The Cyclooxigenase-2 Inhibitor Parecoxib Prevents Epidermal Dysplasia in HPV16-Transgenic Mice: Efficacy and Safety Observations

 , ,
, ,  , ,
, , 


 ,
, 
Abstract
1. Introduction
2. Results
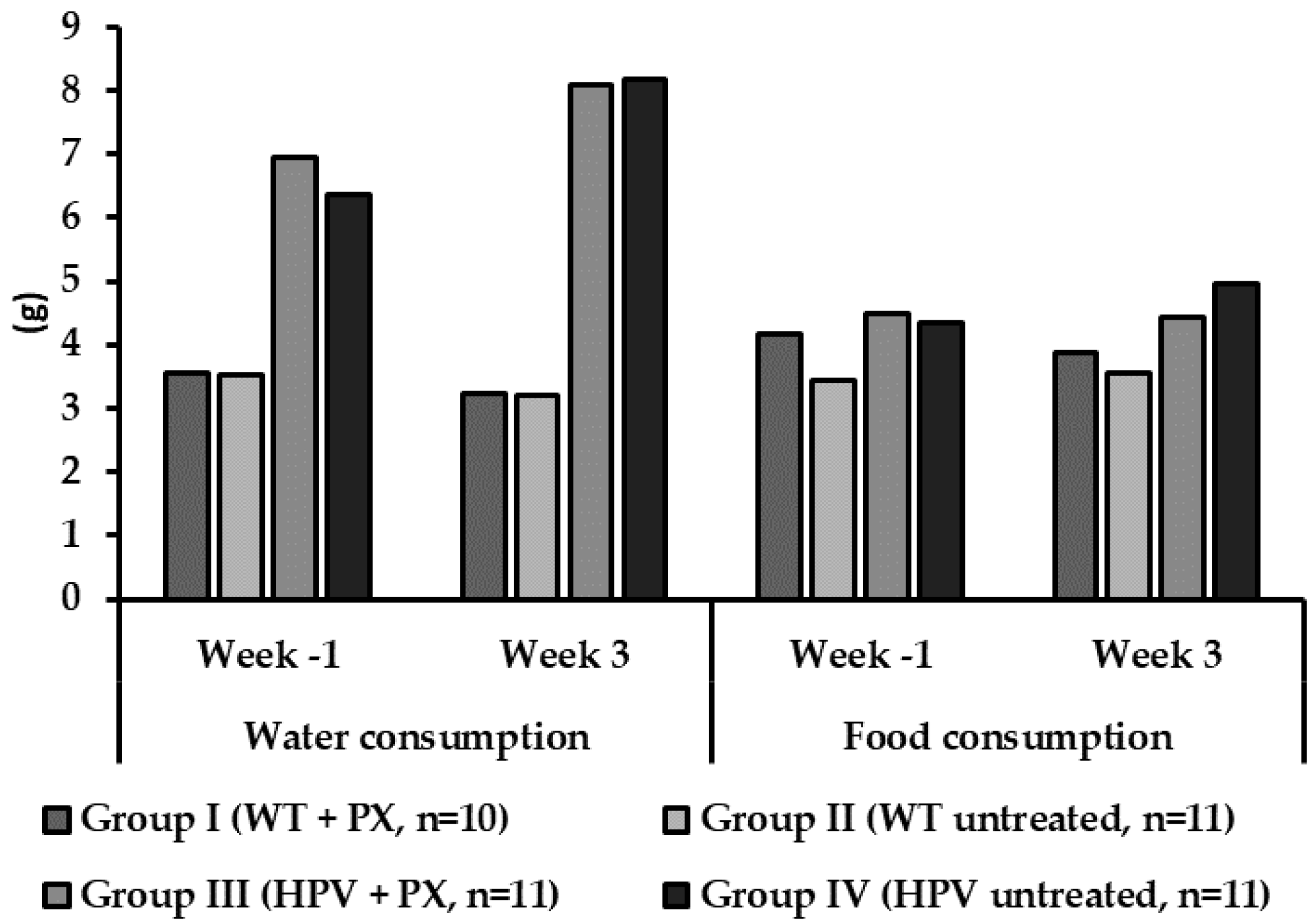
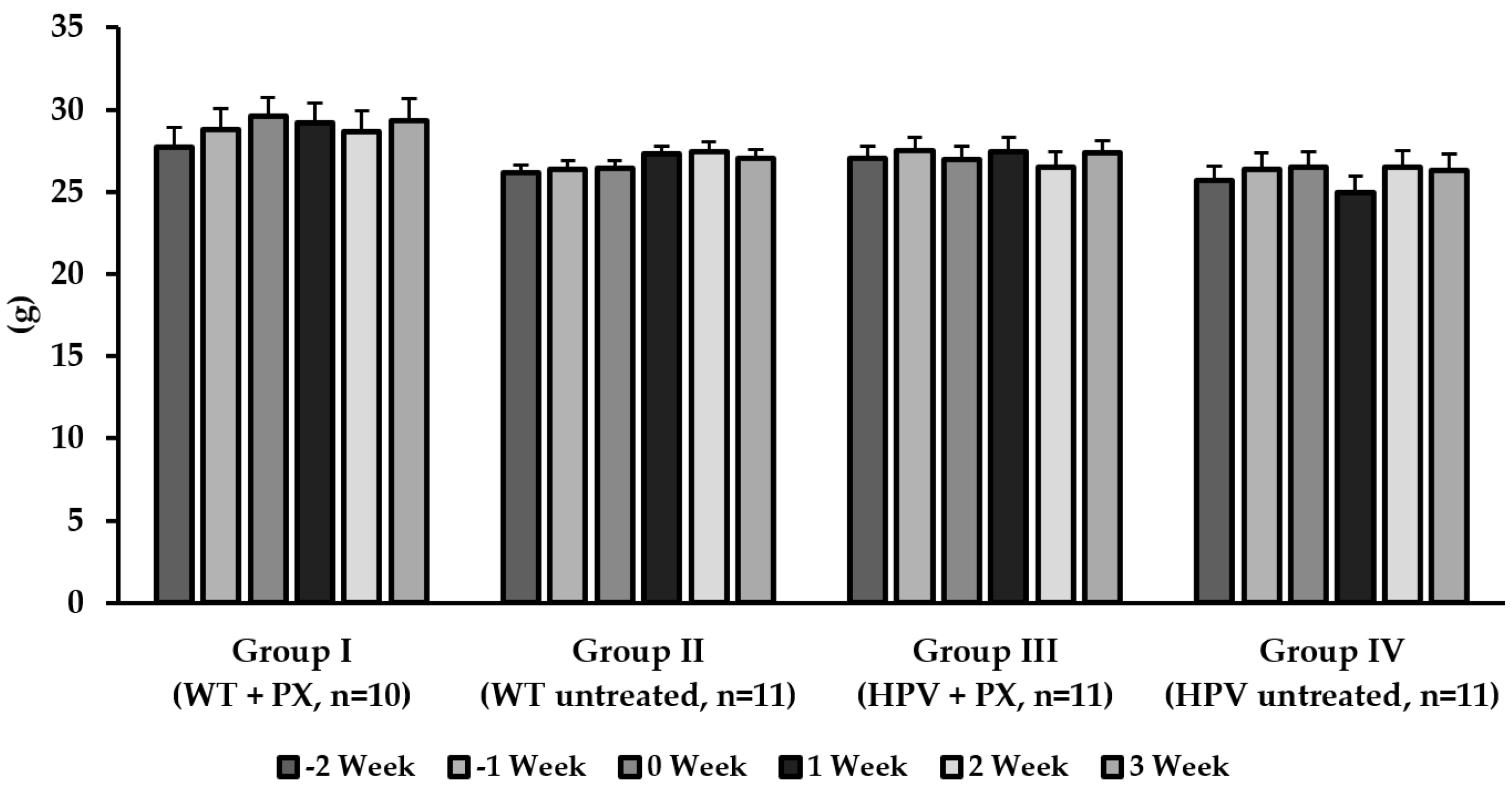
2.1. General Results
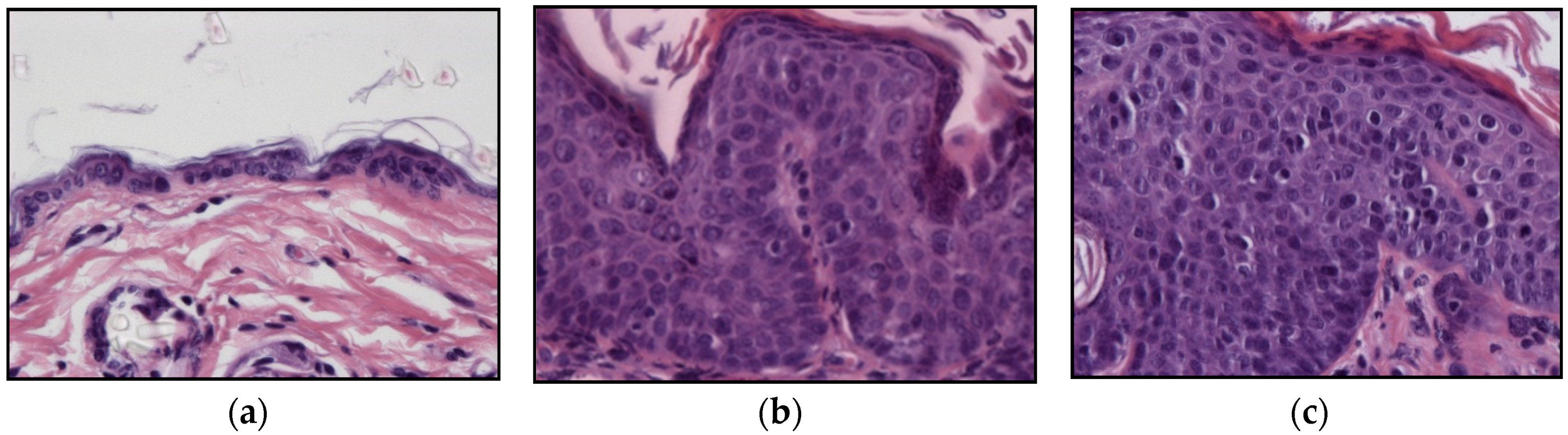
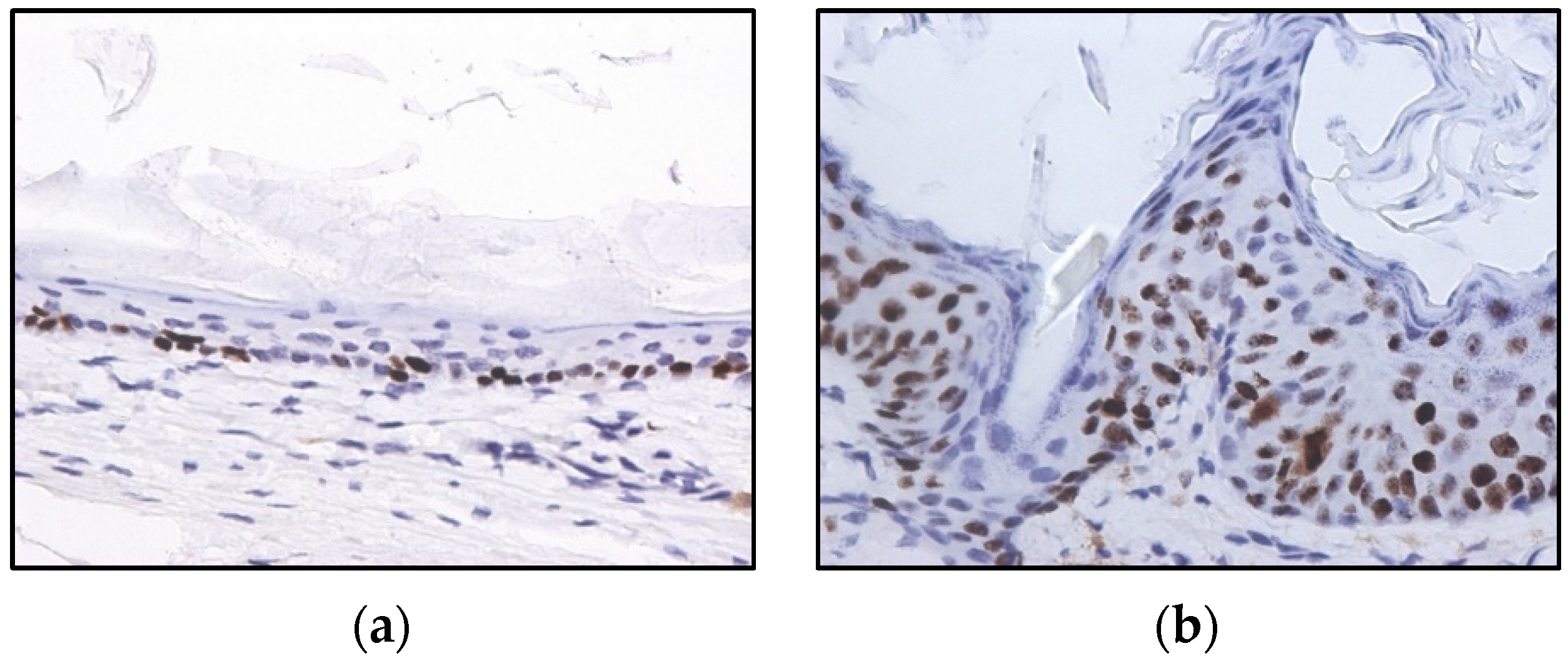
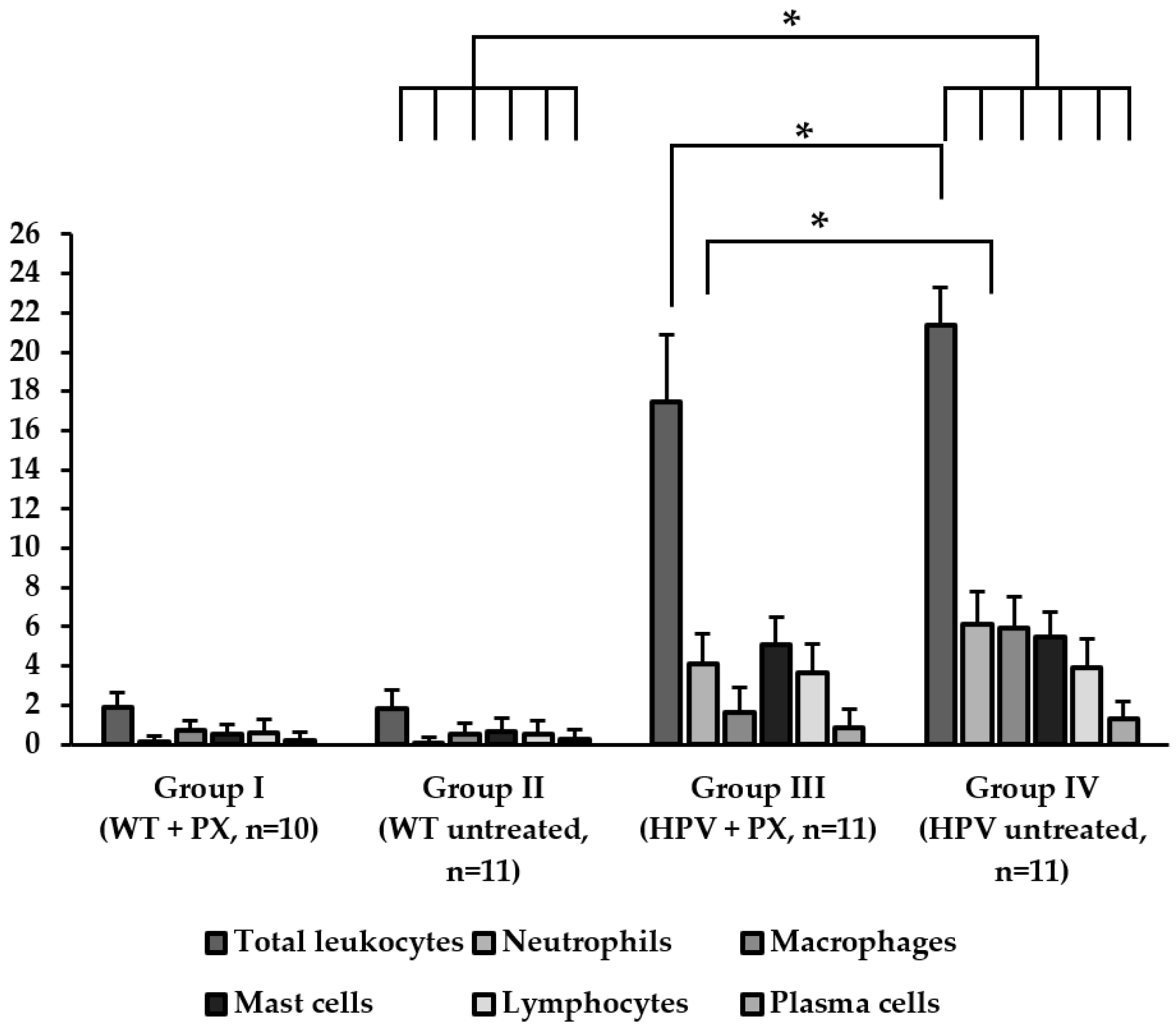
2.2. HPV-Induced Lesions
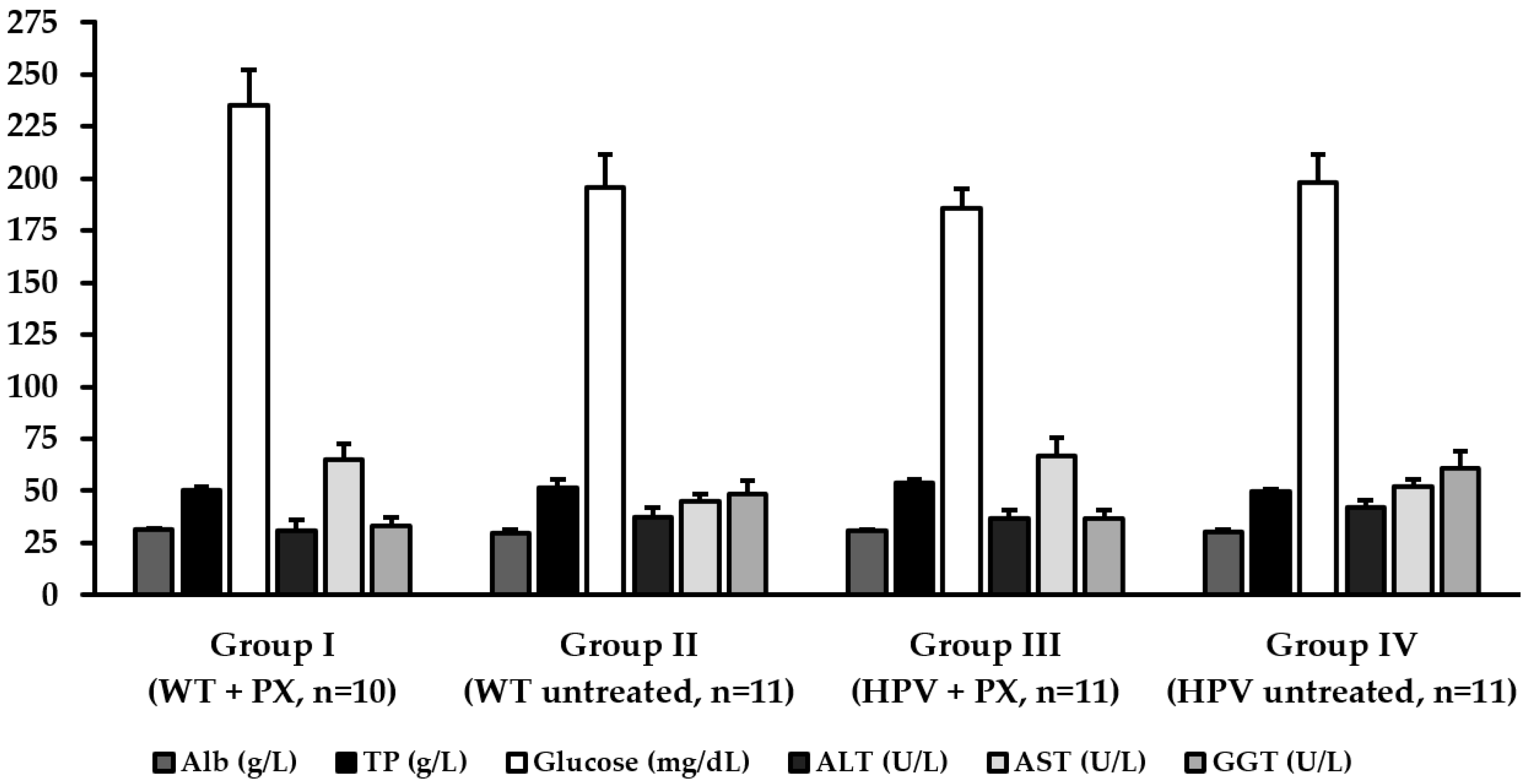
2.3. Hepatic Toxicity
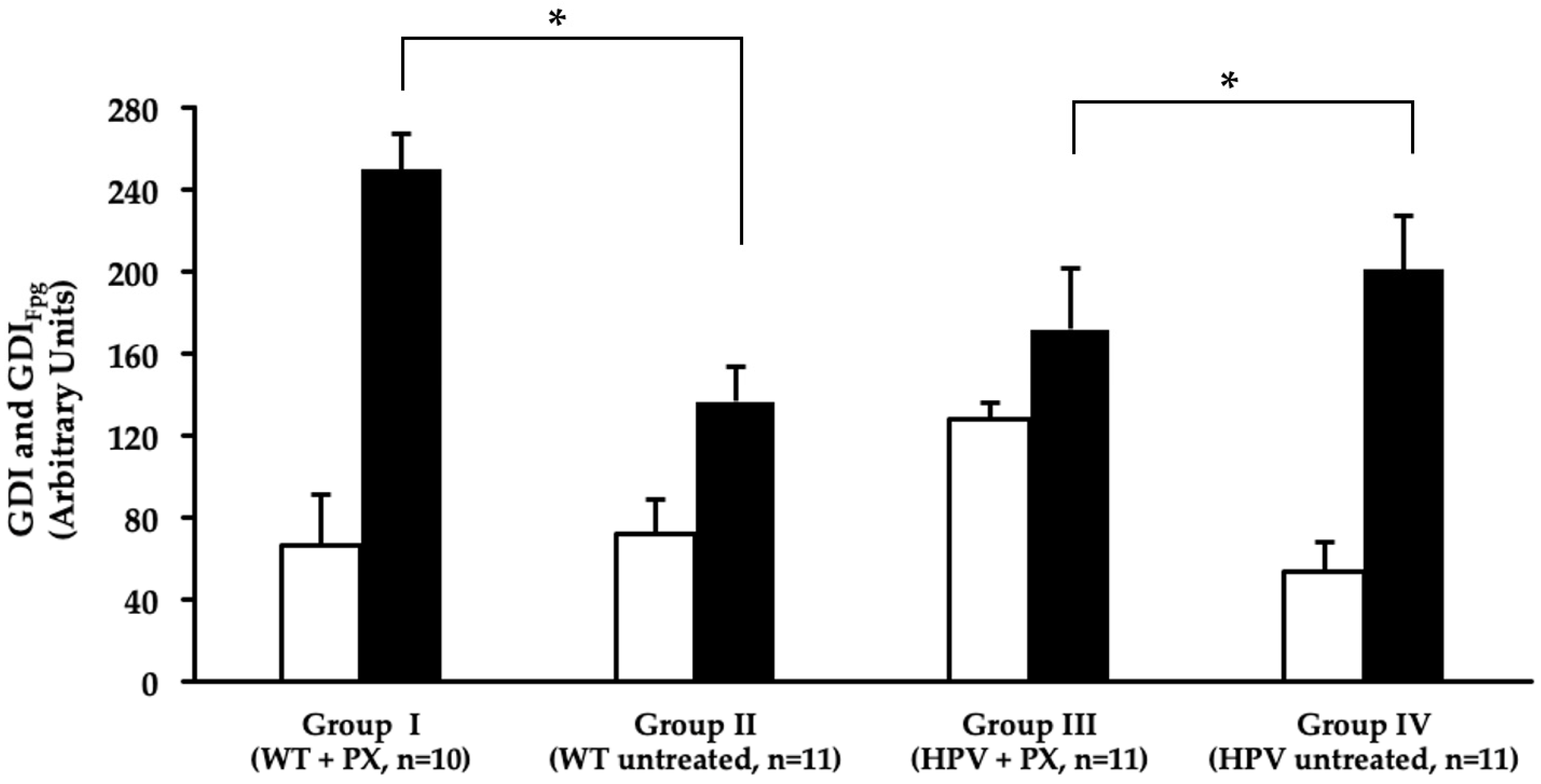
2.4. Oxidative Stress
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Design
4.3. HPV16-Induced Skin Lesions
4.4. Immunochemistry
4.5. Humane Endpoints
4.6. Biochemical Markers and Histology
4.7. Comet Assay
4.8. Oxidative Stress Parameters
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ozcagli, E.; Biri, A.; Dinc, B.; Sardas, S. How Does Infection with Human Papillomavirus 16 and 18 Impact on DNA Damage and Repair in Cervical Cells and Peripheral Blood? OMICS 2018, 22, 1–5. [Google Scholar] [CrossRef]
- Frazer, I.H. Interaction of Human Papillomaviruses with the Host Immune System: A Well Evolved Relationship. Virology 2009, 384, 410–414. [Google Scholar] [CrossRef]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; De Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic Human Papillomavirus Infection. Nat. Rev. Dis. Primers 2016, 2, 1–20. [Google Scholar] [CrossRef]
- Bartsch, H.; Nair, J. Chronic Inflammation and Oxidative Stress in the Genesis and Perpetuation of Cancer: Role of Lipid Peroxidation, DNA Damage, and Repair. Langenbecks Arch. Surg. 2006, 391, 499–510. [Google Scholar] [CrossRef]
- Cruz-Gregorio, A.; Manzo-Merino, J.; Lizano, M. Cellular Redox, Cancer and Human Papillomavirus. Virus Res. 2018, 246, 35–45. [Google Scholar] [CrossRef]
- Miller, C.S.; Johnstone, B.M. Human Papillomavirus as a Risk Factor for Oral Squamous Cell Carcinoma: A Meta-Analysis, 1982–1997. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2001, 91, 622–635. [Google Scholar] [CrossRef]
