A Dual GLP-1/GIP Receptor Agonist Does Not Antagonize Glucagon at Its Receptor but May Act as a Biased Agonist at the GLP-1 Receptor

 , and
, and
Abstract
1. Introduction
2. Results
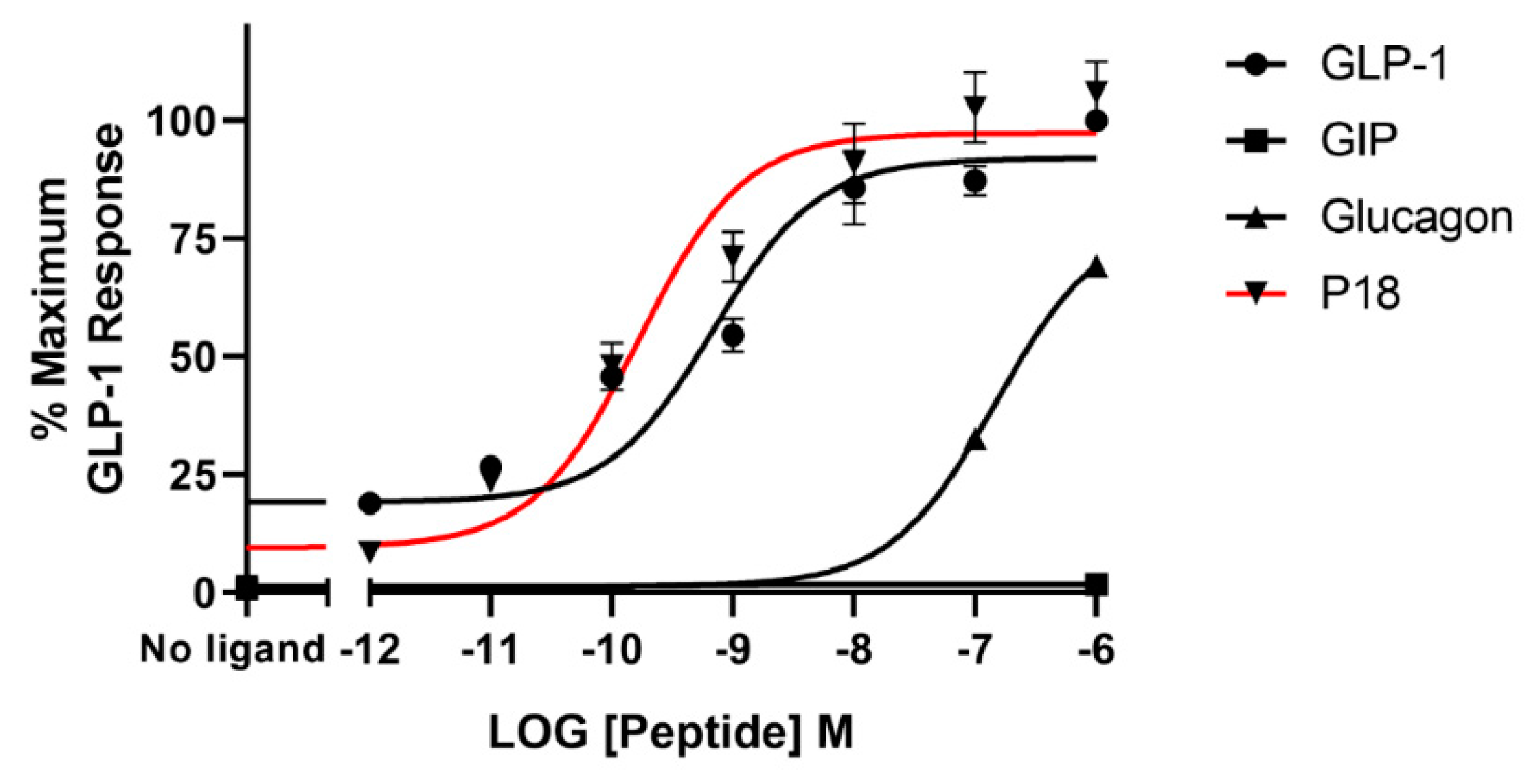
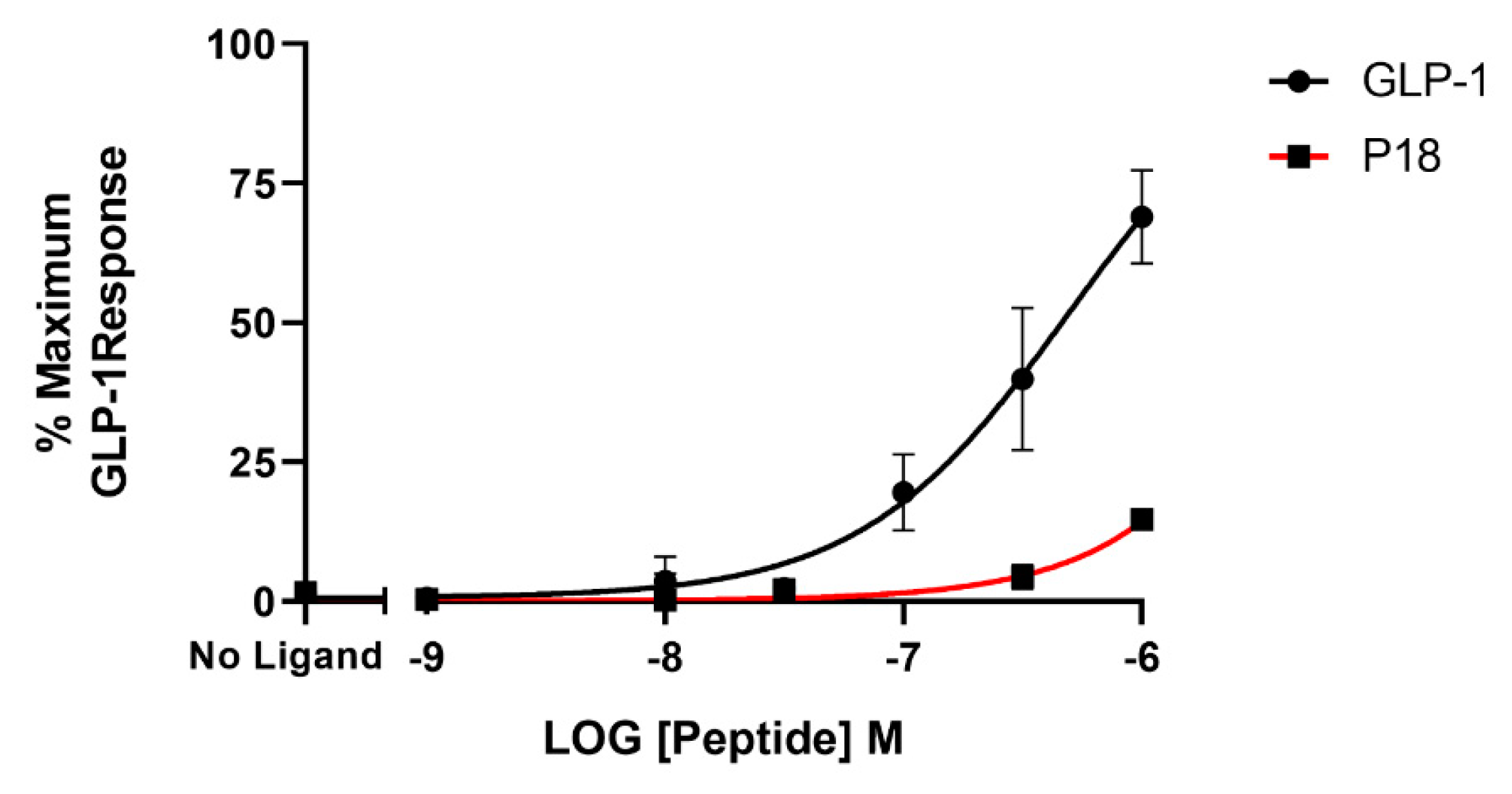
2.1. Activity at the GLP-1 Receptor
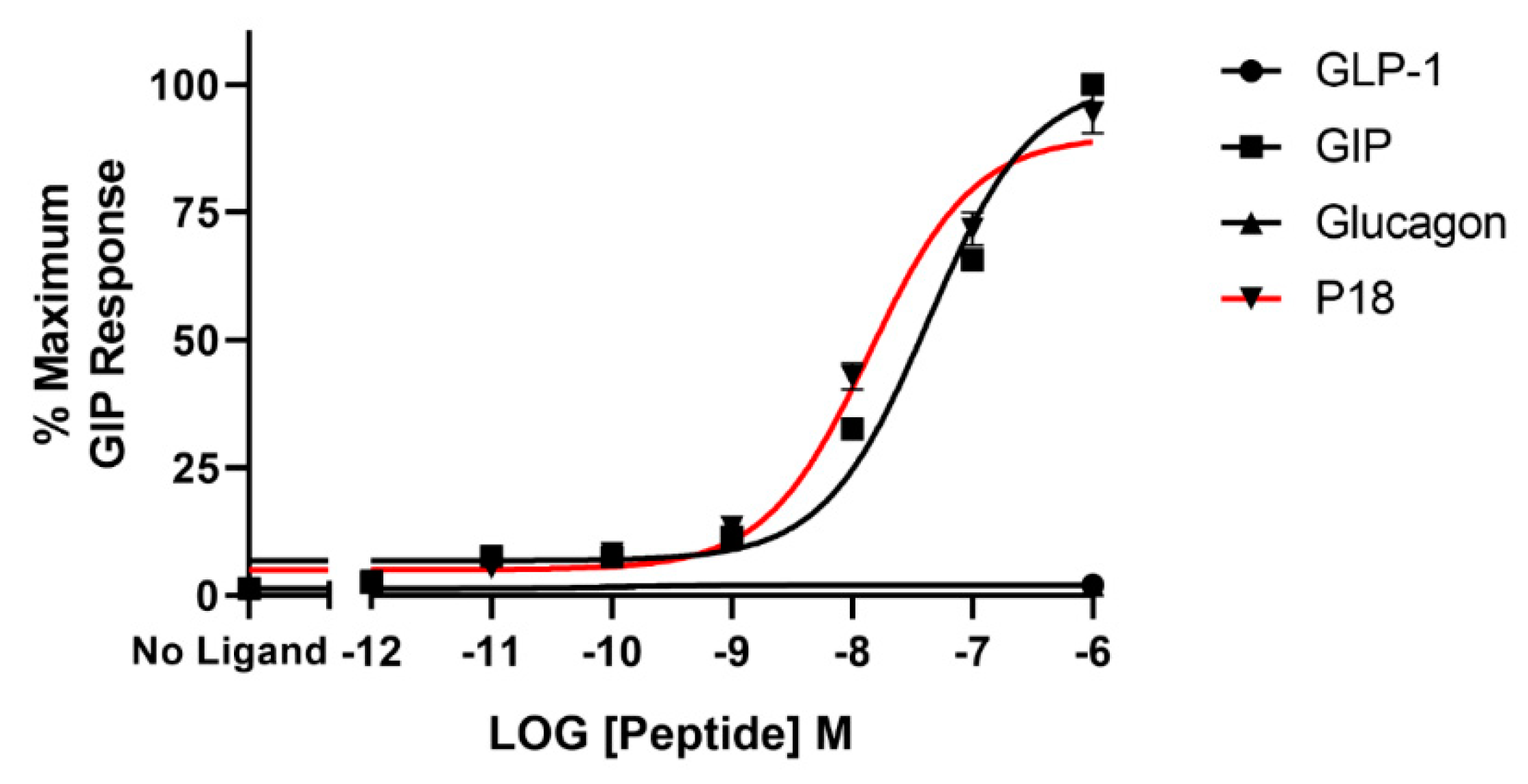
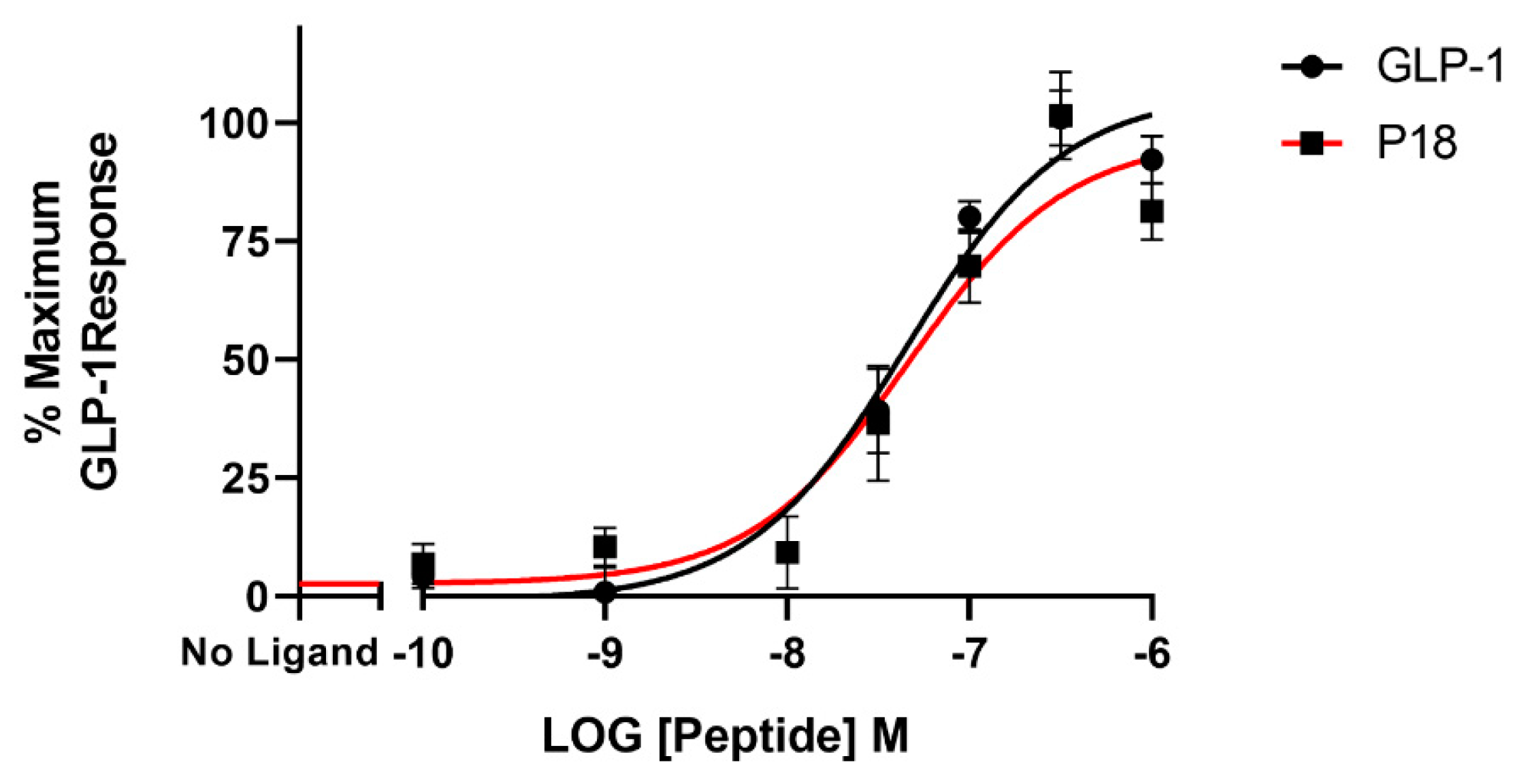
2.2. Activity at the GIP Receptor
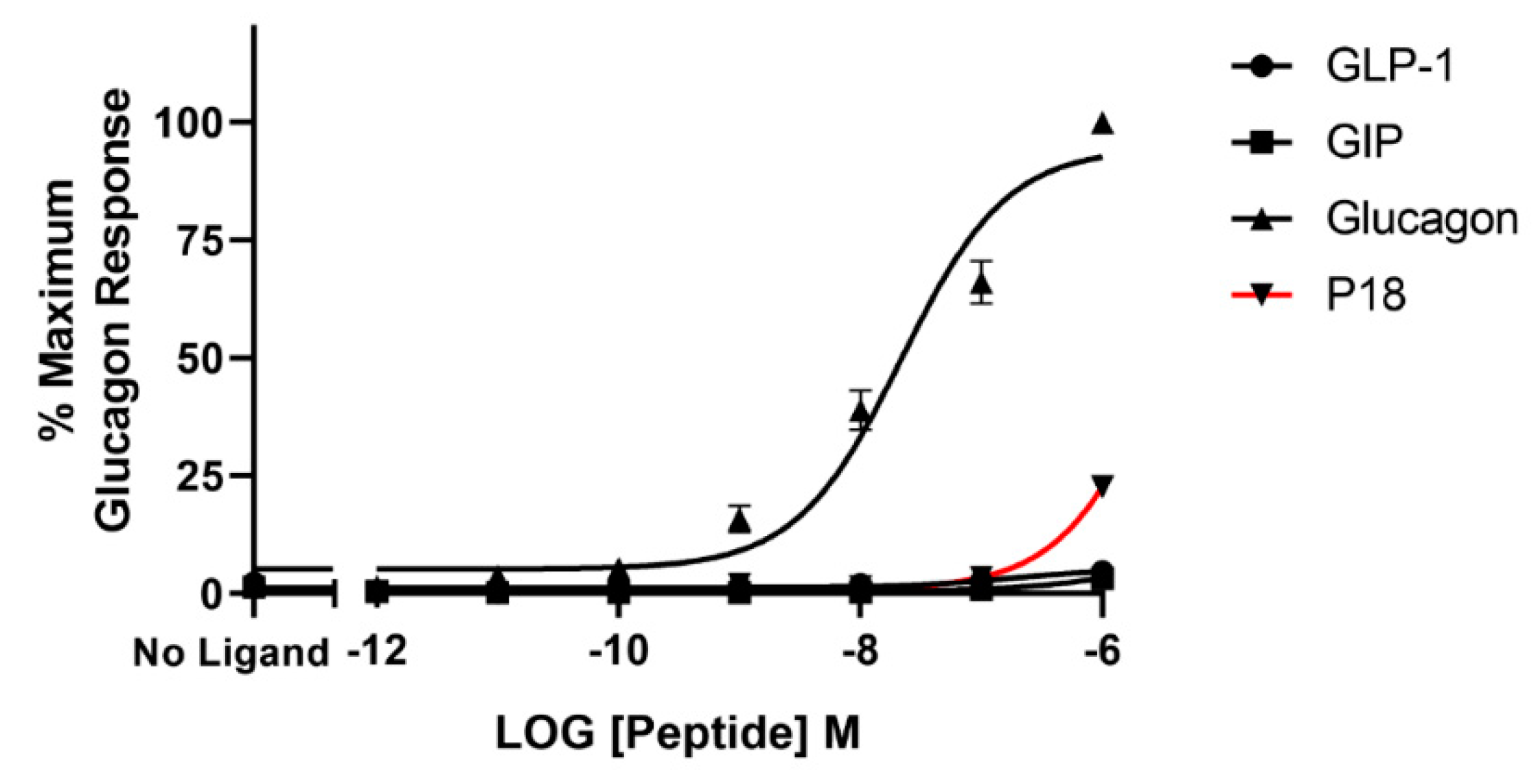
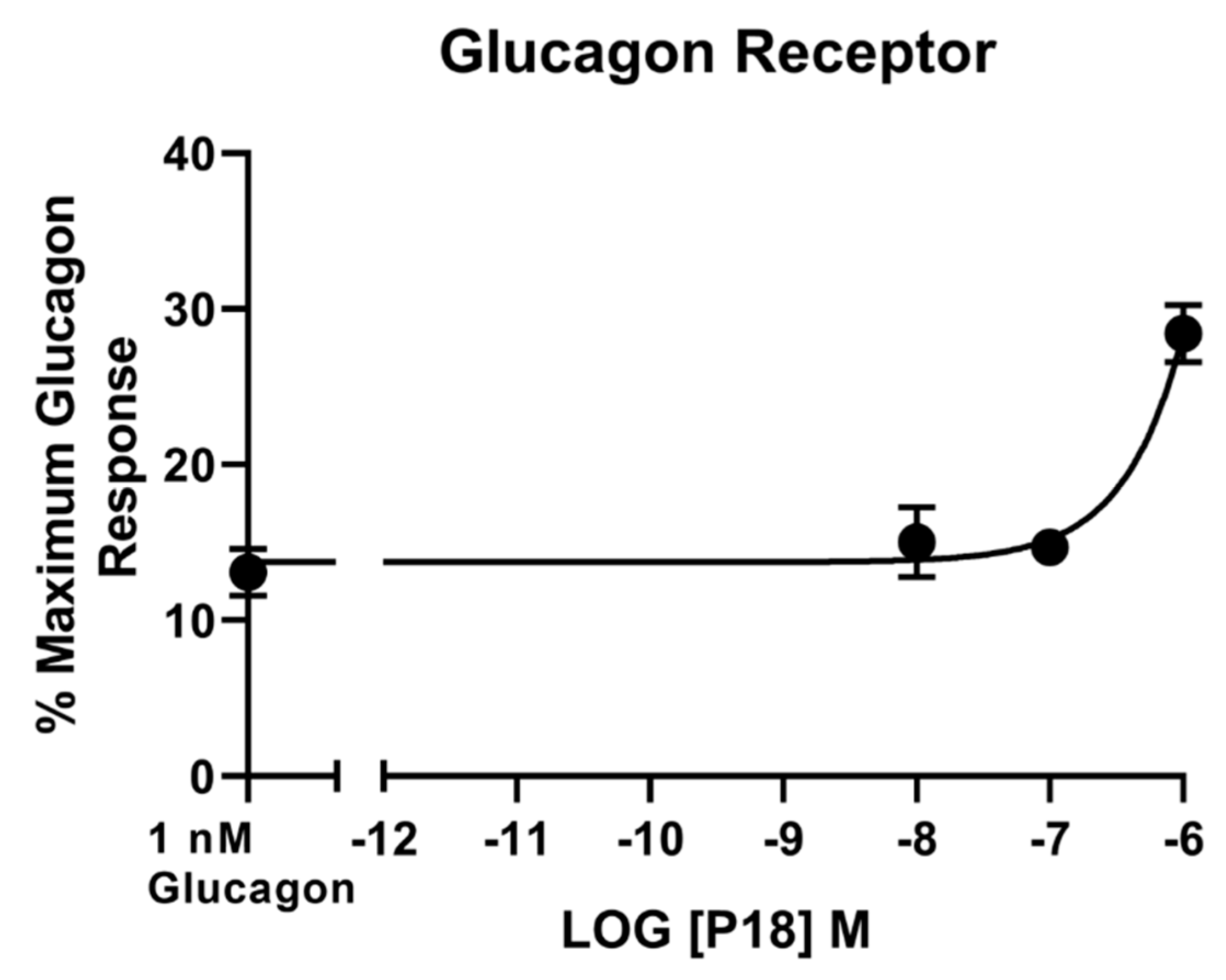
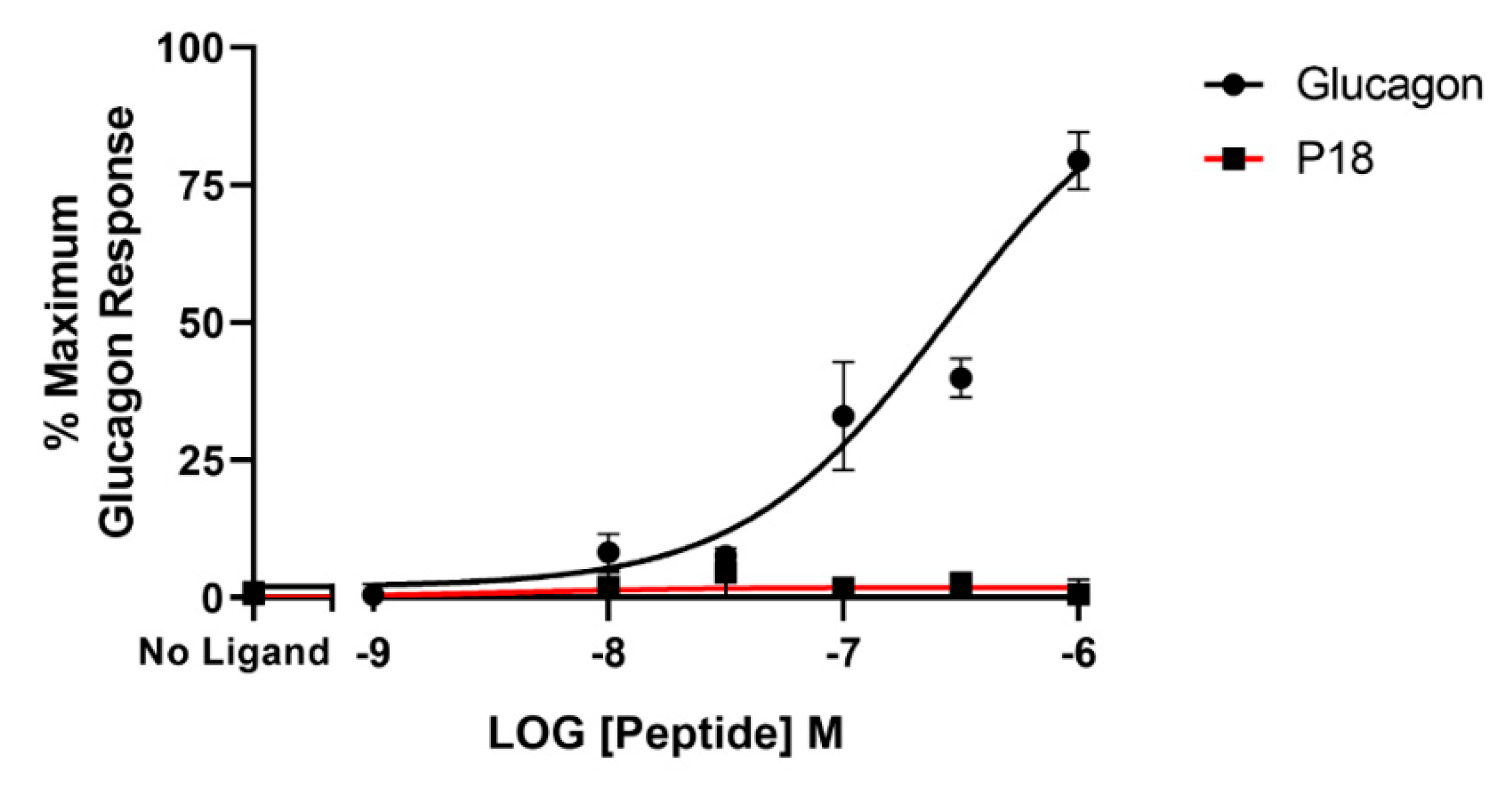
2.3. Activity at the Glucagon Receptor
2.4. Bioluminescence Resonance Energy Transfer (BRET) Assays
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Construction of cDNA
4.3. Cell Culture and Transfection of Cells
4.4. Luciferase Assay
4.5. Bioluminescence Resonance Energy Transfer (BRET) Assays
4.6. Data Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Egan, J.M. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol. Rev. 2008, 60, 470–512. [Google Scholar] [CrossRef]
- McIntosh, C.H.; Widenmaier, S.; Kim, S.J. Pleiotropic actions of the incretin hormones. Vitam. Horm. 2010, 84, 21–79. [Google Scholar] [PubMed]
- Ahrén, B. GLP-1 and Extra-islet Effects. Horm. Metab. Res. 2004, 36, 842–845. [Google Scholar] [CrossRef]
- Schiellerup, S.P.; Skov-Jeppesen, K.; Windeløv, J.A.; Svane, M.S.; Holst, J.J.; Hartmann, B.; Rosenkilde, M.M. Gut Hormones and Their Effect on Bone Metabolism. Potential Drug Therapies in Future Osteoporosis Treatment. Front. Endocrinol. 2019, 10, 75. [Google Scholar] [CrossRef]
