Interrogating the Essential Bacterial Cell Division Protein FtsQ with Fragments Using Target Immobilized NMR Screening (TINS)

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Site-Specific Immobilization of FtsQp
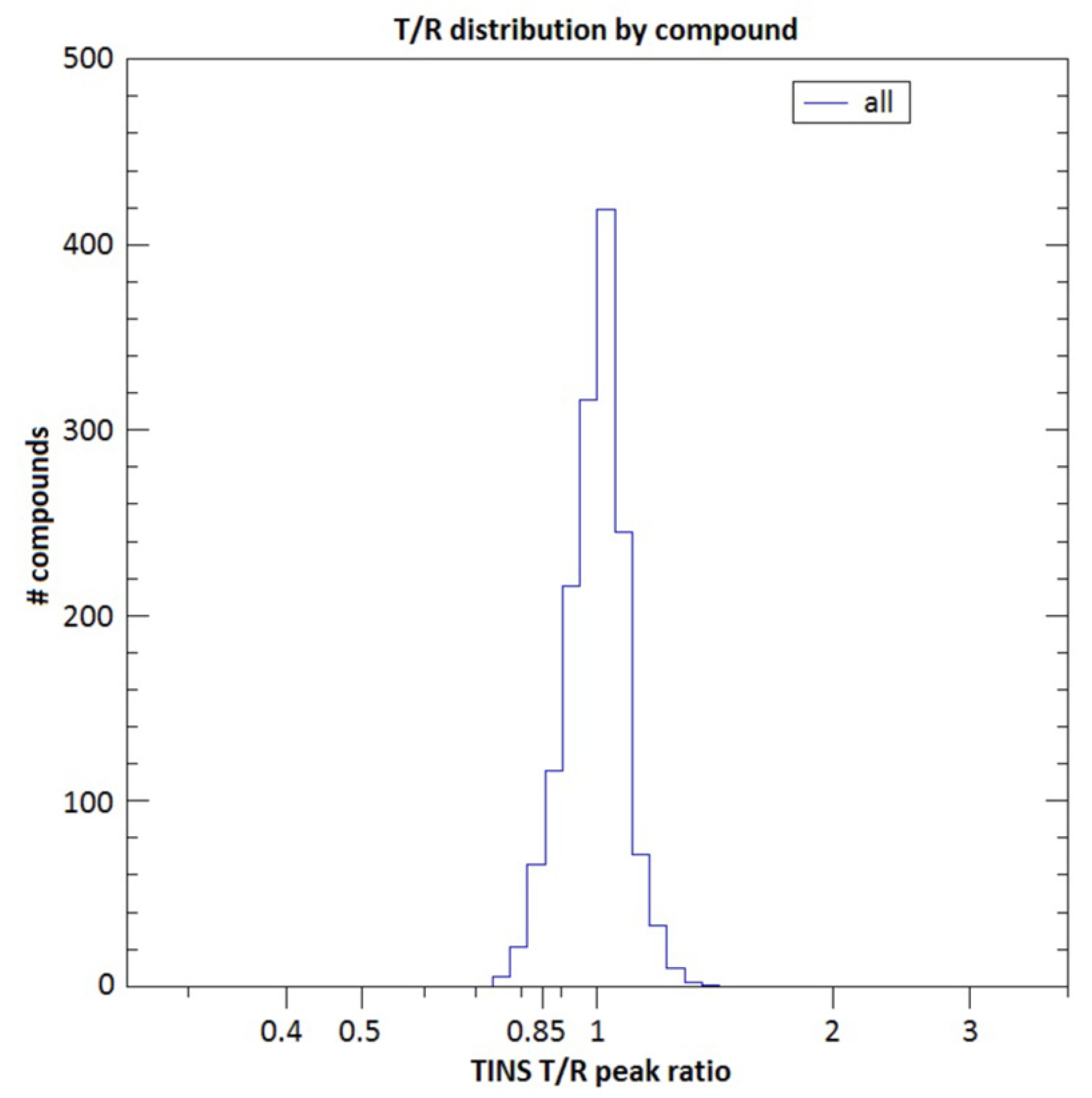
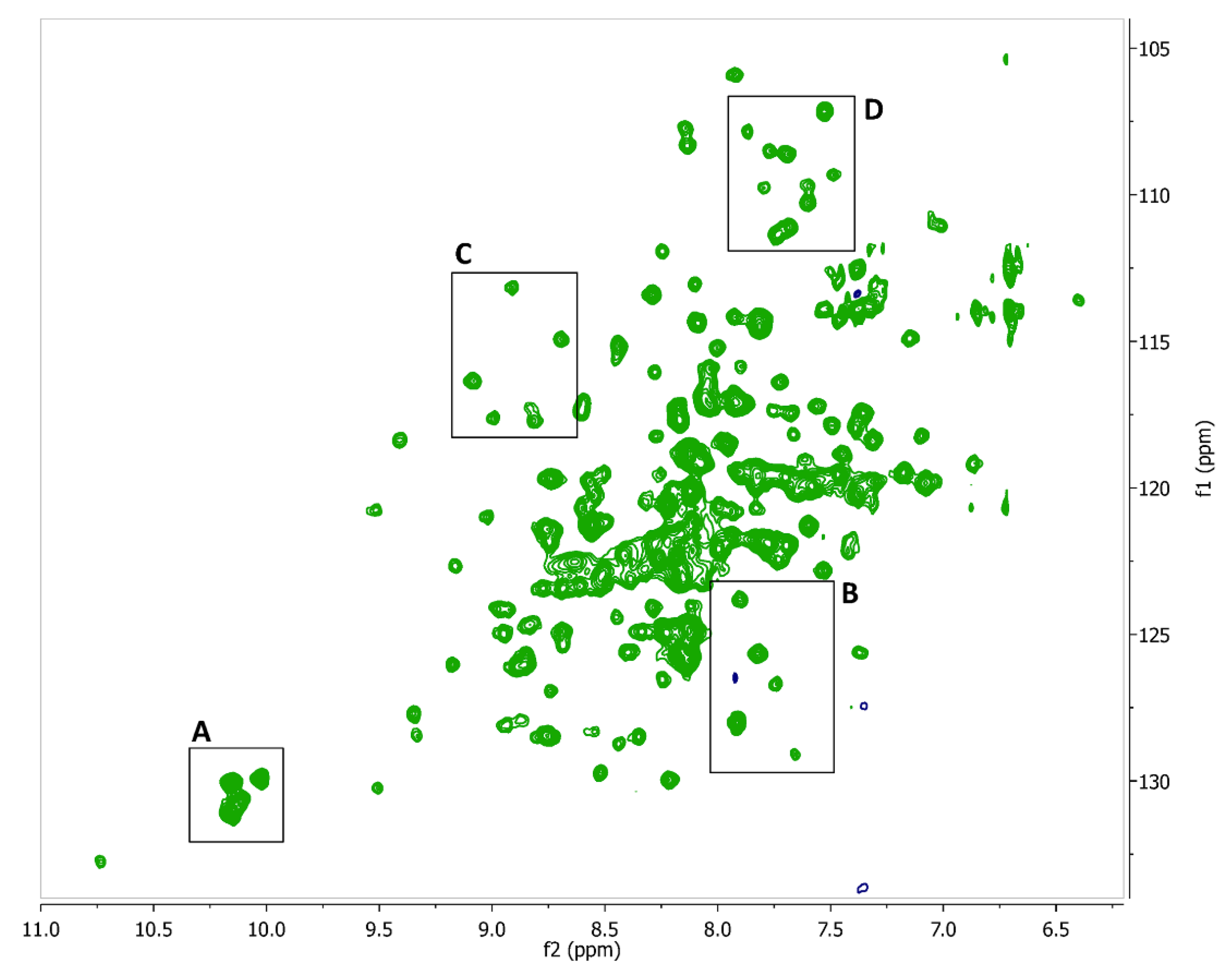
2.2. Fragment Screening Using Target Immobilized NMR Screening (TINS)
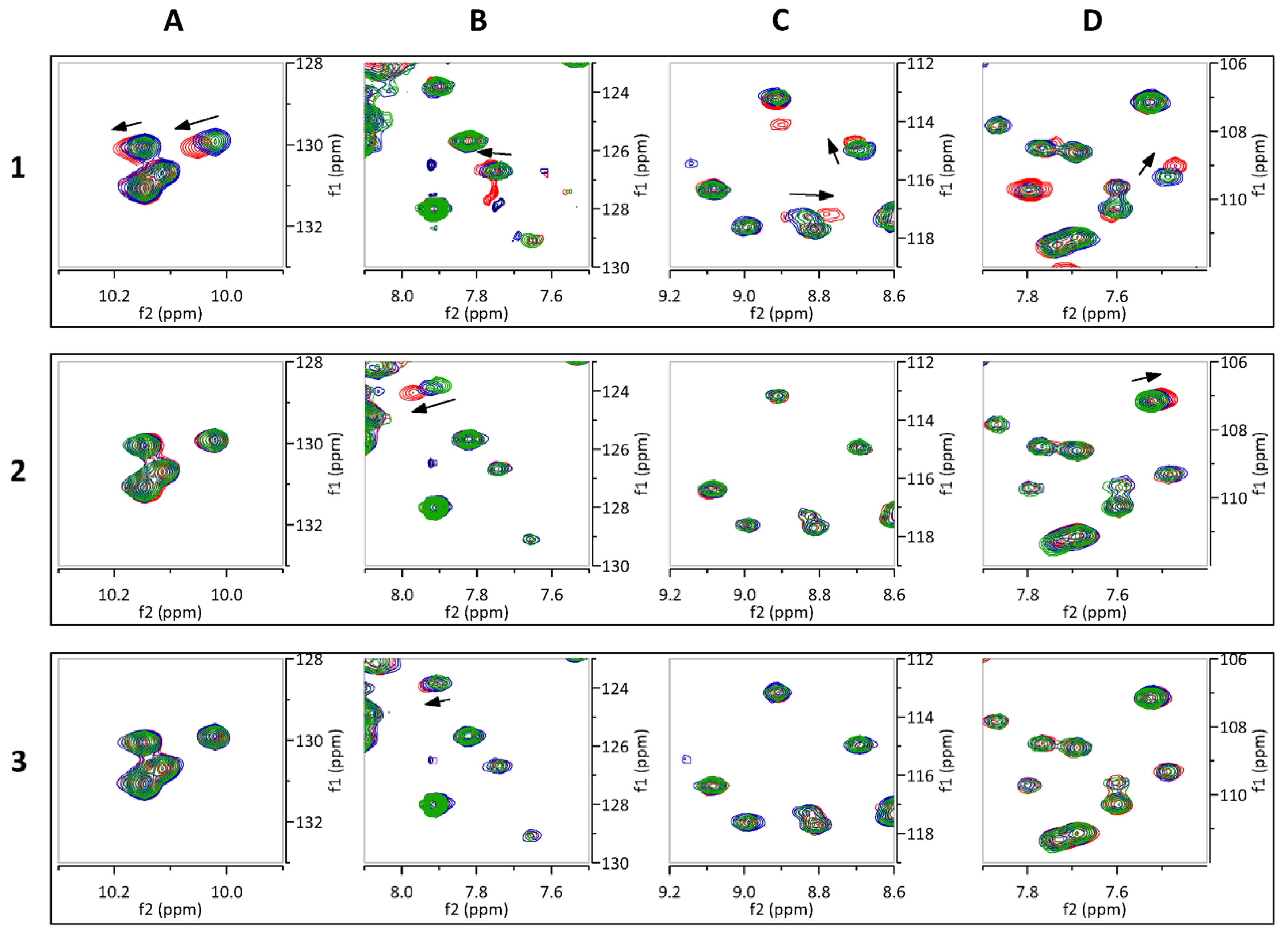
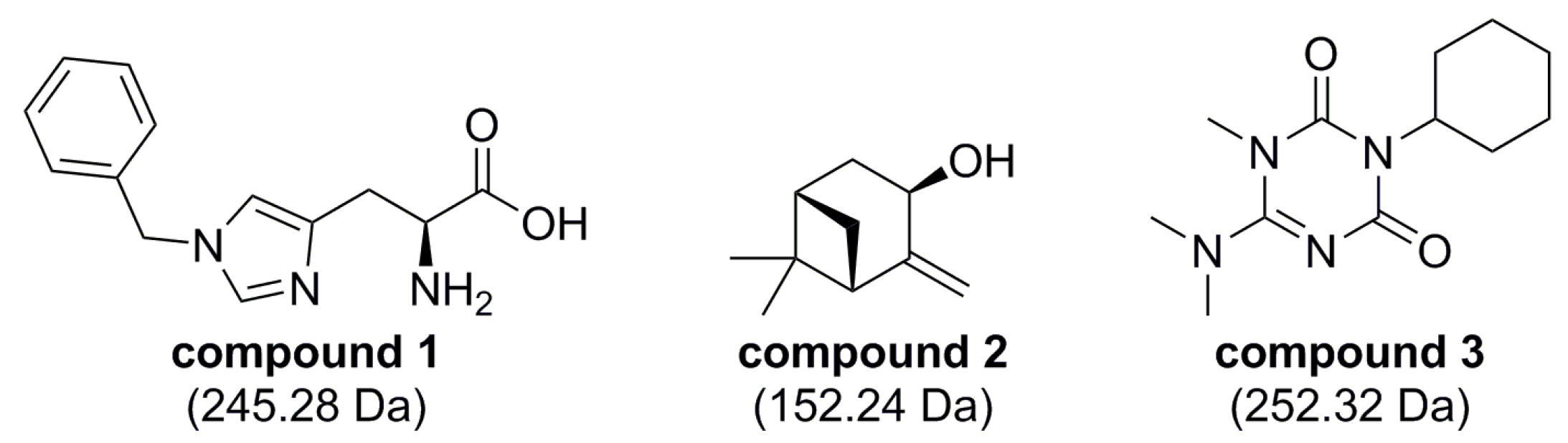
2.3. Hit Validation
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of FtsQp
4.2. Immobilization of FtsQp for TINS
4.3. FtsQp Activity Assay
4.4. Target Immobilized NMR screening
4.5. Production of 15N-Labeled FtsQp
4.6. Biosensor Analysis
4.7. Hit Validation by 1H,15N-HSQC NMR
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PPI | protein-protein interaction |
| NMR | nuclear magnetic resonance |
| TINS | target immobilized NMR screening |
| SPR | surface plasmon resonance |
| HSQC | heteronuclear single quantum coherence |
| CSP | chemical shift perturbation |
| FBDD | fragment-based drug discovery |
| TPSA | total polar surface area |
| HTS | high throughput screening |
| T/R | target spectrum/reference spectrum ratio |
References
- Den Blaauwen, T.; Andreu, J.M.; Monasterio, O. Bacterial cell division proteins as antibiotic targets. Bioorgan. Chem. 2014. [Google Scholar] [CrossRef]