- Mork, J.; Lie, A.K.; Glattre, E.; Clark, S.; Hallmans, G.; Jellum, E.; Koskela, P.; Møller, B.; Pukkala, E.; Schiller, J.T.; et al. Human Papillomavirus Infection as a Risk Factor for Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2001, 344, 1125–1131. [Google Scholar] [CrossRef]
- Ndiaye, C.; Mena, M.; Alemany, L.; Arbyn, M.; Castellsagué, X.; Laporte, L.; Bosch, F.X.; de Sanjosé, S.; Trottier, H. HPV DNA, E6/E7 MRNA, and P16INK4a Detection in Head and Neck Cancers: A Systematic Review and Meta-Analysis. Lancet Oncol. 2014, 15, 1319–1331. [Google Scholar] [CrossRef]
- Andreu, P.; Johansson, M.; Affara, N.I.; Pucci, F.; Tan, T.; Junankar, S.; Korets, L.; Lam, J.; Tawfik, D.; Denardo, D.G.; et al. FcRgamma Activation Regulates Inflammation-Associated Squamous Carcinogenesis. Cancer Cell 2010, 17, 121–134. [Google Scholar] [CrossRef]
- Soslow, R.A.; Dannenberg, A.J.; Rush, D.; Woerner, B.M.; Nasir Khan, K.; Masferrer, J.; Koki, A.T. COX-2 Is Expressed in Human Pulmonary, Colonic, and Mammary Tumors. Cancer 2000, 89, 2637–2645. [Google Scholar] [CrossRef]
- Xu, L.; Stevens, J.; Hilton, M.B.; Seaman, S.; Conrads, T.P.; Veenstra, T.D.; Logsdon, D.; Morris, H.; Swing, D.A.; Patel, N.L.; et al. COX-2 Inhibition Potentiates Antiangiogenic Cancer Therapy and Prevents Metastasis in Preclinical Models. Sci. Transl. Med. 2014, 6, 1–12. [Google Scholar] [CrossRef]
- Pang, L.Y.; Hurst, E.A.; Argyle, D.J. Cyclooxygenase-2: A Role in Cancer Stem Cell Survival and Repopulation of Cancer Cells during Therapy. Stem Cells Int. 2016, 2016, 1–11. [Google Scholar] [CrossRef]
- Bando, T.; Fujita, S.; Nagano, N.; Yoshikawa, S.; Yamanishi, Y.; Minami, M.; Karasuyama, H. Differential Usage of COX-1 and COX-2 in Prostaglandin Production by Mast Cells and Basophils. Biochem. Biophys. Rep. 2017, 10, 82–87. [Google Scholar] [CrossRef]
- Santos, C.; Neto, T.; Ferreirinha, P.; Sousa, H.; Ribeiro, J.; Bastos, M.M.S.M.; Faustino-Rocha, A.I.; Oliveira, P.A.; Medeiros, R.; Vilanova, M.; et al. Celecoxib Promotes Degranulation of CD8 + T Cells in HPV-Induced Lesions of K14-HPV16 Transgenic Mice. Life Sci. 2016, 157, 67–73. [Google Scholar] [CrossRef]
- Padi, S.S.V.; Jain, N.K.; Singh, S.; Kulkarni, S.K. Pharmacological Profile of Parecoxib: A Novel, Potent Injectable Selective Cyclooxygenase-2 Inhibitor. Eur. J. Pharmacol. 2004, 491, 69–76. [Google Scholar] [CrossRef]
