Biological Functions and Molecular Mechanisms of Antibiotic Tigecycline in the Treatment of Cancers

 ,
,
Abstract
1. Introduction
2. Biological Function of Tigecycline in Tumors
2.1. Tigecycline Inhibits Mitochondrial Oxidative Phosphorylation
2.2. Tigecycline Induces Mitochondrial Oxidative Damage
2.3. Tigecycline Affects Mitochondrial Biogenesis
2.4. Tigecycline Induces Cell Cycle Arrest
2.5. Tigecycline Induces Autophagy
2.6. Tigecycline Induces Apoptosis
2.7. Tigecycline Inhibits Migration/Invasion and Angiogenesis
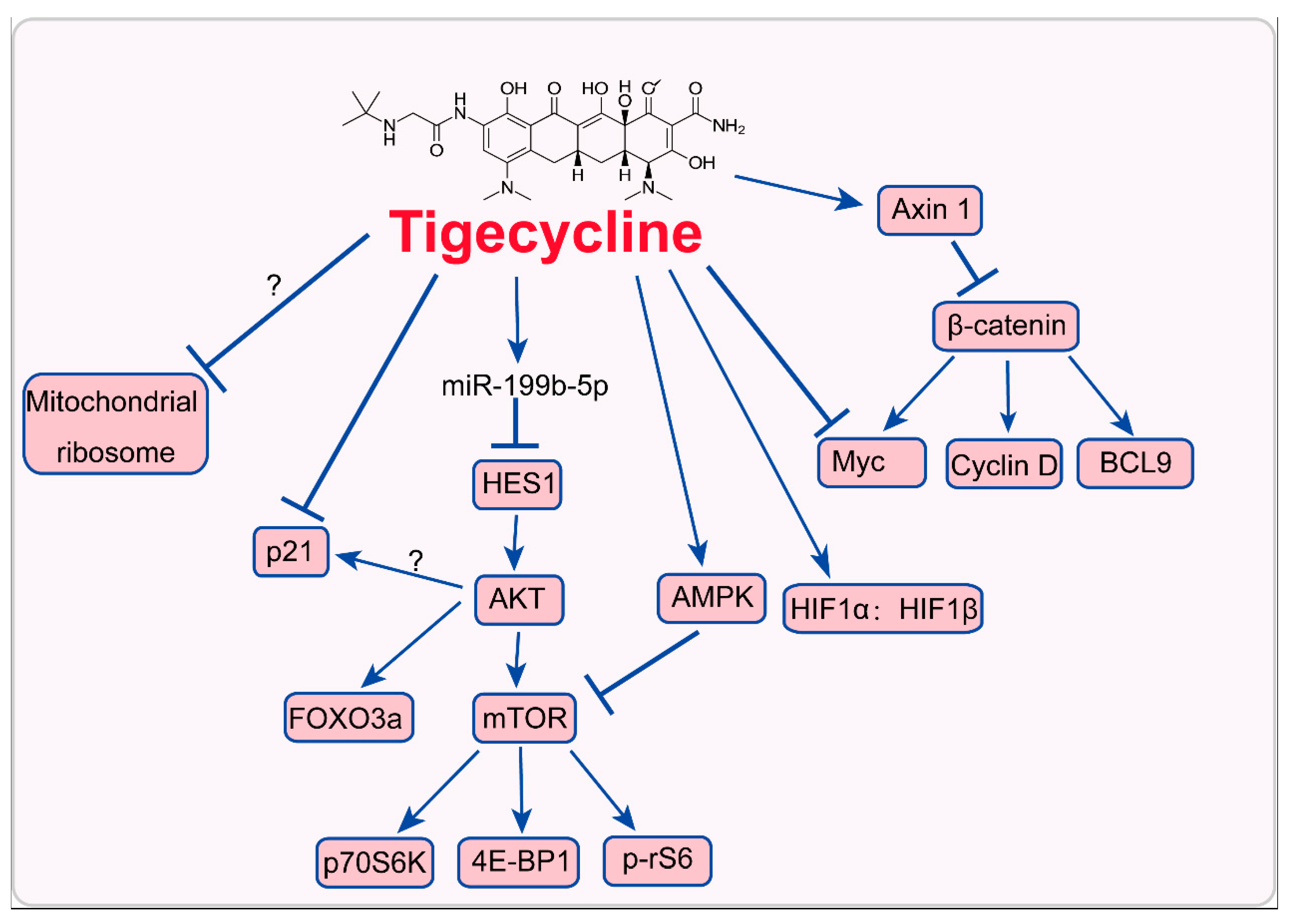
3. Mode of Action of Tigecycline in Tumors
3.1. Inhibition of Mitochondrial Translation
3.2. Inhition of Myc and Activation of HIFs
3.3. Inhibition of PI3K/AKT-FOXO3a/mTOR Signaling
3.4. Activation of AMPK-mTOR Signaling
3.5. Inhibition of Cytoplasmic p21CIP1/Waf1
3.6. Inhibition of Wnt/β-Catenin Signaling
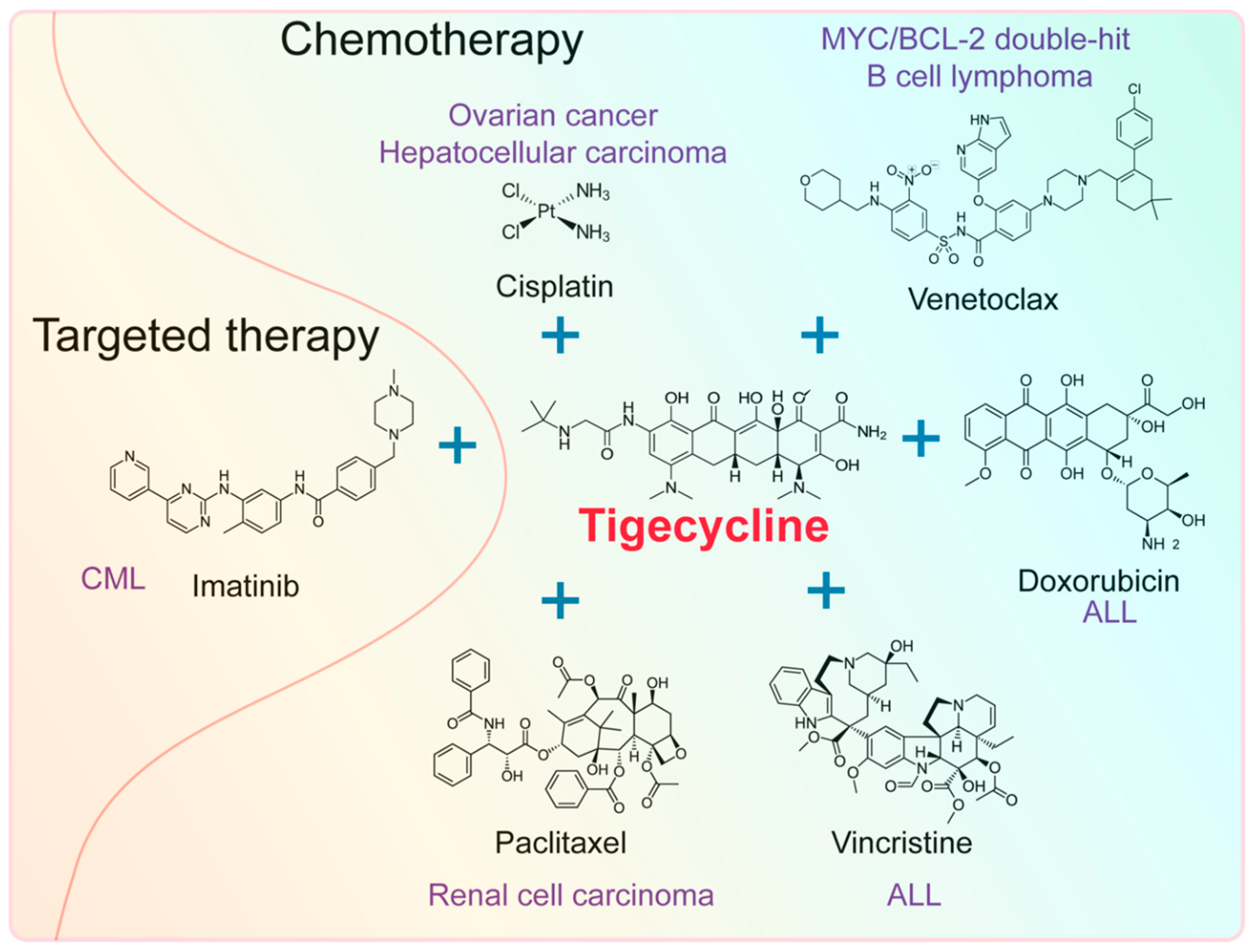
4. Effect of Tigecycline Combined with Other Anticancer Drugs
4.1. Effect of Tigecycline Combined with Chemotherapeutic Drugs
4.2. Effect of Tigecycline Combined with Targeted Drugs
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bhattacharya, B.; Mukherjee, S. Cancer therapy using antibiotics. J. Cancer Ther. 2015, 6, 849. [Google Scholar] [CrossRef]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillere, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Pal, S.K. Further evidence to support judicious use of antibiotics in patients with cancer. Ann. Oncol. 2018, 29, 1349–1351. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Lopez, M.; Joseph, J.; Zielonka, J.; Dwinell, M.B. A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: Therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox Biol. 2018, 14, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef]
- Hortobagyi, G.N. Anthracyclines in the treatment of cancer: An overview. Drugs 1997, 54, 1–7. [Google Scholar] [CrossRef]
- Di Marco, A. Anthracyclines in cancer chemotherapy. In Anthracycline Antibiotics in Cancer Therapy; Muggia, F.M., Young, C.W., Carter, S.K., Eds.; Springer: Drodrecht, Netherland, 1982; Volume 10, pp. 223–258. [Google Scholar]
- Blum, R.H.; Carter, S.K.; Agre, K. A clinical review of bleomycin—a new antineoplastic agent. Cancer 1973, 31, 903–914. [Google Scholar] [CrossRef]
- Volpe, A.; Racioppi, M.; D’Agostino, D.; Cappa, E.; Filianoti, A.; Bassi, P.F. Mitomycin C for the treatment of bladder cancer. Minerva Urol. Nefrol. 2010, 62, 133–144. [Google Scholar]
- Langholz, B.; Skolnik, J.M.; Barrett, J.S.; Renbarger, J.; Seibel, N.L.; Zajicek, A.; Arndt, C.A. Dactinomycin and vincristine toxicity in the treatment of childhood cancer: A retrospective study from the Children’s Oncology Group. Pediatr. Blood Cancer 2011, 57, 252–257. [Google Scholar] [CrossRef]
- Lagler, H.; Kiesewetter, B.; Dolak, W.; Obermueller, M.; Simonitsch-Klupp, I.; Lukas, J.; Neuper, O.; Lamm, W.W.; Mayerhoefer, M.E.; Raderer, M. Treatment of mucosa associated lymphoid tissue lymphoma with a long-term once-weekly regimen of oral azithromycin: Results from the phase II MALT-A trial. Hematol. Oncol. 2019, 37, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Fife, R.S.; Rougraff, B.T.; Proctor, C.; Sledge, G.W., Jr. Inhibition of proliferation and induction of apoptosis by doxycycline in cultured human osteosarcoma cells. J. Lab. Clin. Med. 1997, 130, 530–534. [Google Scholar] [CrossRef]