- Lock, R.L.; Harry, E.J. Cell-division inhibitors: new insights for future antibiotics. Nat. Rev. Drug Discov. 2008, 7, 324–338. [Google Scholar] [CrossRef]
- Sass, P.; Brötz-Oesterhelt, H. Bacterial cell division as a target for new antibiotics. Curr. Opin. Microbiol. 2013, 16, 522–530. [Google Scholar] [CrossRef]
- Lowe, J.; Amos, L.A. Crystal structure of the bacterial cell-division protein FtsZ. Nature 1998, 391, 203–206. [Google Scholar] [CrossRef]
- Nogales, E.; Wolf, S.G.; Downing, K.H. Structure of the alpha beta tubulin dimer by electron crystallography. Nature 1998, 391, 199–203. [Google Scholar] [CrossRef]
- Li, X.; Ma, S. Advances in the discovery of novel antimicrobials targeting the assembly of bacterial cell division protein FtsZ. Eur. J. Med. Chem. 2015, 95, 1–15. [Google Scholar] [CrossRef]
- Haranahalli, K.; Tong, S.; Ojima, I. Recent advances in the discovery and development of antibacterial agents targeting the cell-division protein FtsZ. Bioorgan. Med Chem. 2016, 24, 6354–6369. [Google Scholar] [CrossRef]
- Typas, A.; Sourjik, V. Bacterial protein networks: Properties and functions. Nat. Rev. Microbiol. 2015, 13, 559–572. [Google Scholar] [CrossRef]
- Den Blaauwen, T.; de Pedro, M.A.; Nguyen-Disteche, M.; Ayala, J.A. Morphogenesis of rod-shaped sacculi. FEMS Microbiol. Rev. 2008, 32, 321–344. [Google Scholar] [CrossRef]
- Li, G.W.; Burkhardt, D.; Gross, C.; Weissman, J.S. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 2014, 157, 624–635. [Google Scholar] [CrossRef]