- Holst, J.J.; Knop, F.K.; Vilsboll, T.; Krarup, T.; Madsbad, S. Loss of incretin effect is a specific, important, and early characteristic of type 2 diabetes. Diabetes Care 2011, 34 (Suppl. 2), S251–S257. [Google Scholar] [CrossRef]
- Holst, J.J. From the Incretin Concept and the Discovery of GLP-1 to Today’s Diabetes Therapy. Front. Endocrinol. 2019, 10, 260. [Google Scholar] [CrossRef]
- Finan, B.; Ma, T.; Ottaway, N.; Muller, T.D.; Habegger, K.M.; Heppner, K.M.; Kirchner, H.; Holland, J.; Hembree, J.; Raver, C.; et al. Unimolecular Dual Incretins Maximize Metabolic Benefits in Rodents, Monkeys, and Humans. Sci. Transl. Med. 2013, 5, 209ra151. [Google Scholar] [CrossRef]
- Al-Sabah, S. Molecular Pharmacology of the Incretin Receptors. Med. Princ. Pract. 2016, 25, 15–21. [Google Scholar] [CrossRef]
- Mayo, K.E. International Union of Pharmacology. XXXV. The Glucagon Receptor Family. Pharmacol. Rev. 2003, 55, 167–194. [Google Scholar] [CrossRef]
- Graaf, C.D.; Donnelly, D.; Wootten, D.; Lau, J.; Sexton, P.M.; Miller, L.J.; Ahn, J.-M.; Liao, J.; Fletcher, M.M.; Yang, D.; et al. Glucagon-Like Peptide-1 and Its Class B G Protein-Coupled Receptors: A Long March to Therapeutic Successes. Pharmacol. Rev. 2016, 68, 954–1013. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Wang, J.; Plouffe, B.; Smith, J.S.; Yamani, L.; Kaur, S.; Jean-Charles, P.-Y.; Gauthier, C.; Lee, M.-H.; Pani, B.; et al. Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal. 2018, 11, eaat7650. [Google Scholar] [CrossRef]
- Jorgensen, R.; Martini, L.; Schwartz, T.W.; Elling, C.E. Characterization of glucagon-like peptide-1 receptor beta-arrestin 2 interaction: A high-affinity receptor phenotype. Mol. Endocrinol. 2005, 19, 812–823. [Google Scholar] [CrossRef]
- Al-Sabah, S.; Al-Fulaij, M.; Shaaban, G.; Ahmed, H.A.; Mann, R.J.; Donnelly, D.; Bunemann, M.; Krasel, C. The GIP receptor displays higher basal activity than the GLP-1 receptor but does not recruit GRK2 or arrestin3 effectively. PLoS ONE 2014, 9, e106890. [Google Scholar] [CrossRef]
- Syme, C.A.; Zhang, L.; Bisello, A. Caveolin-1 regulates cellular trafficking and function of the glucagon-like Peptide 1 receptor. Mol. Endocrinol. 2006, 20, 3400–3411. [Google Scholar] [CrossRef]
- Sonoda, N.; Imamura, T.; Yoshizaki, T.; Babendure, J.L.; Lu, J.C.; Olefsky, J.M. Beta-Arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic beta cells. Proc. Natl. Acad. Sci. USA 2008, 105, 6614–6619. [Google Scholar] [CrossRef]
- Jones, B.; Buenaventura, T.; Kanda, N.; Chabosseau, P.; Owen, B.M.; Scott, R.; Goldin, R.; Angkathunyakul, N.; Corrêa, I.R., Jr.; Bosco, D.; et al. Targeting GLP-1 receptor trafficking to improve agonist efficacy. Nat. Commun. 2018, 9, 1602. [Google Scholar] [CrossRef]
