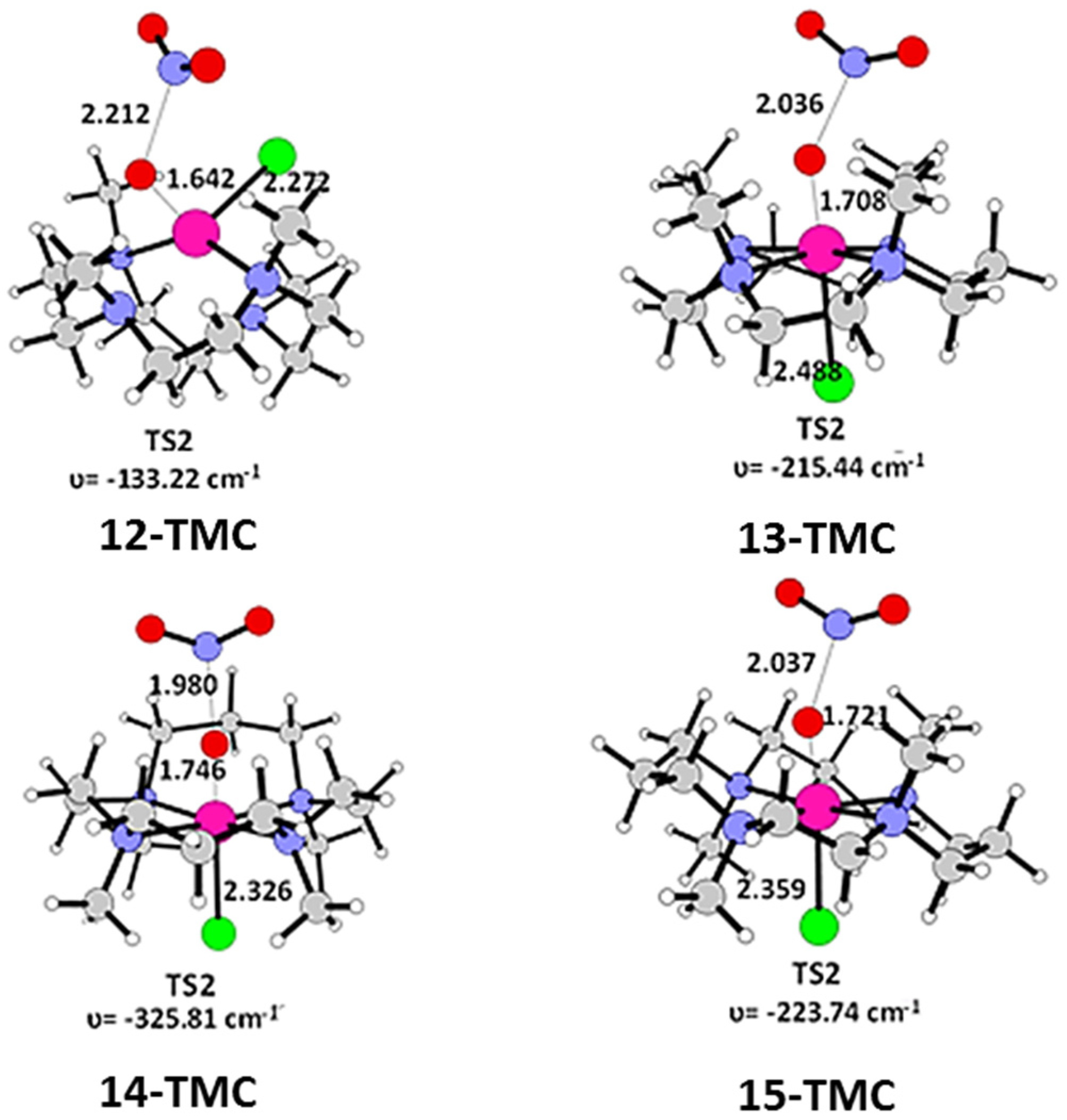
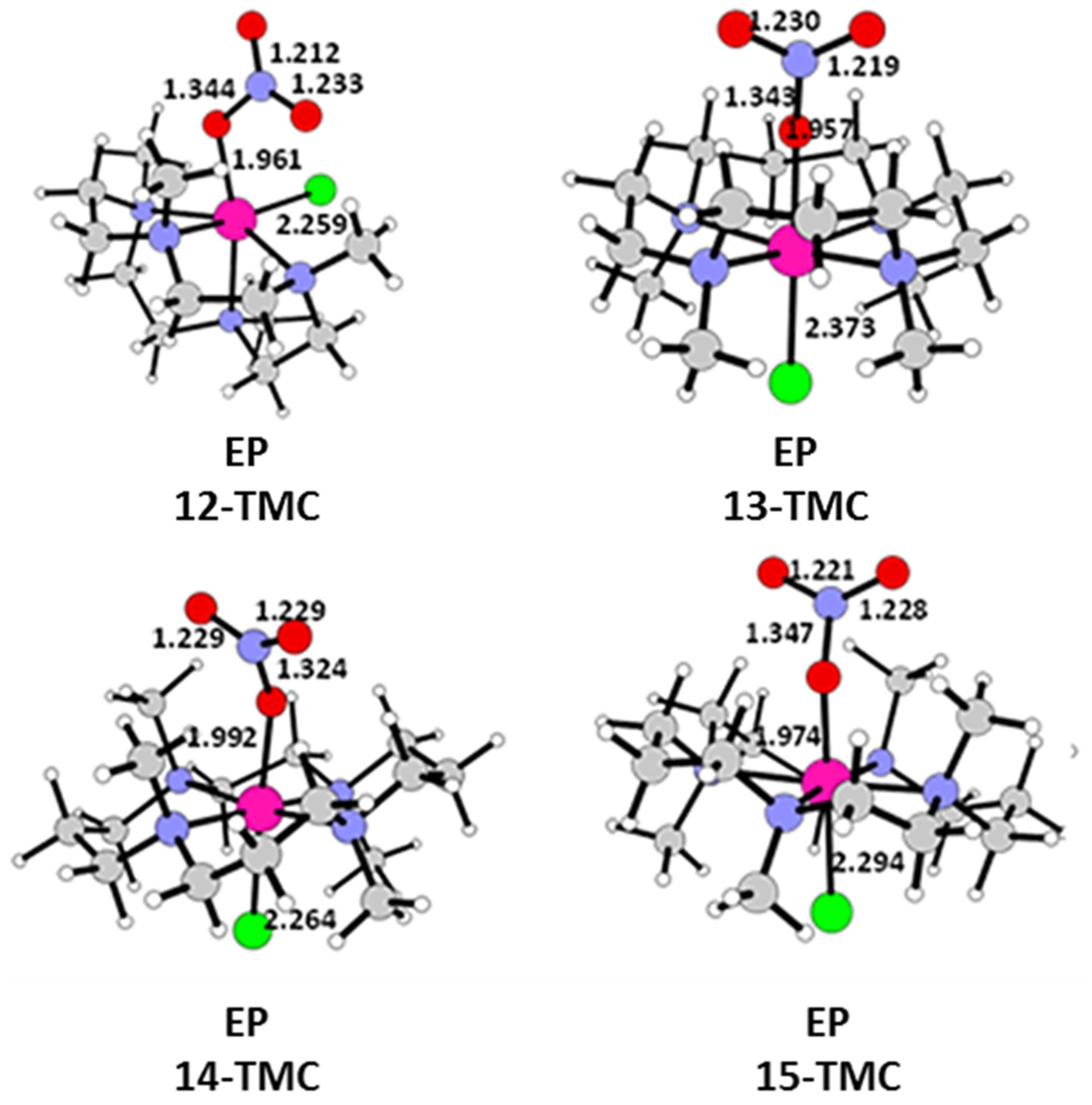
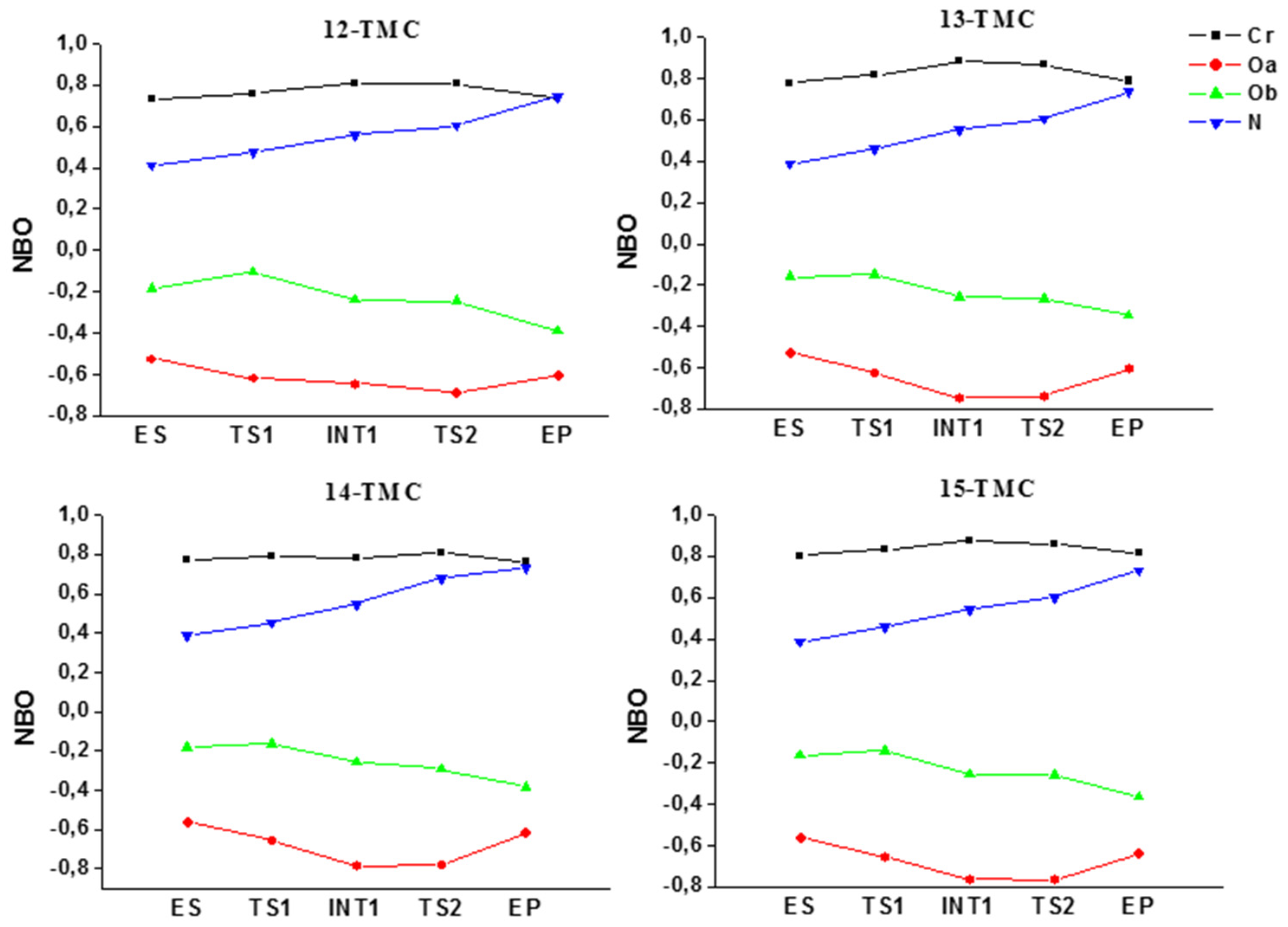
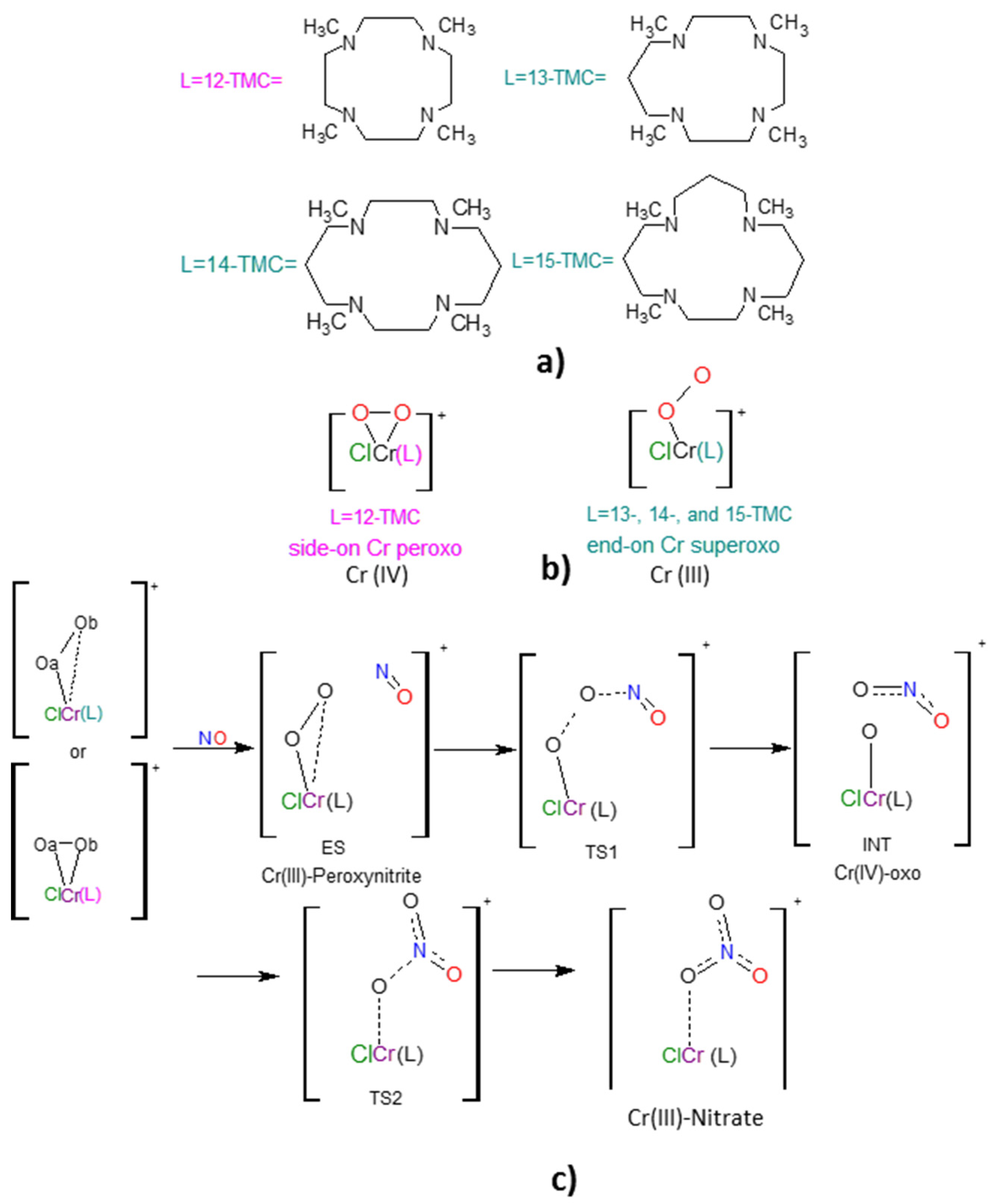
2.1. Structural Aspects
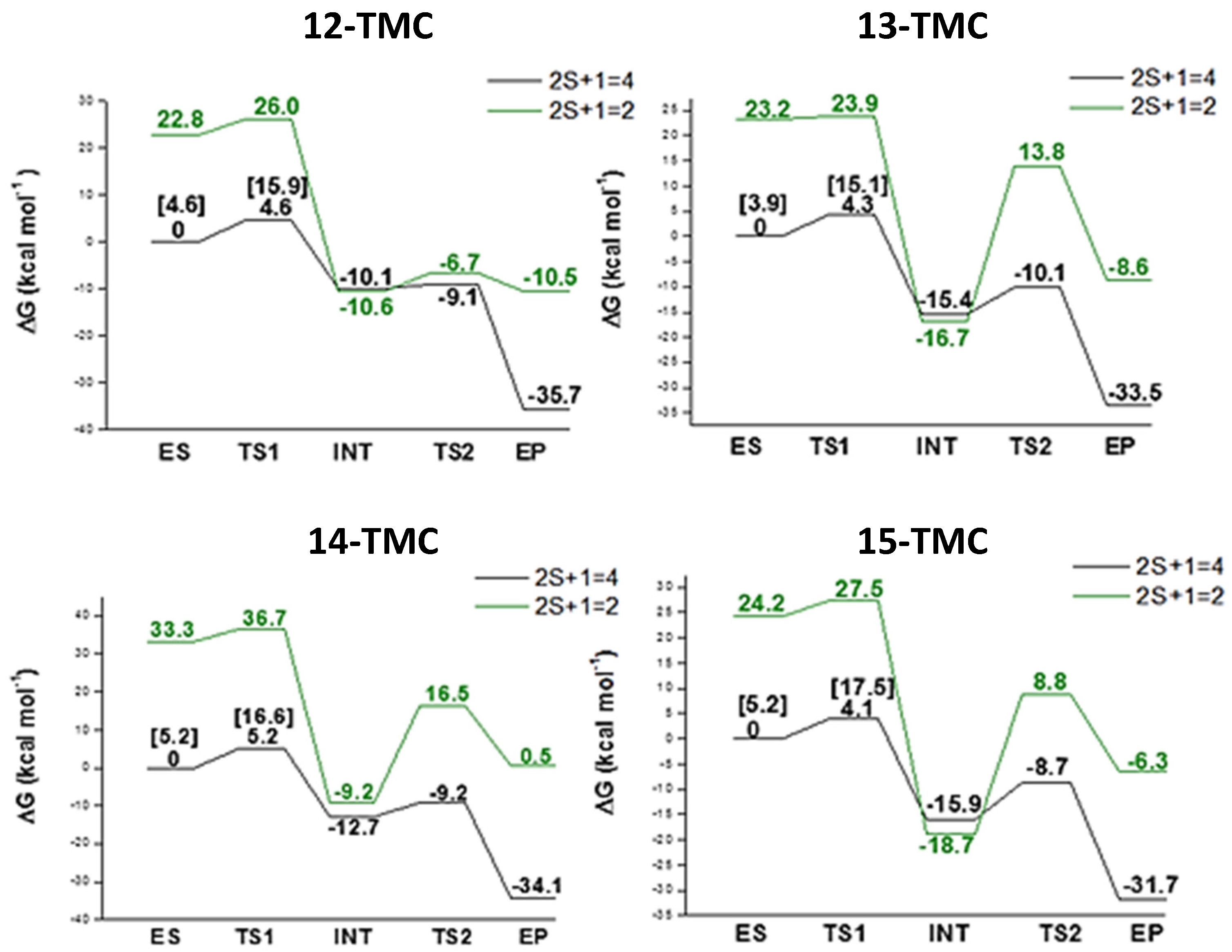
2.2. NO Conversion Mechanism
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TMC | N-tetramethylated cyclam I |
| NODs | Nitric oxide dioxygenases |
| PES | Potential Energy Surface |
| IRC | Intrinsic reaction coordinate |
| SMD | Solvation model based on the quantum mechanical charge density |
| NBO | Natural Bond Orbital |
References
- Cho, J.; Kang, H.Y.; Liu, L.V.; Sarangi, R.; Solomon, E.I.; Nam, W. Mononuclear nickel(II)-superoxo and nickel(III)-peroxo complexes bearing a common macrocyclic TMC ligand. Chem. Sci. 2013, 4, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.; Pfaff, F.F.; Wang, B.; Nam, W. Status of Reactive Non-Heme Metal–Oxygen Intermediates in Chemical and Enzymatic Reactions. J. Am. Chem. Soc. 2014, 136, 13942–13958. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Sarangi, R.; Nam, W. Mononuclear Metal–O2 Complexes Bearing Macrocyclic N-Tetramethylated Cyclam Ligands. Acc. Chem. Res. 2012, 45, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Barefield, E.K. Coordination chemistry of N-tetraalkylated cyclam ligands—A status report. Coord. Chem. Rev. 2010, 254, 1607–1627. [Google Scholar] [CrossRef]
- Cho, J.; Sarangi, R.; Kang, H.Y.; Lee, J.Y.; Kubo, M.; Ogura, T.; Solomon, E.I.; Nam, W. Synthesis, Structural, and Spectroscopic Characterization and Reactivities of Mononuclear Cobalt(III)−Peroxo Complexes. J. Am. Chem. Soc. 2010, 132, 16977–16986. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Sarangi, R.; Annaraj, J.; Kim, S.Y.; Kubo, M.; Ogura, T.; Solomon, E.I.; Nam, W. Geometric and electronic structure and reactivity of a mononuclear ‘side-on’ nickel(iii)–peroxo complex. Nat. Chem. 2009, 1, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cho, K.-B.; Lai, W.; Nam, W.; Shaik, S. Dioxygen Activation by a Non-Heme Iron(II) Complex: Theoretical Study toward Understanding Ferric-Superoxo Complexes. J. Chem. Theory Comput. 