- Vicente, M.; Rico, A.I.; Martinez-Arteaga, R.; Mingorance, J. Septum enlightenment: Assembly of bacterial division proteins. J. Bacteriol. 2006, 188, 19–27. [Google Scholar] [CrossRef]
- Gonzalez, M.D.; Akbay, E.A.; Boyd, D.; Beckwith, J. Multiple interaction domains in FtsL, a protein component of the widely conserved bacterial FtsLBQ cell division complex. J. Bacteriol. 2010, 192, 2757–2768. [Google Scholar] [CrossRef]
- Glas, M.; McLaughlin, S.H.; Roseboom, W.; Liu, F.; Koningstein, G.M.; Fish, A.; den Blaauwen, T.; Heck, A.J.; de Jong, L.; Bitter, W.; et al. The Soluble Periplasmic Domains of Escherichia coli Cell Division Proteins FtsQ/FtsB/FtsL Form a Trimeric Complex with Submicromolar Affinity. J. Biol. Chem. 2015, 290, 21498–21509. [Google Scholar] [CrossRef]
- Buddelmeijer, N.; Beckwith, J. A complex of the Escherichia coli cell division proteins FtsL, FtsB and FtsQ forms independently of its localization to the septal region. Mol. Microbiol. 2004, 52, 1315–1327. [Google Scholar] [CrossRef]
- Hwang, H.; Vreven, T.; Janin, J.; Weng, Z. Protein-Protein Docking Benchmark Version 4.0. Proteins 2010, 78, 3111–3114. [Google Scholar] [CrossRef]
- Arkin Michelle, R.; Tang, Y.; Wells James, A. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef]
- Clackson, T.; Wells, J.A. A hot spot of binding energy in a hormone-receptor interface. Science 1995, 267, 383–386. [Google Scholar] [CrossRef]