- Ismail, S.; Dubois-Vedrenne, I.; Laval, M.; Tikhonova, I.G.; D’Angelo, R.; Sanchez, C.; Clerc, P.; Gherardi, M.-J.; Gigoux, V.; Magnan, R.; et al. Internalization and desensitization of the human glucose-dependent-insulinotropic receptor is affected by N-terminal acetylation of the agonist. Mol. Cell. Endocrinol. 2015, 414, 202–215. [Google Scholar] [CrossRef]
- Gabe, M.B.N.; Sparre-Ulrich, A.H.; Pedersen, M.F.; Gasbjerg, L.S.; Inoue, A.; Bräuner-Osborne, H.; Hartmann, B.; Rosenkilde, M.M. Human GIP(3-30)NH 2 inhibits G protein-dependent as well as G protein-independent signaling and is selective for the GIP receptor with high-affinity binding to primate but not rodent GIP receptors. Biochem. Pharmacol. 2018, 150, 97–107. [Google Scholar] [CrossRef]
- Knudsen, L.B.; Lau, J. The Discovery and Development of Liraglutide and Semaglutide. Front. Endocrinol. 2019, 10, 155. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.E.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Shyangdan, D.S.; Royle, P.; Clar, C.; Sharma, P.; Waugh, N.; Snaith, A. Glucagon-like peptide analogues for type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2011. [Google Scholar] [CrossRef]
- Nauck, M.A.; Heimesaat, M.M.; Orskov, C.; Holst, J.J.; Ebert, R.; Creutzfeldt, W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J. Clin. Investig. 1993, 91, 301–307. [Google Scholar] [CrossRef]