- Richards, C.; Pantanowitz, L.; Dezube, B.J. Antimicrobial and non-antimicrobial tetracyclines in human cancer trials. Pharmacol. Res. 2011, 63, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Lokeshwar, B.L.; Selzer, M.G.; Zhu, B.Q.; Block, N.L.; Golub, L.M. Inhibition of cell proliferation, invasion, tumor growth and metastasis by an oral non-antimicrobial tetracycline analog (COL-3) in a metastatic prostate cancer model. Int. J. Cancer 2002, 98, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Scatena, C.; Roncella, M.; Di Paolo, A.; Aretini, P.; Menicagli, M.; Fanelli, G.; Marini, C.; Mazzanti, C.M.; Ghilli, M.; Sotgia, F.; et al. Doxycycline, an inhibitor of mitochondrial biogenesis, effectively reduces cancer stem cells (CSCs) in early breast cancer patients: A clinical pilot study. Front. Oncol. 2018, 8, 452. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, L.; Zhang, F.; Vlashi, E. Doxycycline inhibits the cancer stem cell phenotype and epithelial-to-mesenchymal transition in breast cancer. Cell Cycle 2017, 16, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, X.; Zhao, Y.Y.; Curtis, J.M.; Brindley, D.N. Doxycycline attenuates breast cancer related inflammation by decreasing plasma lysophosphatidate concentrations and inhibiting NF-kappaB activation. Mol. Cancer 2017, 16, 36. [Google Scholar] [CrossRef]
- Olson, M.W.; Ruzin, A.; Feyfant, E.; Rush, T.S., 3rd; O’Connell, J.; Bradford, P.A. Functional, biophysical, and structural bases for antibacterial activity of tigecycline. Antimicrob. Agents Chemother. 2006, 50, 2156–2166. [Google Scholar] [CrossRef]
- Wenzel, R.; Bate, G.; Kirkpatrick, P. Tigecycline. Nat. Rev. Drug Discov. 2005, 4, 809–810. [Google Scholar] [CrossRef]
- Cunha, A.B.; Baron, J.; Cunha, B.C. Once daily high dose tigecycline—pharmacokinetic/pharmacodynamic based dosing for optimal clinical effectiveness: Dosing matters, revisited. Expert Rev. Anti Infect. Ther. 2017, 15, 257–267. [Google Scholar] [CrossRef]
- Skrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Yang, L.; Jiang, X.; Xu, C.; Wang, M.; Wang, Q.; Zhou, Z.; Xiang, Z.; Cui, H. Antibiotic drug tigecycline inhibited cell proliferation and induced autophagy in gastric cancer cells. Biochem. Biophys. Res. Commun. 2014, 446, 105–112. [Google Scholar] [CrossRef]
- Ren, A.; Qiu, Y.; Cui, H.; Fu, G. Tigecycline exerts an antitumoral effect in oral squamous cell carcinoma. Oral Dis. 2015, 21, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Dong, Z.; Tan, P.; Zhang, Y.; Liu, L.; Yang, L.; Liu, Y.; Cui, H. Antibiotic drug tigecycline inhibits melanoma progression and metastasis in a p21CIP1/Waf1-dependent manner. Oncotarget 2016, 7, 3171–3185. [Google Scholar] [CrossRef]
- Zhong, X.; Zhao, E.; Tang, C.; Zhang, W.; Tan, J.; Dong, Z.; Ding, H.F.; Cui, H. Antibiotic drug tigecycline reduces neuroblastoma cells proliferation by inhibiting Akt activation in vitro and in vivo. Tumour Biol. 2016, 37, 7615–7623. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Yi, L.; Dong, Z.; Ouyang, Q.; Zhou, J.; Pang, Y.; Wu, Y.; Xu, L.; Cui, H. Tigecycline inhibits glioma growth by regulating miRNA-199b-5p-HES1-AKT pathway. Mol. Cancer Ther. 2016, 15, 421–429. [Google Scholar] [CrossRef]