- Jin, X.; Zhou, F.; Liu, Y.; Cheng, C.; Yao, L.; Jia, Y.; Wang, G.; Zhang, J. Simultaneous Determination of Parecoxib and Its Main Metabolites Valdecoxib and Hydroxylated Valdecoxib in Mouse Plasma with a Sensitive LC–MS/MS Method to Elucidate the Decreased Drug Metabolism of Tumor Bearing Mice. J. Pharm. Biomed. Anal. 2018, 158, 1–7. [Google Scholar] [CrossRef]
- Brunton, L.; Hilal Dandan, R.; Knollmann, B. Lipid-Derived Autacoids: Eicosanoids and Platelet-Activating Factor. In Goodman & Gilman’s—The Pharmacological Basis of Therapeutics, 12th ed.; Education, M.H., Ed.; McGraw-Hill Education: New York, NY, USA, 2011; pp. 937–1002. [Google Scholar]
- Xiong, W.; Li, W.H.; Jiang, Y.X.; Liu, S.; Ai, Y.Q.; Liu, R.; Chang, L.; Zhang, M.; Wang, X.L.; Bai, H.; et al. Parecoxib: An Enhancer of Radiation Therapy for Colorectal Cancer. Asian Pac. J. Cancer Prev. 2015, 16, 627–633. [Google Scholar] [CrossRef]
- Fernandes, J.V.; Fernandes, T.A.A.D.M.; de Azevedo, J.C.V.; Cobucci, R.N.O.; de Carvalho, M.G.F.; Andrade, V.S.; De Araújo, J.M.G. Link between Chronic Inflammation and Human Papillomavirus-Induced Carcinogenesis (Review). Oncol. Lett. 2015, 9, 1015–1026. [Google Scholar] [CrossRef]
- Moutinho, M.S.S.; Aragão, S.; Carmo, D.; Casaca, F.; Silva, S.; Ribeiro, J.; Sousa, H.; Pires, I.; Queiroga, F.; Colaço, B.; et al. Curcumin and Rutin Down-Regulate Cyclooxygenase-2 and Reduce Tumor-Associated Inflammation in HPV16-Transgenic Mice. Anticancer Res. 2018, 38, 1461–1466. [Google Scholar] [CrossRef]
- Onder, G.; Pellicciotti, F.; Gambassi, G.; Bernabei, R. NSAID-Related Psychiatric Adverse Events: Who Is at Risk? Drugs 2004, 64, 2619–2627. [Google Scholar] [CrossRef]
- Abdulla, A.; Adams, N.; Bone, M.; Elliott, A.M.; Gaffin, J.; Jones, D.; Knaggs, R.; Martin, D.; Sampson, L.; Schofield, P.; et al. Guidance on the Management of Pain in Older People. Age Ageing 2013, 42, 1–57. [Google Scholar] [CrossRef]