- Glas, M.; Vernooij, I.G.; Bitter, W.; den Blaauwen, T.; Luirink, J. Fine-mapping the contact sites of the Escherichia coli cell division proteins FtsB and FtsL on the FtsQ protein. J. Biol. Chem. 2013, 288, 24340–24350. [Google Scholar]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘rule of three’ for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Erlanson, D.A.; McDowell, R.S.; O’Brien, T. Fragment-based drug discovery. J. Med. Chem. 2004, 47, 3463–3482. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.M.; Leach, A.R.; Harper, G. Molecular Complexity and Its Impact on the Probability of Finding Leads for Drug Discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Siegal, G.; AB, E.; Schultz, J. Integration of fragment screening and library design. Drug Discov. Today 2007, 12, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Vanwetswinkel, S.; Heetebrij, R.J.; van Duynhoven, J.; Hollander, J.G.; Filippov, D.V.; Hajduk, P.J.; Siegal, G. TINS, target immobilized NMR screening: An efficient and sensitive method for ligand discovery. Chem. Biol. 2005, 12, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Marquardsen, T.; Hofmann, M.; Hollander, J.G.; Loch, C.M.; Kiihne, S.R.; Engelke, F.; Siegal, G. Development of a dual cell, flow-injection sample holder, and NMR probe for comparative ligand-binding studies. J. Magn. Reson. 