- Jia, X.; Gu, Z.; Chen, W.; Jiao, J. Tigecycline targets nonsmall cell lung cancer through inhibition of mitochondrial function. Fundam. Clin. Pharmacol. 2016, 30, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.A.; Robinson, T.J.; Liu, J.C.; Shrestha, M.; Voisin, V.; Ju, Y.; Chung, P.E.; Pellecchia, G.; Fell, V.L.; Bae, S.; et al. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J. Clin. Invest. 2016, 126, 3739–3757. [Google Scholar] [CrossRef] [PubMed]
- Norberg, E.; Lako, A.; Chen, P.H.; Stanley, I.A.; Zhou, F.; Ficarro, S.B.; Chapuy, B.; Chen, L.; Rodig, S.; Shin, D.; et al. Differential contribution of the mitochondrial translation pathway to the survival of diffuse large B-cell lymphoma subsets. Cell Death Differ. 2017, 24, 251–262. [Google Scholar] [CrossRef]
- Tan, J.; Song, M.; Zhou, M.; Hu, Y. Antibiotic tigecycline enhances cisplatin activity against human hepatocellular carcinoma through inducing mitochondrial dysfunction and oxidative damage. Biochem. Biophys. Res. Commun. 2017, 483, 17–23. [Google Scholar] [CrossRef]
- Li, H.; Jiao, S.; Li, X.; Banu, H.; Hamal, S.; Wang, X. Therapeutic effects of antibiotic drug tigecycline against cervical squamous cell carcinoma by inhibiting Wnt/beta-catenin signaling. Biochem. Biophys. Res. Commun. 2015, 467, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, N.; He, B.; Pan, C.; Lan, Y.; Zhou, H.; Liu, X. Inhibition of autophagy enhances the selective anti-cancer activity of tigecycline to overcome drug resistance in the treatment of chronic myeloid leukemia. J. Exp. Clin. Cancer Res. 2017, 36, 43. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Liu, W.; Huang, Q.; Wang, Y.; Li, H.; Xiong, Y. Targeting mitochondrial respiration selectively sensitizes pediatric acute lymphoblastic leukemia cell lines and patient samples to standard chemotherapy. Am. J. Cancer Res. 2017, 7, 2395–2405. [Google Scholar] [PubMed]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.D.; Cook, D.R.; Choi, M.Y.; Li, M.Z.; Haigis, K.M.; Elledge, S.J. A role for mitochondrial translation in promotion of viability in K-Ras mutant cells. Cell Rep. 2017, 20, 427–438. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.; Gritti, I.; Nicoli, P.; Giorgio, M.; Doni, M.; Conti, A.; Bianchi, V.; Casoli, L.; Sabo, A.; Mironov, A.; et al. The mitochondrial translation machinery as a therapeutic target in Myc-driven lymphomas. Oncotarget 2016, 7, 72415–72430. [Google Scholar] [CrossRef] [PubMed]
- Jhas, B.; Sriskanthadevan, S.; Skrtic, M.; Sukhai, M.A.; Voisin, V.; Jitkova, Y.; Gronda, M.; Hurren, R.; Laister, R.C.; Bader, G.D.; et al. Metabolic adaptation to chronic inhibition of mitochondrial protein synthesis in acute myeloid leukemia cells. PLoS ONE 2013, 8, e58367. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, Y. Inhibition of mitochondrial translation as a therapeutic strategy for human ovarian cancer to overcome chemoresistance. Biochem. Biophys. Res. Commun. 2019, 509, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Zhang, Y.; Wang, W.; Wu, J.; Yang, Q.; Xu, W.; Jiang, S.; Han, Y.; Yu, K.; Zhang, S. Inhibition of autophagy enhances the antitumour activity of tigecycline in multiple myeloma. J. Cell Mol. Med. 2018, 22, 5955–5963. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, W.; Huang, Q.; Wang, J.; Wang, Y.; Li, H.; Fu, X. Tigecycline as a dual inhibitor of retinoblastoma and angiogenesis via inducing mitochondrial dysfunctions and oxidative damage. Sci. Rep. 2018, 8, 11747. [Google Scholar] [CrossRef]
