Pathway-Centric Structure-Based Multi-Target Compound Screening for Anti-Virulence Drug Repurposing


Abstract
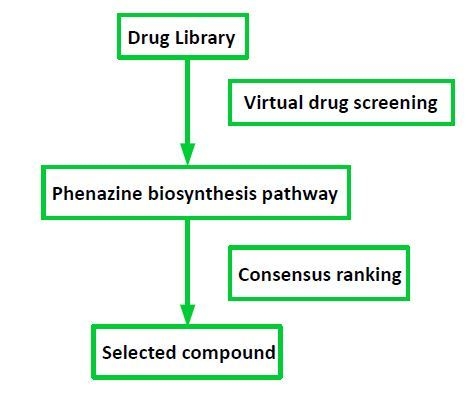
:
1. Introduction
2. Result
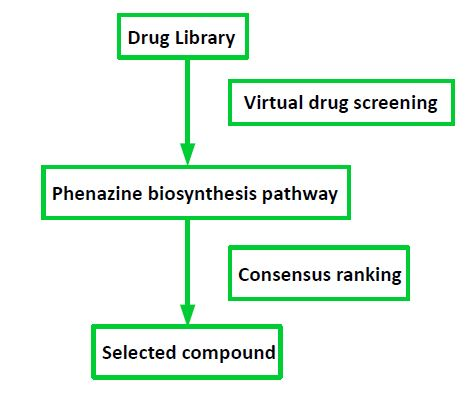