- Veettil, S.K.; Lim, K.G.; Ching, S.M.; Saokaew, S.; Phisalprapa, P. Effects of Aspirin and Non-Aspirin Nonsteroidal Anti-Inflammatory Drugs on the Incidence of Recurrent Colorectal Adenomas: A Systematic Review with Meta-Analysis and Trial Sequential Analysis of Randomized Clinical Trials. BMC Cancer 2017, 17, 763. [Google Scholar] [CrossRef]
- Grabosch, M.S.; Shariff, O.M.; Wulff, J.L.; Helm, C.W. Non-Steroidal Anti-Inflammatory Agents to Induce Regression and Prevent the Progression of Cervical Intraepithelial Neoplasia (Review). Cochrane Database Syst. Rev. 2014, 9, 1–33. [Google Scholar] [CrossRef]
- Arbeit, J.M.; Münger, K.; Howley, P.M.; Hanahan, D. Progressive Squamous Epithelial Neoplasia in K14-Human Papillomavirus Type 16 Transgenic Mice. J. Virol. 1994, 68, 4358–4368. [Google Scholar]
- Coussens, L.M.; Hanahan, D.; Arbeit, J.M. Genetic Predisposition and Parameters of Malignant Progression in K14- HPV16 Transgenic Mice. Am. J. Pathol. 1996, 149, 1899–1917. [Google Scholar]
- Herber, R.; Liem, A.; Pitot, H.; Lambert, P.F. Squamous Epithelial Hyperplasia and Carcinoma in Mice Transgenic for the Human Papillomavirus Type 16 E7 Oncogene. J. Virol. 1996, 70, 1873–1881. [Google Scholar]
- Gil da Costa, R.M.; Aragão, S.; Moutinho, M.; Alvarado, A.; Carmo, D.; Casaca, F.; Silva, S.; Ribeiro, J.; Sousa, H.; Ferreira, R.; et al. HPV16 Induces a Wasting Syndrome in Transgenic Mice: Amelioration by Dietary Polyphenols via NF-ΚB Inhibition. Life Sci. 2017, 169, 11–19. [Google Scholar] [CrossRef]
- O’Donoghue, G.T.; Roche-Nagel, G.; Harmey, J.H.; Bouchier-Hayes, D.J. Cyclooxygenase-2 Inhibition Attenuates Surgically Induced Residual Tumour Growth and Metatases Following Cytoreductive Surgery in a Murine Model of Breast Cancer. J. Surg. Res. 2003, 114, 227. [Google Scholar] [CrossRef]
- Santander, S.; Cebrián, C.; Esquivias, P.; Conde, B.; Esteva, F.; Jiménez, P.; Ortego, J.; Lanas, Á.; Piazuelo, E. Cyclooxygenase Inhibitors Decrease the Growth and Induce Regression of Human Esophageal Adenocarcinoma Xenografts in Nude Mice. Int. J. Oncol. 2012, 40, 527–534. [Google Scholar] [CrossRef]
- Smakman, N.; Kranenburg, O.; Vogten, J.M.; Bloemendaal, A.L.A.; Van Diest, P.; Rinkes, I.H.M.B. Cyclooxygenase-2 Is a Target of KRAS D12, Which Facilitates the Outgrowth of Murine C26 Colorectal Liver Metastases. Clin. Cancer Res. 2005, 11, 41–48. [Google Scholar]