- Wang, B.; Ao, J.; Yu, D.; Rao, T.; Ruan, Y.; Yao, X. Inhibition of mitochondrial translation effectively sensitizes renal cell carcinoma to chemotherapy. Biochem. Biophys. Res. Commun. 2017, 490, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Liemburg-Apers, D.C.; Willems, P.H.G.M.; Koopman, W.J.H.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.G.; Piskounova, E.; Morrison, S.J. Cancer, oxidative stress, and metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Sotgia, F.; Lisanti, M.P. Mitochondrial biogenesis drives tumor cell proliferation. Am. J. Pathol. 2011, 178, 1949–1952. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Dong, Z.; Cui, H. The autophagy-lysosomal pathways and their emerging roles in modulating proteostasis in tumors. Cells 2018, 8, 4. [Google Scholar] [CrossRef]
- Bold, R.J.; Termuhlen, P.M.; McConkey, D.J. Apoptosis, cancer and cancer therapy. Surg. Oncol. 1997, 6, 133–142. [Google Scholar] [CrossRef]
- Koff, J.L.; Ramachandiran, S.; Bernal-Mizrachi, L. A time to kill: Targeting apoptosis in cancer. Int. J. Mol. Sci. 2015, 16, 2942–2955. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, S.B. Cellular basis of cancer metastasis: A review of fundamentals and new advances. Acta Histochem. 2006, 108, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Adjei, A.A. Targeting angiogenesis in cancer therapy: Moving beyond vascular endothelial growth factor. Oncologist 2015, 20, 660–673. [Google Scholar] [CrossRef]
- Rajabi, M.; Mousa, S.A. The role of angiogenesis in cancer treatment. Biomedicines 2017, 5, 34. [Google Scholar] [CrossRef]
- Smits, P.; Smeitink, J.; van den Heuvel, L. Mitochondrial translation and beyond: Processes implicated in combined oxidative phosphorylation deficiencies. J. Biomed. Biotech. 2010, 2010, 24. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Xochitl, P.-M.; Soledad, F.; Yolanda, C.-V.; Sanna, M.; Faviola, T.-C.; Miguel, S.-V. Protein synthesis and assembly in mitochondrial disorders. Curr. Top. Med. Chem. 2008, 8, 1335–1350. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Heerma van Voss, M.R.; Vesuna, F.; Bol, G.M.; Afzal, J.; Tantravedi, S.; Bergman, Y.; Kammers, K.; Lehar, M.; Malek, R.; Ballew, M.; et al. Targeting mitochondrial translation by inhibiting DDX3: A novel radiosensitization strategy for cancer treatment. Oncogene 2017, 37, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Reed, G.A.; Schiller, G.J.; Kambhampati, S.; Tallman, M.S.; Douer, D.; Minden, M.D.; Yee, K.W.; Gupta, V.; Brandwein, J.; Jitkova, Y.; et al. A Phase 1 study of intravenous infusions of tigecycline in patients with acute myeloid leukemia. Cancer Med. 2016, 5, 3031–3040. [Google Scholar] [CrossRef] [PubMed]