2012, 8, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Cho, J.; Lee, Y.-M.; Sarangi, R.; Nam, W. Synthesis, Characterization, and Reactivity of Cobalt(III)–Oxygen Complexes Bearing a Macrocyclic N-Tetramethylated Cyclam Ligand. Chem. Eur. J. 2013, 19, 14112–14118. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Han, J.E.; Karlin, K.D.; Nam, W. An isoelectronic NO dioxygenase reaction using a nonheme iron(III)-peroxo complex and nitrosonium ion. Chem. Commun. 2014, 50, 1742–1744. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Han, J.E.; Cho, J.; Kubo, M.; Ogura, T.; Siegles, M.A.; Karlin, K.D.; Nam, W. Chromium(IV)–Peroxo Complex Formation and Its Nitric Oxide Dioxygenase Reactivity. J. Am. Chem. Soc. 2012, 134, 15269–15272. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Cho, K.-B.; Karlin, K.D.; Nam, W. Reactions of a Chromium(III)-Superoxo Complex and Nitric Oxide That Lead to the Formation of Chromium(IV)-Oxo and Chromium(III)-Nitrito Complexes. J. Am. Chem. Soc. 2013, 135, 14900–14903. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Woo, J.; Nam, W. An “End-On” Chromium(III)-Superoxo Complex: Crystallographic and Spectroscopic Characterization and Reactivity in C−H Bond Activation of Hydrocarbons. J. Am. Chem. Soc. 2010, 132, 5958–5959. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-B.; Kang, H.; Woo, J.; Park, Y.J.; Seo, M.S.; Cho, J.; Nam, W. Mechanistic Insights into the C–H Bond Activation of Hydrocarbons by Chromium(IV) Oxo and Chromium(III) Superoxo Complexes. Inorg. Chem. 2014, 53, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed]
- Valero, R.; Costa, R.; Moreira, I.D.P.R.; Truhlar, D.G.; Illas, F. Performance of the M06 family of exchange-correlation functionals for predicting magnetic coupling in organic and inorganic molecules. J. Chem. Phys. 2008, 128, 114103. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Fukui, K.J. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Z.; Cheng, R.; He, X.; Liu, B. Mechanistic DFT Study on Ethylene Trimerization of Chromium Catalysts Supported by a Versatile Pyrrole Ligand System. Organometallics 2014, 33, 2599–2607. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO, version 3.1.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 12-TMC | 13-TMC | 14-TMC | 15-TMC | |
|---|---|---|---|---|
| Cr-Cl | 2.277 | 2.424 | 2.311 | 2.347 |
| Cr-Oa | 1.887 | 1.972 | 1.919 | 1.894 |
| Cr-Ob | 2.598 | 2.716 | 2.999 | 2.902 |
| O-O | 1.299 | 1.315 | 1.307 | 1.304 |
| Cr-O-O | 107.8 | 109.9 | 135.9 | 129.4 |
| 12-TMC | 13-TMC | 14-TMC | 15-TMC | |
|---|---|---|---|---|
| ES_trans | 3.7502 | 3.7502 | 3.7502 | 3.7502 |
| ES_cis | 3.7502 | 3.7502 | 3.7502 | 3.7502 |
| TS1_trans | 3.7502 | 3.7524 | 3.7502 | 3.7502 |
| TS1_cis | 3.7507 | 3.7507 | 3.7506 | 3.7507 |
| INT | 3.7519 | 3.8107 | 3.7519 | 3.7520 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marino, T.; Fortino, M.G.; Russo, N.; Toscano, M.; Alberto, M.E. Computational Mechanistic Insights on the NO Oxidation Reaction Catalyzed by Non-Heme Biomimetic Cr-N-Tetramethylated Cyclam Complexes. Int. J. Mol. Sci. 2019, 20, 3955. https://doi.org/10.3390/ijms20163955
Marino T, Fortino MG, Russo N, Toscano M, Alberto ME. Computational Mechanistic Insights on the NO Oxidation Reaction Catalyzed by Non-Heme Biomimetic Cr-N-Tetramethylated Cyclam Complexes. International Journal of Molecular Sciences. 2019; 20(16):3955. https://doi.org/10.3390/ijms20163955
Chicago/Turabian StyleMarino, Tiziana, Maria Grazia Fortino, Nino Russo, Marirosa Toscano, and Marta Erminia Alberto. 2019. "Computational Mechanistic Insights on the NO Oxidation Reaction Catalyzed by Non-Heme Biomimetic Cr-N-Tetramethylated Cyclam Complexes" International Journal of Molecular Sciences 20, no. 16: 3955. https://doi.org/10.3390/ijms20163955
APA StyleMarino, T., Fortino, M. G., Russo, N., Toscano, M., & Alberto, M. E. (2019). Computational Mechanistic Insights on the NO Oxidation Reaction Catalyzed by Non-Heme Biomimetic Cr-N-Tetramethylated Cyclam Complexes. International Journal of Molecular Sciences, 20(16), 3955. https://doi.org/10.3390/ijms20163955