- Eberstål, S.; Badn, W.; Fritzell, S.; Esbjörnsson, M.; Darabi, A.; Visse, E.; Siesjö, P. Inhibition of Cyclooxygenase-2 Enhances Immunotherapy against Experimental Brain. Cancer Immunol. Immunother. 2012, 61, 1191–1199. [Google Scholar] [CrossRef]
- Ebrahimi, S.; Soltani, A.; Hashemy, S.I. Oxidative Stress in Cervical Cancer Pathogenesis and Resistance to Therapy. J. Cell. Biochem. 2019, 120, 6868–6877. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and Cancer. Nature 2012, 90, 58–73. [Google Scholar] [CrossRef]
- Bastos-Pereira, A.L.; Lugarini, D.; De Oliveira-Christoff, A.; Ávila, T.V.; Teixeira, S.; Pires, A.D.R.A.; Muscará, M.N.; Cadena, S.M.S.C.; Donatti, L.; Da Silva De Assis, H.C.; et al. Celecoxib Prevents Tumor Growth in an Animal Model by a COX-2 Independent Mechanism. Cancer Chemother. Pharmacol. 2010, 65, 267–276. [Google Scholar] [CrossRef]
- Paiva, I.; Gil da Costa, R.M.; Ribeiro, J.; Sousa, H.; Bastos, M.M.S.M.; Faustino-Rocha, A.; Lopes, C.; Oliveira, P.A.; Medeiros, R. MicroRNA-21 Expression and Susceptibility to HPV-Induced Carcinogenesis—Role of Microenvironment in K14-HPV16 Mice Model. Life Sci. 2015, 128, 8–14. [Google Scholar] [CrossRef]
- Paiva, I.; Gil da Costa, R.M.; Ribeiro, J.; Sousa, H.; Bastos, M.; Faustino-Rocha, A.; Oliveira, P.A.; Medeiros, R. A Role for MicroRNA-155 Expression in Microenvironment Associated to HPV-Induced Carcinogenesis in K14-HPV16 Transgenic Mice. PLoS ONE 2015, 10, e0116868. [Google Scholar] [CrossRef]
- Forbes, D.; Blom, H.; Kostomitsopulos, N.; Moore, G.; Perretta, G. Euroguide: On the Accommodation and Care of Animals Used for Experimental and Other Scientific Purposes; Federation of European Laboratory Animal Science Associations: London, UK, 2007. [Google Scholar]
- Oliveira, M.; Nascimento-Gonçalves, E.; Silva, J.; Oliveira, P.A.; Ferreira, R.; Antunes, L.; Arantes-Rodrigues, R.; Faustino-Rocha, A.I. Implementation of Human Endpoints in a Urinary Bladder Carcinogenesis Study in Rats. In Vivo 2017, 31, 1073–1080. [Google Scholar] [CrossRef][Green Version]