- Kurland, C.G. Molecular characterization of ribonucleic acid from Escherichia coli ribosomes: I. Isolation and molecular weights. J. Mol. Biol. 1960, 2, 83–91. [Google Scholar] [CrossRef]
- Wilson, D.N. The A-Z of bacterial translation inhibitors. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 393–433. [Google Scholar] [CrossRef] [PubMed]
- Jenner, L.; Starosta, A.L.; Terry, D.S.; Mikolajka, A.; Filonava, L.; Yusupov, M.; Blanchard, S.C.; Wilson, D.N.; Yusupova, G. Structural basis for potent inhibitory activity of the antibiotic tigecycline during protein synthesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3812–3816. [Google Scholar] [CrossRef] [PubMed]
- Schedlbauer, A.; Kaminishi, T.; Ochoa-Lizarralde, B.; Dhimole, N.; Zhou, S.; López-Alonso, J.P.; Connell, S.R.; Fucini, P. Structural characterization of an alternative mode of tigecycline binding to the bacterial ribosome. Antimicrob. Agents Chemother. 2015, 59, 2849–2854. [Google Scholar] [CrossRef]
- Greber, B.J.; Bieri, P.; Leibundgut, M.; Leitner, A.; Aebersold, R.; Boehringer, D.; Ban, N. Ribosome: The complete structure of the 55S mammalian mitochondrial ribosome. Science 2015, 348, 303–308. [Google Scholar] [CrossRef]
- Kondo, J.; Westhof, E. The bacterial and mitochondrial ribosomal A-site molecular switches possess different conformational substates. Nucleic Acids Res. 2008, 36, 2654–2666. [Google Scholar] [CrossRef]
- O’Brien, T.W. Properties of human mitochondrial ribosomes. IUBMB Life 2003, 55, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Cavdar Koc, E.; Burkhart, W.; Blackburn, K.; Moseley, A.; Spremulli, L.L. The small subunit of the mammalian mitochondrial ribosome: Identification of the full complement of ribosomal proteins present. J. Biol. Chem. 2001, 276, 19363–19374. [Google Scholar] [CrossRef] [PubMed]
- Koc, E.C.; Burkhart, W.; Blackburn, K.; Moyer, M.B.; Schlatzer, D.M.; Moseley, A.; Spremulli, L.L. The large subunit of the mammalian mitochondrial ribosome: Analysis of the complement of ribosomal proteins present. J. Boil. Chem. 2001, 276, 43958–43969. [Google Scholar] [CrossRef] [PubMed]
- Ling, M.; Merante, F.; Chen, H.S.; Duff, C.; Duncan, A.M.; Robinson, B.H. The human mitochondrial elongation factor tu (EF-Tu) gene: cDNA sequence, genomic localization, genomic structure, and identification of a pseudogene. Gene 1997, 197, 325–336. [Google Scholar] [CrossRef]
- Fu, J.; Momčilović, I.; Prasad, P.V.V. Roles of protein synthesis elongation factor Ef-Tu in heat tolerance in plants. J. Bot. 2012, 2012, 8. [Google Scholar] [CrossRef]
- Cai, Y.-C.; Bullard, J.M.; Thompson, N.L.; Spremulli, L.L. Interaction of mitochondrial elongation factor Tu with aminoacyl-tRNA and elongation factor Ts. J. Biol Chem. 2000, 275, 20308–20314. [Google Scholar] [CrossRef]
- D’Souza, A.R.; Minczuk, M. Mitochondrial transcription and translation: Overview. Essays Biochem. 2018, 62, 309–320. [Google Scholar] [CrossRef]
- Oran, A.R.; Adams, C.M.; Zhang, X.Y.; Gennaro, V.J.; Pfeiffer, H.K.; Mellert, H.S.; Seidel, H.E.; Mascioli, K.; Kaplan, J.; Gaballa, M.R.; et al. Multi-focal control of mitochondrial gene expression by oncogenic MYC provides potential therapeutic targets in cancer. Oncotarget 2016, 7, 72395–72414. [Google Scholar] [CrossRef]
- Dang, C.V.; Kim, J.-W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef]