2006, 182, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Huth, J.R.; Fesik, S.W. Druggability Indices for Protein Targets Derived from NMR-Based Screening Data. J. Med. Chem. 2005, 48, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Murali, N.; Miller, W.M.; John, B.K.; Avizonis, D.A.; Smallcombe, S.H. Spectral unraveling by space-selective Hadamard spectroscopy. J. Magn. Reson. 2006, 179, 182–189. [Google Scholar] [CrossRef]
- Früh, V.; Heetebrij, R.J.; Siegal, G. Target-Immobilized NMR Screening: Validation and Extension to Membrane Proteins. In Fragment-Based Drug Discovery; John Wiley & Sons, Ltd.: Chichester, UK, 2008; pp. 135–157. [Google Scholar]
- Leach, A.R.; Hann, M.M. Molecular complexity and fragment-based drug discovery: Ten years on. Curr. Opin. Chem. Biol. 2011, 15, 489–496. [Google Scholar] [CrossRef]
- Kureisaite-Ciziene, D.; Varadajan, A.; McLaughlin, S.H.; Glas, M.; Silva, A.M.; Luirink, R.; Mueller, C.; Den Blaauwen, T.; Grossmann, T.N.; Luirink, J.; et al. Structural Analysis of the Interaction between the Bacterial Cell Division Proteins FtsQ and FtsB. MBio 2018, 9. [Google Scholar] [CrossRef]
- O’Shannessy, D.J.; Brigham-Burke, M.; Peck, K. Immobilization chemistries suitable for use in the BIAcore surface plasmon resonance detector. Anal. Biochem. 1992, 205, 132–136. [Google Scholar] [CrossRef]