- Collins, A.R. The Comet Assay for DNA Damage and Repair. Mol. Biotechnol. 2004, 26, 249–261. [Google Scholar] [CrossRef]
- Shaposhnikov, S.; Azqueta, A.; Henriksson, S.; Meier, S.; Gaivão, I.; Huskisson, N.H.; Smart, A.; Brunborg, G.; Nilsson, M.; Collins, A.R. Twelve-Gel Slide Format Optimised for Comet Assay and Fluorescent in Situ Hybridisation. Toxicol. Lett. 2010, 195, 31–34. [Google Scholar] [CrossRef]
- Azqueta, A.; Shaposhnikov, S.; Collins, A.R. DNA Oxidation: Investigating Its Key Role in Environmental Mutagenesis with the Comet Assay. Mutat. Res. 2009, 674, 101–108. [Google Scholar] [CrossRef]
- Gornall, G.A.; Bardawill, C.J.; David, M.M. Determination of Serum Proteins by Means of the Biuret Reaction. J. Biol. Chem. 1949, 177, 751–766. [Google Scholar]
- Payá, M.; Halliwell, B.; Hoult, J.R.S. Interactions of a Series of Coumarins with Reactive Oxygen Species. Biochem. Pharmacol. 2002, 44, 205–214. [Google Scholar] [CrossRef]
- Peixoto, F.; Alves-Fernandes, D.; Santos, D.; Fontaínhas-Fernandes, A. Toxicological Effects of Oxyfluorfen on Oxidative Stress Enzymes in Tilapia Oreochromis Niloticus. Pestic. Biochem. Physiol. 2006, 85, 91–96. [Google Scholar] [CrossRef]
- Oliveira, M.M.; Teixeira, J.C.; Vasconcelos-Nóbrega, C.; Felix, L.M.; Sardão, V.A.; Colaço, A.A.; Oliveira, P.A.; Peixoto, F.P. Mitochondrial and Liver Oxidative Stress Alterations Induced by N-Butyl-N-(4-Hydroxybutyl)Nitrosamine: Relevance for Hepatotoxicity. J. Appl. Toxicol. 2013, 33, 434–443. [Google Scholar] [CrossRef]
- Monteiro-Cardoso, V.; Castro, M.; Oliveira, M.M.; Moreira, P.; Peixoto, F.; Videira, R. Age-Dependent Biochemical Dysfunction in Skeletal Muscle of Triple- Transgenic Mouse Model of Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 100–115. [Google Scholar] [CrossRef]
- Félix, L.M.; Correia, F.; Pinto, P.A.; Campos, S.P.; Fernandes, T.; Videira, R.; Oliveira, M.M.; Peixoto, F.P.; Antunes, L.M. Propofol Affinity to Mitochondrial Membranes Does Not Alter Mitochondrial Function. Eur. J. Pharmacol. 2017, 803, 48–56. [Google Scholar] [CrossRef]
- Hatton, P.J.; Dixon, D.; Cole, D.J.; Edwards, R. Glutathione Transferase Activities and Herbicide Selectivity in Maize and Associated Weed Species. Pestic. Sci. 1996, 46, 267–275. [Google Scholar] [CrossRef]