- Rava, M.; D’Andrea, A.; Nicoli, P.; Gritti, I.; Donati, G.; Doni, M.; Giorgio, M.; Olivero, D.; Amati, B. Therapeutic synergy between tigecycline and venetoclax in a preclinical model of MYC/BCL2 double-hit B cell lymphoma. Sci. Transl. Med. 2018, 10, eaan8723. [Google Scholar] [CrossRef]
- Bisso, A.; Sabo, A.; Amati, B. MYC in Germinal Center-derived lymphomas: Mechanisms and therapeutic opportunities. Immunol. Rev. 2019, 288, 178–197. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Cui, H. Epigenetic modulation of metabolism in glioblastoma. Sem. Cancer Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Dong, Z.; Ke, X.; Hou, J.; Zhao, E.; Zhang, K.; Wang, F.; Yang, L.; Xiang, Z.; Cui, H. The roles of sirtuins family in cell metabolism during tumor development. Sem. Cancer Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Vara, J.Á.F.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Iso, T.; Kedes, L.; Hamamori, Y. HES and HERP families: Multiple effectors of the Notch signaling pathway. J. Cell. Physiol. 2003, 194, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Vincent, E.E.; Poffenberger, M.C.; Jones, R.G. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer lett. 2015, 356, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Dulic, V.; Stein, G.H.; Far, D.F.; Reed, S.I. Nuclear accumulation of p21Cip1 at the onset of mitosis: A role at the G2/M-phase transition. Mol. Cell. Boil. 1998, 18, 546–557. [Google Scholar] [CrossRef]
- Lee, S.; Helfman, D.M. Cytoplasmic p21Cip1 is involved in Ras-induced inhibition of the ROCK/LIMK/cofilin pathway. J. Boil. Chem. 2004, 279, 1885–1891. [Google Scholar] [CrossRef]
- Sestakova, B.; Ondrusova, L.; Vachtenheim, J. Cell cycle inhibitor p21/WAF1/CIP1 as a cofactor of MITF expression in melanoma cells. Pigm. Cell Melanoma Res. 2010, 23, 238–251. [Google Scholar] [CrossRef]
- de Andrade, B.A.; Leon, J.E.; Carlos, R.; Delgado-Azanero, W.; Mosqueda-Taylor, A.; de Almeida, O.P. Immunohistochemical expression of p16, p21, p27 and cyclin D1 in oral nevi and melanoma. Head Neck Pathol. 2012, 6, 297–304. [Google Scholar] [CrossRef]
- Gorospe, M.; Cirielli, C.; Wang, X.; Seth, P.; Capogrossi, M.C.; Holbrook, N.J. p21(Waf1/Cip1) protects against p53-mediated apoptosis of human melanoma cells. Oncogene 1997, 14, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Liao, Y.; Xia, W.; Spohn, B.; Lee, M.-H.; Hung, M.-C. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat. Cell Biol. 2001, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Üren, A.; Fallen, S.; Yuan, H.; Usubütün, A.; Küçükali, T.; Schlegel, R.; Toretsky, J.A. Activation of the canonical Wnt pathway during genital keratinocyte transformation: A model for cervical cancer progression. Cancer Res. 2005, 65, 6199–6206. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jho, E.-H. The protein stability of axin, a negative regulator of wnt signaling, is regulated by smad ubiquitination regulatory factor 2 (Smurf2). J. Biol. Chem. 2010, 285, 36420–36426. [Google Scholar] [CrossRef] [PubMed]
- Schmalhofer, O.; Brabletz, S.; Brabletz, T.J.C. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009, 28, 151–166. [Google Scholar] [CrossRef]