- Miyawaki, K.; Yamada, Y.; Ban, N.; Ihara, Y.; Tsukiyama, K.; Zhou, H.; Fujimoto, S.; Oku, A.; Tsuda, K.; Toyokuni, S.; et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat. Med. 2002, 8, 738–742. [Google Scholar] [CrossRef]
- Gault, V.A.; O’Harte, F.P.; Harriott, P.; Flatt, P.R. Characterization of the cellular and metabolic effects of a novel enzyme-resistant antagonist of glucose-dependent insulinotropic polypeptide. Biochem. Biophys. Res. Commun. 2002, 290, 1420–1426. [Google Scholar] [CrossRef]
- McClean, P.L.; Irwin, N.; Cassidy, R.S.; Holst, J.J.; Gault, V.A.; Flatt, P.R. GIP receptor antagonism reverses obesity, insulin resistance, and associated metabolic disturbances induced in mice by prolonged consumption of high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1746–E1755. [Google Scholar] [CrossRef]
- Al-Sabah, S.; Al-Fulaij, M.; Ahmed, H.A. Selectivity of peptide ligands for the human incretin receptors expressed in HEK-293 cells. Eur. J. Pharmacol. 2014, 741, 311–315. [Google Scholar] [CrossRef]
- Sparre-Ulrich, A.H.; Hansen, L.S.; Svendsen, B.; Christensen, M.; Knop, F.K.; Hartmann, B.; Holst, J.J.; Rosenkilde, M.M. Species-specific action of (Pro3)GIP–A full agonist at human GIP receptors, but a partial agonist and competitive antagonist at rat and mouse GIP receptors: Species-specific activity of (Pro3)GIP. Br. J. Pharmacol. 2016, 173, 27–38. [Google Scholar] [CrossRef]