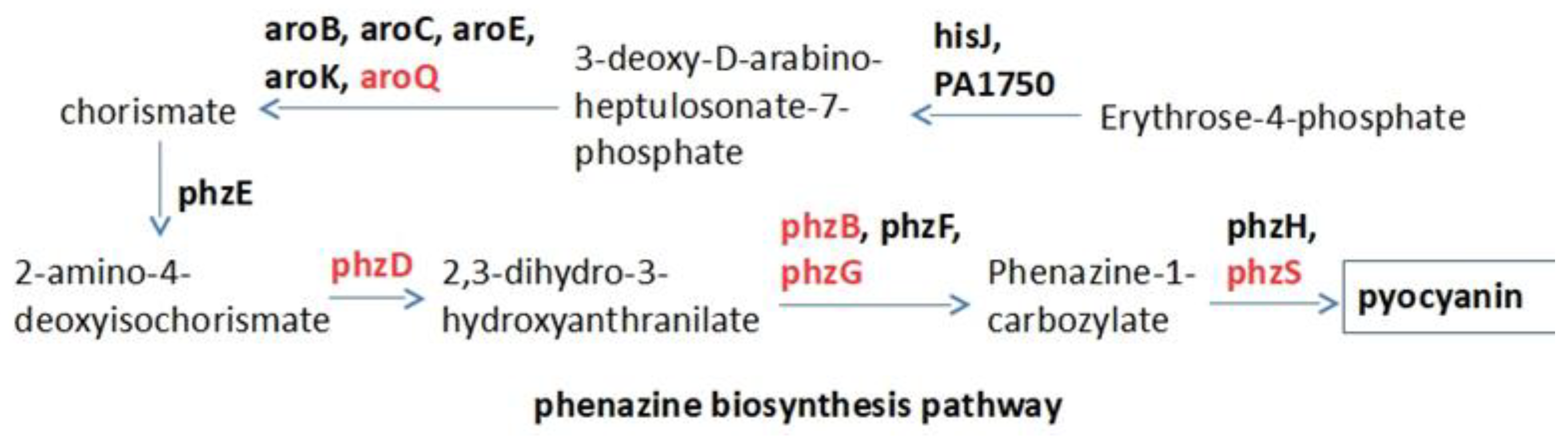
2.1. Target Selection and Compound Library
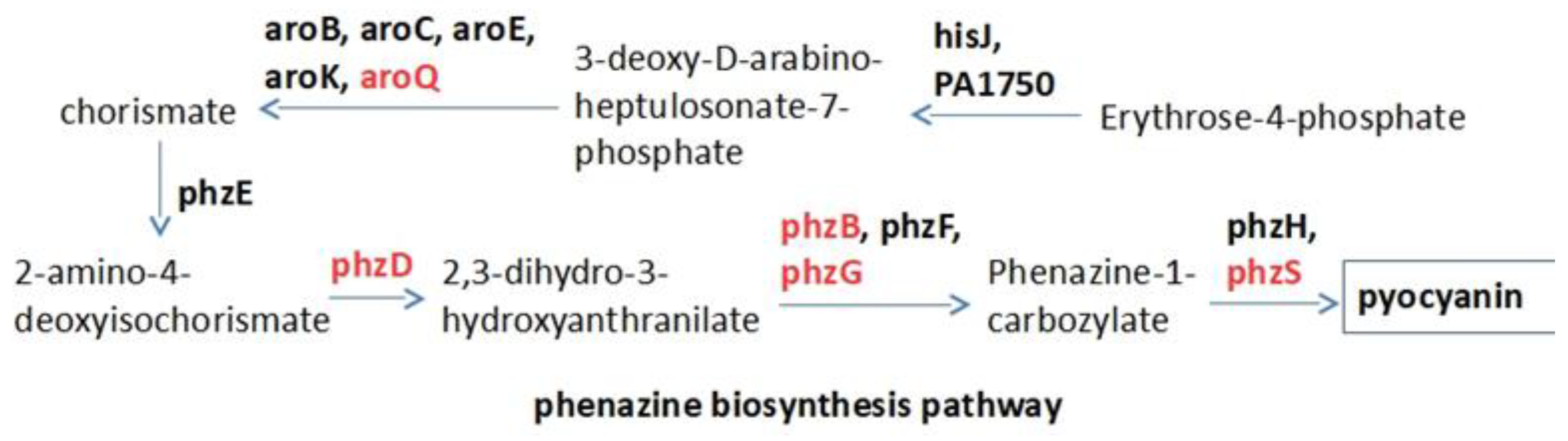