- Roolf, C.; Krohn, S.; Kretzschmar, C.; Huebner, R.; Rolfs, A.; Freund, M.; Junghanss, C. PI3K/Akt signaling interacts with Wnt/β-Catenin signaling but does not induce an accumulation of β-catenin in the nucleus of acute lymphoblastic leukemia cell lines. Blood 2013, 122, 4886. [Google Scholar]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Hakeam, H.A.; Al Duhailib, Z.; Salahuddin, N.; Amin, T. Impact of tigecycline versus imipenem-cilastatin on fibrinogen levels following cytoreductive surgery (CRS) and hyperthermic intraperitoneal chemotherapy (HIPEC): A randomized-controlled study. J. Chemoth. 2018, 30, 224–232. [Google Scholar] [CrossRef]
- Yilmaz Duran, F.; Yildirim, H.; Sen, E.M. A lesser known side effect of tigecycline: Hypofibrinogenemia. Turk. J. Haematol. 2018, 35, 83–84. [Google Scholar] [CrossRef]
- Vandecasteele, S.J.; Seneca, S.; Smet, J.; Reynders, M.; De Ceulaer, J.; Vanlander, A.V.; van Coster, R. Tigecycline-induced inhibition of mitochondrial DNA translation may cause lethal mitochondrial dysfunction in humans. Clin. Microbiol. Infect. 2018, 24, 431.e1–431.e3. [Google Scholar] [CrossRef]
- Myojin, S.; Fukuoka, K.; Kanemaru, A.; Baba, S.; Okamoto, Y.; Suzuki, H.; Kamada, K.; Yoshida, A.; Kikuchi, K.; Horikoshi, Y. Chronic otitis media caused by Mycobacterium abscessus spp. massiliense treated with tigecycline in a 10-year-old child. Int. J. Infect. Dis. 2018, 74, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Kosmidis, C.I.; Chandrasekar, P.H. Management of gram-positive bacterial infections in patients with cancer. Leuk. Lymphoma 2012, 53, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yan, Y.; Li, Z.; Qian, L.; Gong, Z. The antibiotic drug tigecycline: A focus on its promising anticancer properties. Front. Pharmacol. 2016, 7, 473. [Google Scholar] [CrossRef] [PubMed]
- Cunha, B.A.; McDermott, B.; Nausheen, S. Single daily high-dose tigecycline therapy of a multidrug-resistant (MDR) Klebsiella pneumoniae and Enterobacter aerogenes nosocomial urinary tract infection. J. Chemother. 2007, 19, 753–754. [Google Scholar] [CrossRef] [PubMed]
- Jitkova, Y.; Gronda, M.; Hurren, R.; Wang, X.; Goard, C.A.; Jhas, B.; Schimmer, A.D. A novel formulation of tigecycline has enhanced stability and sustained antibacterial and antileukemic activity. PLoS ONE 2014, 9, e95281. [Google Scholar] [CrossRef] [PubMed]
- Bucaneve, G.; Micozzi, A.; Picardi, M.; Ballanti, S.; Cascavilla, N.; Salutari, P.; Specchia, G.; Fanci, R.; Luppi, M.; Cudillo, L.; et al. Results of a multicenter, controlled, randomized clinical trial evaluating the combination of piperacillin/tazobactam and tigecycline in high-risk hematologic patients with cancer with febrile neutropenia. J. Clin. Oncol. 2014, 32, 1463–1471. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Cell Line Name | IC50 (Treatment Time) | Methods | References |
|---|---|---|---|---|
| Melanoma | A375 | 7.24 μM (48 h) | MTT assay | [25] |
| MV3 | 10.9 μM (48 h) | |||
| Non-small cell lung cancer | A549 | 5.8 μM (14 d) | Colony formation | [28] |
| PC19 | 8.7 μM (14 d) | |||
| H157 | 6.8 μM (14 d) | |||
| EBC-1 | 5.9 μM (14 d) | |||
| CML | K562 | 51.4 μM (48 h) | Cell Counting Kit-8 assay | [33] |