- Kim, S.-J.; Nian, C.; Karunakaran, S.; Clee, S.M.; Isales, C.M.; McIntosh, C.H.S. GIP-Overexpressing Mice Demonstrate Reduced Diet-Induced Obesity and Steatosis, and Improved Glucose Homeostasis. PLoS ONE 2012, 7, e40156. [Google Scholar] [CrossRef]
- Højberg, P.V.; Vilsbøll, T.; Rabøl, R.; Knop, F.K.; Bache, M.; Krarup, T.; Holst, J.J.; Madsbad, S. Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia 2009, 52, 199–207. [Google Scholar] [CrossRef]
- Frias, J.P.; Nauck, M.A.; Van, J.; Kutner, M.E.; Cui, X.; Benson, C.; Urva, S.; Gimeno, R.E.; Milicevic, Z.; Robins, D.; et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: A randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018, 392, 2180–2193. [Google Scholar] [CrossRef]
- Runge, S.; Wulff, B.S.; Madsen, K.; Bräuner-Osborne, H.; Knudsen, L.B. Different domains of the glucagon and glucagon-like peptide-1 receptors provide the critical determinants of ligand selectivity. Br. J. Pharmacol. 2003, 138, 787–794. [Google Scholar] [CrossRef]
- Pearson, M.J.; Unger, R.H.; Holland, W.L. Clinical Trials, Triumphs, and Tribulations of Glucagon Receptor Antagonists. Diabetes Care 2016, 39, 1075–1077. [Google Scholar] [CrossRef]
- Kelly, E. Efficacy and ligand bias at the μ-opioid receptor: Efficacy and ligand bias at the μ-opioid receptor. Br. J. Pharmacol. 2013, 169, 1430–1446. [Google Scholar] [CrossRef]