- Gartaganis, S.P.; Patsoukis, N.E.; Nikolopoulos, D.K.; Georgiou, C.D. Evidence for Oxidative Stress in Lens Epithelial Cells in Pseudoexfoliation Syndrome. Eye 2007, 21, 1406–1411. [Google Scholar] [CrossRef]
- Hissin, P.J.; Hilf, R. A Fluorometric Method for Determination of Oxidized and Reduced Glutathione in Tissues. Anal. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Groups | Thymus | Heart | Lungs | Urinary Bladder | Spleen | Liver | Left Kidney | Right Kidney |
|---|---|---|---|---|---|---|---|---|
| Group I (WT + PX, n = 10) | 0.0009 ± 0.0001 | 0.0041 ± 0.0002 | 0.0061 ± 0.0002 | 0.0009 ± 0.0002 | 0.0043 ± 0.0002 | 0.0520 ± 0.0018 | 0.0059 ± 0.0002 | 0.0059 ± 0.0002 |
| Group II (WT untreated, n = 11) | 0.0012 ± 0.0002 | 0.0042 ± 0.0002 a | 0.0063 ± 0.0003 | 0.0003 ± 0.0002 | 0.0047 ± 0.0002 a | 0.0574 ± 0.0012 a | 0.0057 ± 0.0002 a | 0.0062 ± 0.0002 |
| Group III (HPV + PX, n = 11) | 0.0009 ± 0.0001 | 0.0044 ± 0.0002 | 0.0054 ± 0.0001 a | 0.0009 ± 0.0002 | 0.0053 ± 0.0003 a | 0.0626 ± 0.0011 a | 0.0061 ± 0.0002 a | 0.0058 ± 0.0002 a |
| Group IV (HPV untreated, n = 11) | 0.0014 ± 0.0001 | 0.0051 ± 0.0002 | 0.0071 ± 0.0002 | 0.0008 ± 0.0001 | 0.0083 ± 0.0010 | 0.0717 ± 0.0019 | 0.0069 ± 0.0002 | 0.0068 ± 0.0002 |
| Ear Skin Incidence/n (%) | Chest Skin Incidence/n (%) | |||||
|---|---|---|---|---|---|---|
| Experimental Groups | Normal | Hyperplasia | Dysplasia | Normal | Hyperplasia | Dysplasia |
| Group I (WT + PX, n = 10) | 10/10 (100.0%) | 0/10 (0%) | 0/10 (0%) | 10/10 (100.0%) | 0/10 (0%) | 0/10 (0%) |
| Group II (WT untreated, n = 11) | 11/11 (100.0%) | 0/10 (0%) | 0/10 (0%) | 11/11 (100.0%) | 0/10 (0%) | 0/10 (0%) |
| Group III (HPV + PX, n = 11) | 0/10 (0%) | 10/10 (100.0%) | 0/10 (0%) a | 0/11 (0%) | 10/10 (100.0%) | 0/10 (0%) a |
| Group IV (HPV untreated, n = 11) | 0/10 (0%) | 11/11 (100.0%) | 7/11 (63.6%) | 0/11 (0%) | 11/11 (100.0%) | 7/11 (63.6%) |
| Experimental Groups | CAT (mmol H2O2 min−1mg−1) | SOD (U min−1mg−1) | GST (mM CDNB min−1mg−1) | GR (µM NADPHox min−1mg−1) | GSH:GSSG | LPO (nM MDA mg−1) |
|---|---|---|---|---|---|---|