| RB1/TP53-mutant human TNBC cells | BT549; MDA-MB-436; Du4475 | average IC50 = 3 μM (72 h) | MTT assay | [29] |
| RB1-proficient/TP53-mutant TNBC cells | HCC38; Hs578t; MDA-MB-231 | average IC50 ≈ 20 μM (72 h) * | ||
| Mouse colon cancer | CT26 | 33 μM (72 h) | CyQuant direct cell proliferation assay | [36] |
| Classification | Cancer Type | Main Biological Phenotypes | Pathways or Molecular Mechanisms | References |
|---|---|---|---|---|
| Hematologic tumors | Myc-driven lymphomas | Abnormally swollen mitochondria, OxPhos↓, ETC↓, intrinsic apoptosis | Mitochondrial translation↓ | [37] |
| CML | OxPhos↓, autophagy, intrinsic apoptosis | PI3K-AKT-mTOR↓, mitochondrial translation↓ | [33,35] | |
| ALL | OxPhos↓, oxidative damages | -- | [34] | |
| DLBCLs | OxPhos↓, ETC↓, ROS↑ | Mitochondrial translation↓ | [30] | |
| AML | Abnormally swollen mitochondria, OxPhos↓ | Mitochondrial translation↓, EF-Tu↓, HIFs↑ | [22,38] | |
| Solid tumors | NSCLC | OxPhos↓, intrinsic apoptosis, ROS↑, MMP↓, ATP levels↓ | -- | [28] |
| Ovarian cancer | OxPhos↓, ETC↓, ROS↑, oxidative damage, cell cycle arrest at G2/M phase, intrinsic apoptosis | Mitochondrial translation↓, Myc↓ | [6,39] | |
| HCC | ATP levels↓, OxPhos↓, ROS↑, oxidative damage | Mitochondrial translation↓ | [31] | |
| RB1-deficient TNBC | ATP levels↓, OxPhos↓ | Mitochondrial translation↓ | [29] | |
| Melanoma | Cell cycle arrest at G0/G1 phase, migration/invasion↓ | Cytoplasmic p21↓ | [25] | |
| Glioma | Cell cycle arrest at G0/G1 phase | miR-199b-5p-HES1-AKT↑ | [27] | |
| Neuroblastoma | Cell cycle arrest at G0/G1 phase | AKT-FOXO3a↓ | [26] | |
| Oral squamous cell carcinoma | Cell cycle arrest at G0/G1 phase | CDK4-CCNE2↓ | [24] | |
| Multiple myeloma | Cell cycle arrest at G0/G1 phase, autophagy | AMPK-mTOR↑ | [40] | |
| Gastric cancer | Autophagy | AMPK-mTOR↑ | [23] | |
| Retinoblastoma | Intrinsic apoptosis, oxidative damage, angiogenesis↓, ATP levels↓ | -- | [41] | |
| Cervical squamous cell carcinoma | Intrinsic apoptosis | Wnt/β-catenin↓ | [32] | |
| Renal cell carcinoma | Intrinsic apoptosis | Mitochondrial translation↓, EF-Tu↓, PI3K/AKT-mTOR↓ | [42] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Z.; Abbas, M.N.; Kausar, S.; Yang, J.; Li, L.; Tan, L.; Cui, H. Biological Functions and Molecular Mechanisms of Antibiotic Tigecycline in the Treatment of Cancers. Int. J. Mol. Sci. 2019, 20, 3577. https://doi.org/10.3390/ijms20143577
Dong Z, Abbas MN, Kausar S, Yang J, Li L, Tan L, Cui H. Biological Functions and Molecular Mechanisms of Antibiotic Tigecycline in the Treatment of Cancers. International Journal of Molecular Sciences. 2019; 20(14):3577. https://doi.org/10.3390/ijms20143577
Chicago/Turabian StyleDong, Zhen, Muhammad Nadeem Abbas, Saima Kausar, Jie Yang, Lin Li, Li Tan, and Hongjuan Cui. 2019. "Biological Functions and Molecular Mechanisms of Antibiotic Tigecycline in the Treatment of Cancers" International Journal of Molecular Sciences 20, no. 14: 3577. https://doi.org/10.3390/ijms20143577
APA StyleDong, Z., Abbas, M. N., Kausar, S., Yang, J., Li, L., Tan, L., & Cui, H. (2019). Biological Functions and Molecular Mechanisms of Antibiotic Tigecycline in the Treatment of Cancers. International Journal of Molecular Sciences, 20(14), 3577. https://doi.org/10.3390/ijms20143577