- Rajagopal, S.; Rajagopal, K.; Lefkowitz, R.J. Teaching old receptors new tricks: Biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 2010, 9, 373–386. [Google Scholar] [CrossRef]
- Kremers, G.-J.; Goedhart, J.; van Munster, E.B.; Gadella, T.W.J. Cyan and Yellow Super Fluorescent Proteins with Improved Brightness, Protein Folding, and FRET Förster Radius‡. Biochemistry 2006, 45, 6570–6580. [Google Scholar] [CrossRef]
- Krasel, C.; Bunemann, M.; Lorenz, K.; Lohse, M.J. beta-Arrestin Binding to the beta2-Adrenergic Receptor Requires Both Receptor Phosphorylation and Receptor Activation. J. Biol. Chem. 2005, 280, 9528–9535. [Google Scholar] [CrossRef]
- Wan, Q.; Okashah, N.; Inoue, A.; Nehmé, R.; Carpenter, B.; Tate, C.G.; Lambert, N.A. Mini G protein probes for active G protein–coupled receptors (GPCRs) in live cells. J. Biol. Chem. 2018, 293, 7466–7473. [Google Scholar] [CrossRef]
- Shaaban, G.; Oriowo, M.; Al-Sabah, S. Rate of Homologous Desensitization and Internalization of the GLP-1 Receptor. Molecules 2016, 22, 22. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GLP-1 Receptor | GIP Receptor | Glucagon Receptor | ||||
|---|---|---|---|---|---|---|
| pEC50 | Emax (% GLP-1) | pEC50 | Emax (% GIP) | pEC50 | Emax (% Glucagon) | |
| GLP-1 | 9.4 ± 0.14 | 100 | ND | ND | ND | ND |
| GIP | ND | ND | 7.1 ± 0.11 | 100 | ND | ND |
| Glucagon | 6.4 ± 0.1 a | 62.8 ± 7.4 a | ND | ND | 7.5 ± 0.28 | 100 |