- Boes, A.; Olatunji, S.; Breukink, E.; Terrak, M. Regulation of the Peptidoglycan Polymerase Activity of PBP1b by Antagonist Actions of the Core Divisome Proteins FtsBLQ and FtsN. MBio 2019, 10, e01912-18. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glas, M.; AB, E.; Hollander, J.; Siegal, G.; Luirink, J.; de Esch, I. Interrogating the Essential Bacterial Cell Division Protein FtsQ with Fragments Using Target Immobilized NMR Screening (TINS). Int. J. Mol. Sci. 2019, 20, 3684. https://doi.org/10.3390/ijms20153684
Glas M, AB E, Hollander J, Siegal G, Luirink J, de Esch I. Interrogating the Essential Bacterial Cell Division Protein FtsQ with Fragments Using Target Immobilized NMR Screening (TINS). International Journal of Molecular Sciences. 2019; 20(15):3684. https://doi.org/10.3390/ijms20153684
Chicago/Turabian StyleGlas, Marjolein, Eiso AB, Johan Hollander, Gregg Siegal, Joen Luirink, and Iwan de Esch. 2019. "Interrogating the Essential Bacterial Cell Division Protein FtsQ with Fragments Using Target Immobilized NMR Screening (TINS)" International Journal of Molecular Sciences 20, no. 15: 3684. https://doi.org/10.3390/ijms20153684
APA StyleGlas, M., AB, E., Hollander, J., Siegal, G., Luirink, J., & de Esch, I. (2019). Interrogating the Essential Bacterial Cell Division Protein FtsQ with Fragments Using Target Immobilized NMR Screening (TINS). International Journal of Molecular Sciences, 20(15), 3684. https://doi.org/10.3390/ijms20153684