2.2. Raw Docking Score for aroQ, phzB, phzD, phzG, phzS
2.3. Normalized Docking Score with Linear Regression Method
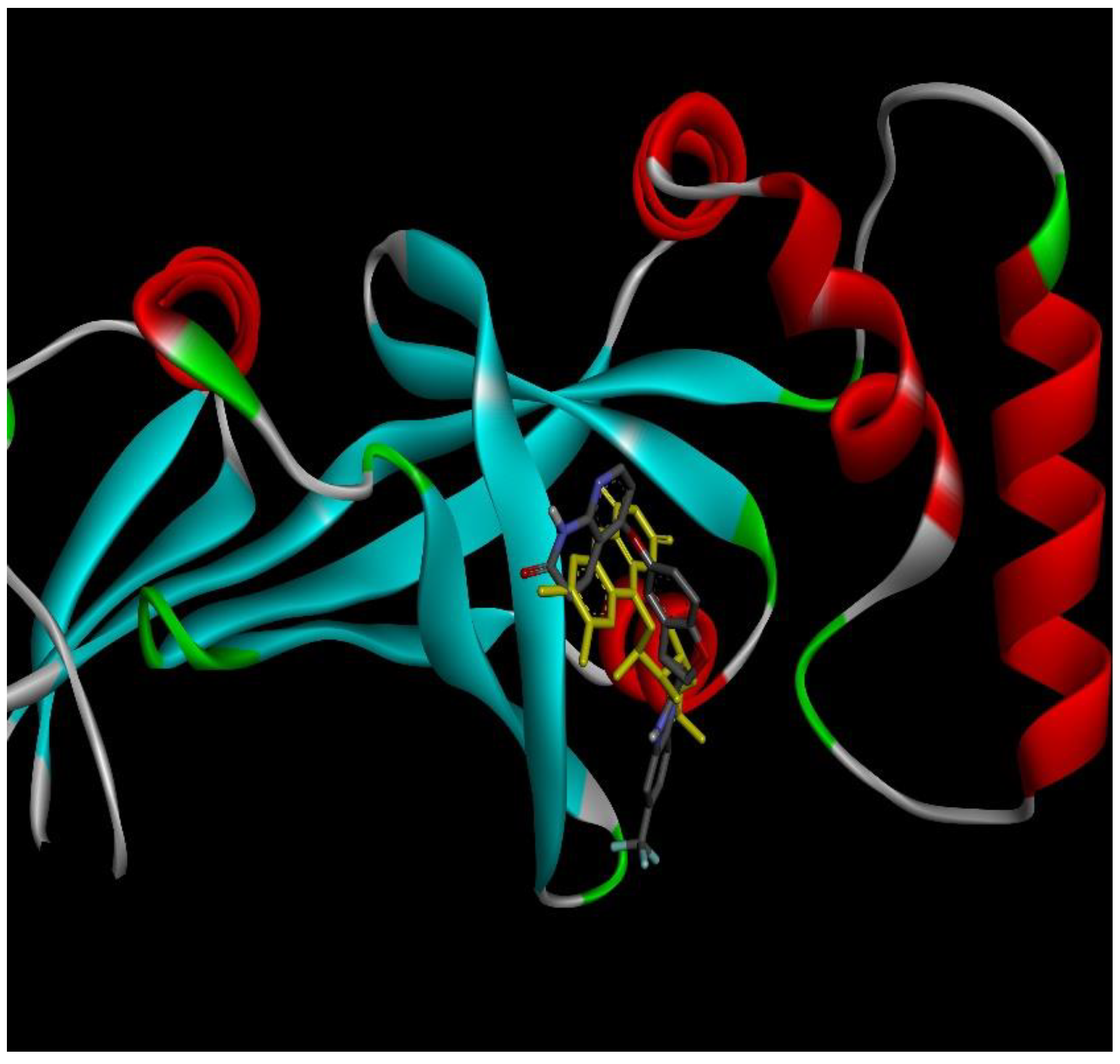
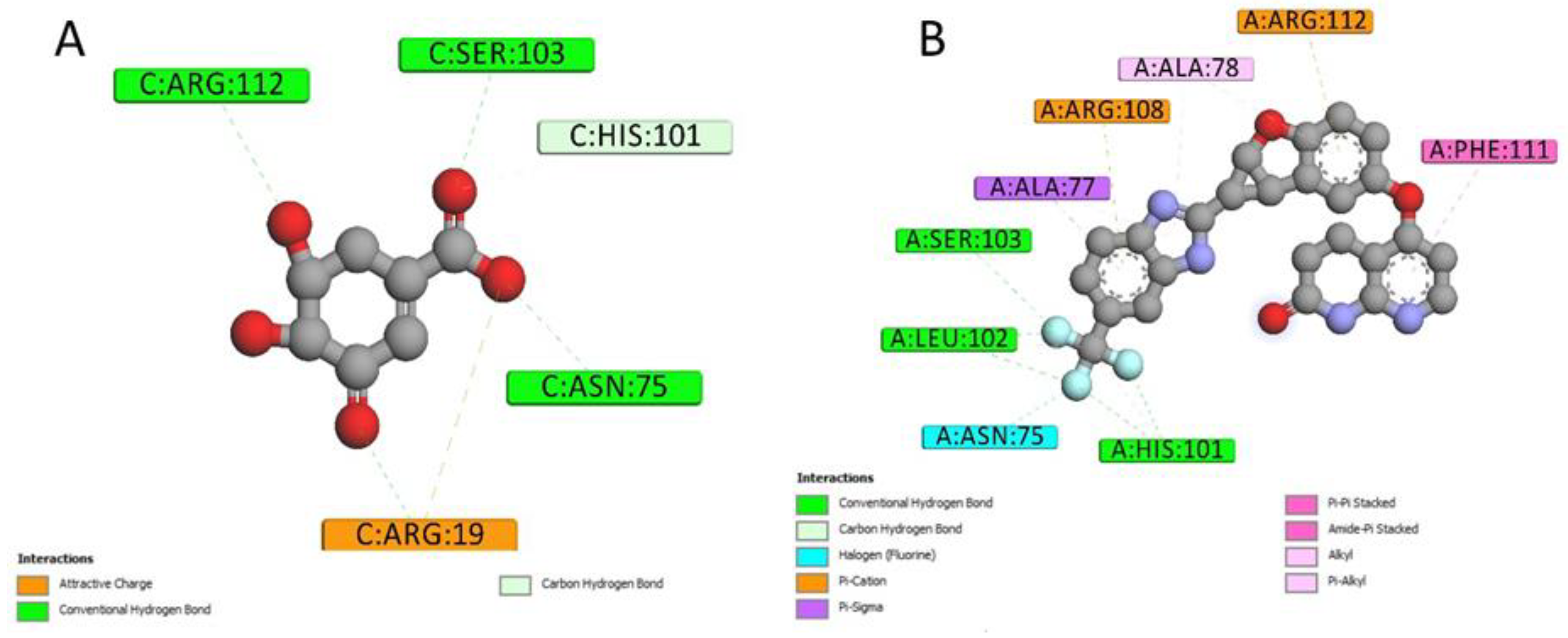
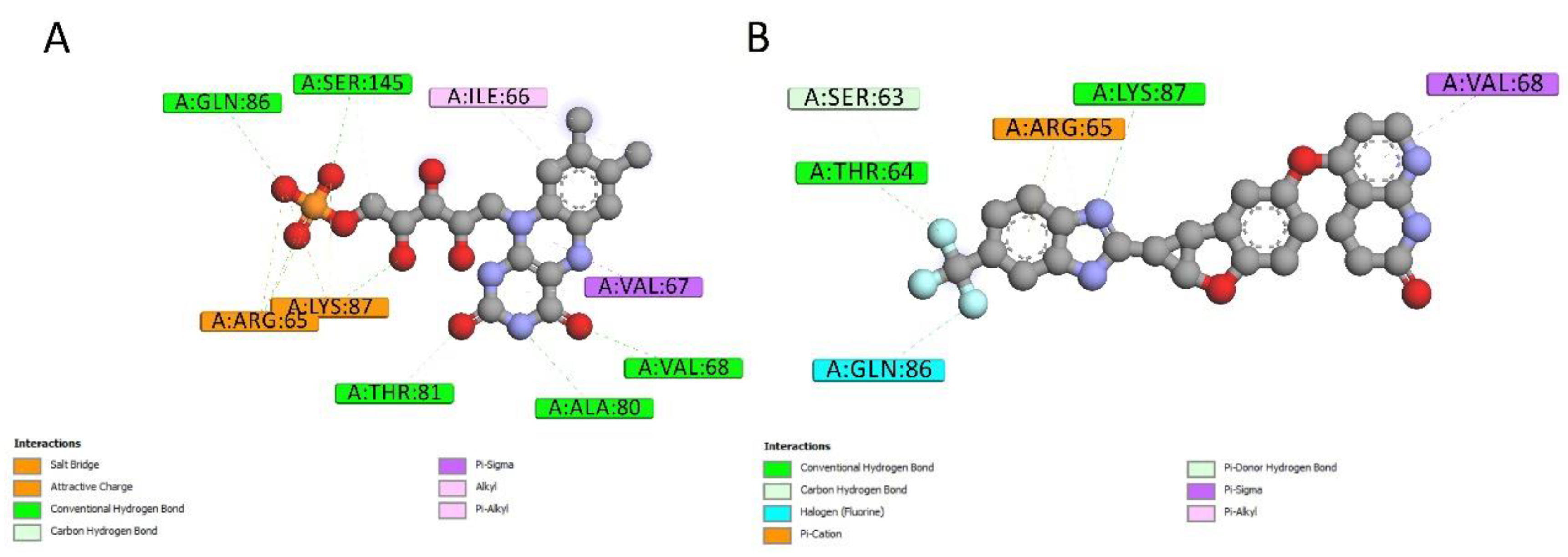