| P18 | 9.7 ± 0.2 | 106 ± 10.7 | 7.8 ± 0.03 b | 94.4 ± 4.09 | 4.9 ± 0.50 c | 22.6 ± 1.96 d |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Zamel, N.; Al-Sabah, S.; Luqmani, Y.; Adi, L.; Chacko, S.; Schneider, T.D.; Krasel, C. A Dual GLP-1/GIP Receptor Agonist Does Not Antagonize Glucagon at Its Receptor but May Act as a Biased Agonist at the GLP-1 Receptor. Int. J. Mol. Sci. 2019, 20, 3532. https://doi.org/10.3390/ijms20143532
Al-Zamel N, Al-Sabah S, Luqmani Y, Adi L, Chacko S, Schneider TD, Krasel C. A Dual GLP-1/GIP Receptor Agonist Does Not Antagonize Glucagon at Its Receptor but May Act as a Biased Agonist at the GLP-1 Receptor. International Journal of Molecular Sciences. 2019; 20(14):3532. https://doi.org/10.3390/ijms20143532
Chicago/Turabian StyleAl-Zamel, Noura, Suleiman Al-Sabah, Yunus Luqmani, Lobna Adi, Siby Chacko, Tom Dario Schneider, and Cornelius Krasel. 2019. "A Dual GLP-1/GIP Receptor Agonist Does Not Antagonize Glucagon at Its Receptor but May Act as a Biased Agonist at the GLP-1 Receptor" International Journal of Molecular Sciences 20, no. 14: 3532. https://doi.org/10.3390/ijms20143532
APA StyleAl-Zamel, N., Al-Sabah, S., Luqmani, Y., Adi, L., Chacko, S., Schneider, T. D., & Krasel, C. (2019). A Dual GLP-1/GIP Receptor Agonist Does Not Antagonize Glucagon at Its Receptor but May Act as a Biased Agonist at the GLP-1 Receptor. International Journal of Molecular Sciences, 20(14), 3532. https://doi.org/10.3390/ijms20143532