| Group I (WT + PX, n = 10) | 0.026 ± 0.007 | 2.40 ± 0.24 | 0.73 ± 0.18 | 26 11 ± 2.90 a | 0.98 ± 0.14 a | 3.82 ± 1.94 a |
| Group II (WT untreated, n = 11) | 0.028 ± 0.003 | 2.77 ± 0.30 | 0.81 ± 0.30 | 20.67 ± 1.88 | 0.72 ± 0.12 | 10.39 ± 1.86 |
| Group III (HPV + PX, n = 11) | 0.022 ± 0.003 | 2.40 ± 0.41 b | 0.68 ± 0.22 | 23.82 ± 1.88 | 0.77 ± 0.14 | 7.96 ± 3.32 |
| Group IV (HPV untreated, n = 11) | 0.024 ± 0.001 | 3.30 ± 0.63 | 0.76 ± 0.22 | 23.50 ± 1.60 | 0.68 ± 0.11 | 12.07 ± 2.08 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, T.; Campos, S.; Silva, M.G.; Ribeiro, R.; Santos, S.; Almeida, J.; Pires, M.J.; Gil da Costa, R.M.; Córdova, C.; Nogueira, A.; et al. The Cyclooxigenase-2 Inhibitor Parecoxib Prevents Epidermal Dysplasia in HPV16-Transgenic Mice: Efficacy and Safety Observations. Int. J. Mol. Sci. 2019, 20, 3902. https://doi.org/10.3390/ijms20163902
Ferreira T, Campos S, Silva MG, Ribeiro R, Santos S, Almeida J, Pires MJ, Gil da Costa RM, Córdova C, Nogueira A, et al. The Cyclooxigenase-2 Inhibitor Parecoxib Prevents Epidermal Dysplasia in HPV16-Transgenic Mice: Efficacy and Safety Observations. International Journal of Molecular Sciences. 2019; 20(16):3902. https://doi.org/10.3390/ijms20163902
Chicago/Turabian StyleFerreira, Tiago, Sandra Campos, Mónica G. Silva, Rita Ribeiro, Susana Santos, José Almeida, Maria João Pires, Rui Miguel Gil da Costa, Cláudia Córdova, António Nogueira, and et al. 2019. "The Cyclooxigenase-2 Inhibitor Parecoxib Prevents Epidermal Dysplasia in HPV16-Transgenic Mice: Efficacy and Safety Observations" International Journal of Molecular Sciences 20, no. 16: 3902. https://doi.org/10.3390/ijms20163902
APA StyleFerreira, T., Campos, S., Silva, M. G., Ribeiro, R., Santos, S., Almeida, J., Pires, M. J., Gil da Costa, R. M., Córdova, C., Nogueira, A., Neuparth, M. J., Medeiros, R., Monteiro Bastos, M. M. d. S., Gaivão, I., Peixoto, F., Oliveira, M. M., & Oliveira, P. A. (2019). The Cyclooxigenase-2 Inhibitor Parecoxib Prevents Epidermal Dysplasia in HPV16-Transgenic Mice: Efficacy and Safety Observations. International Journal of Molecular Sciences, 20(16), 3902. https://doi.org/10.3390/ijms20163902