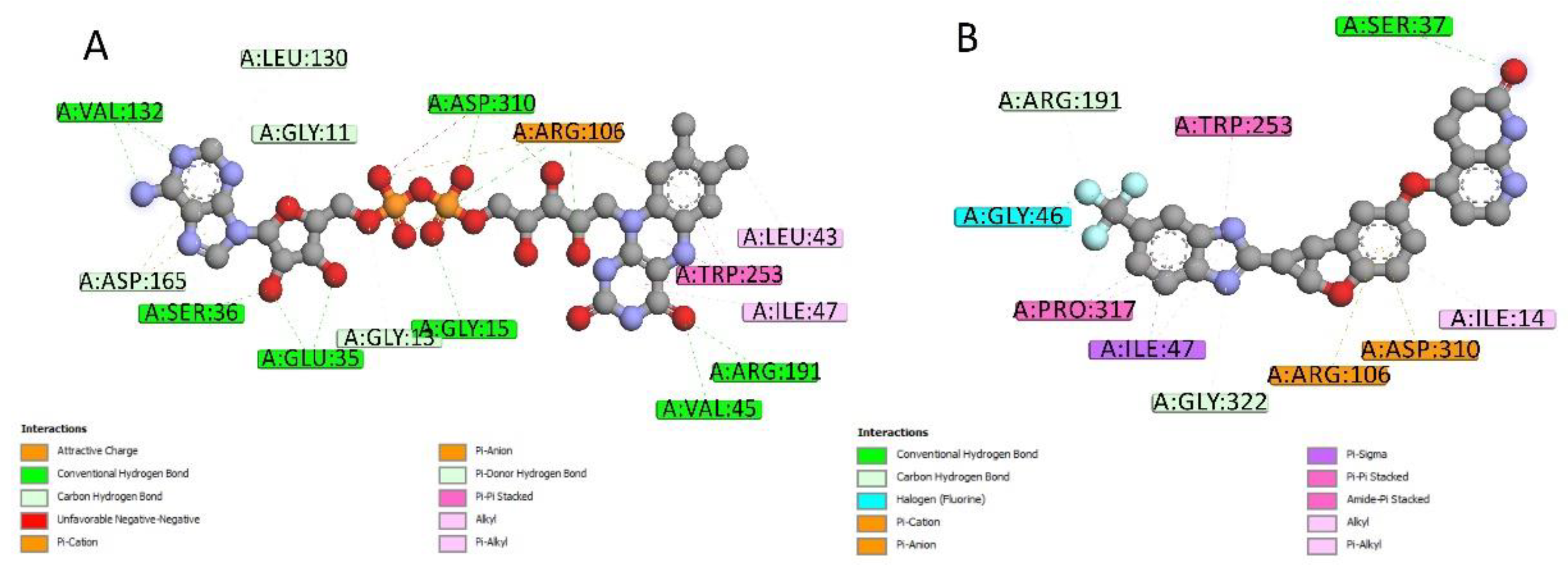
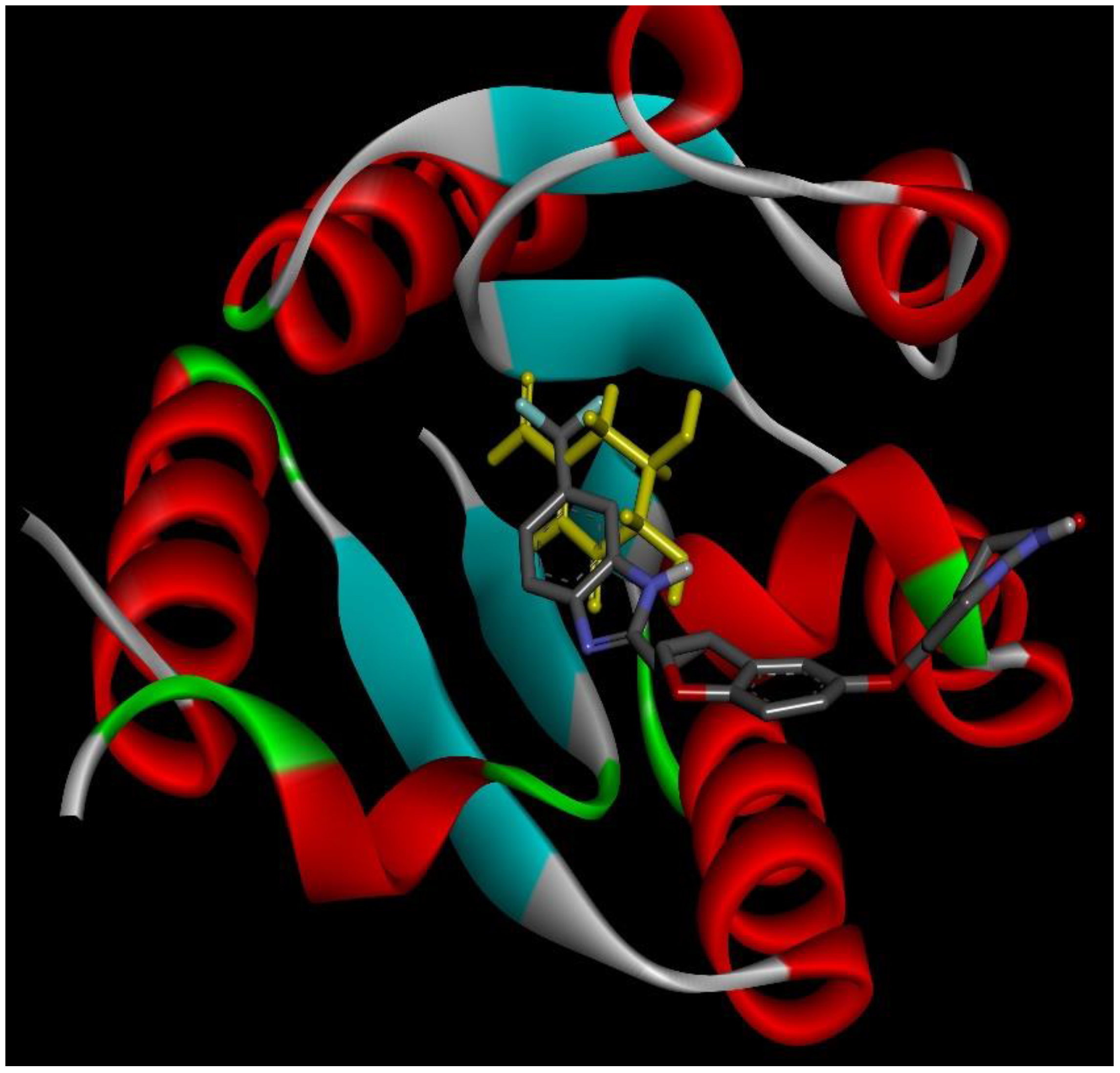
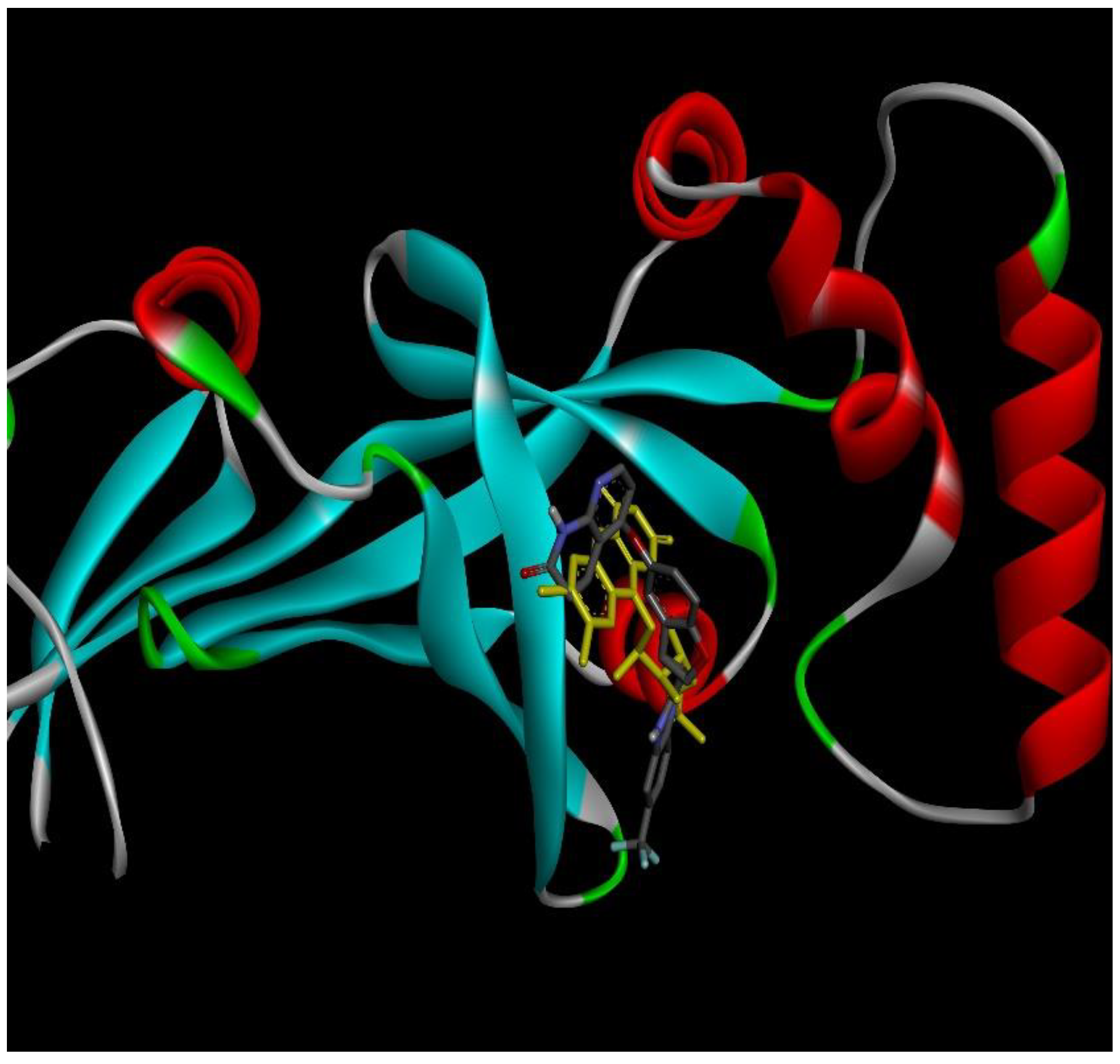
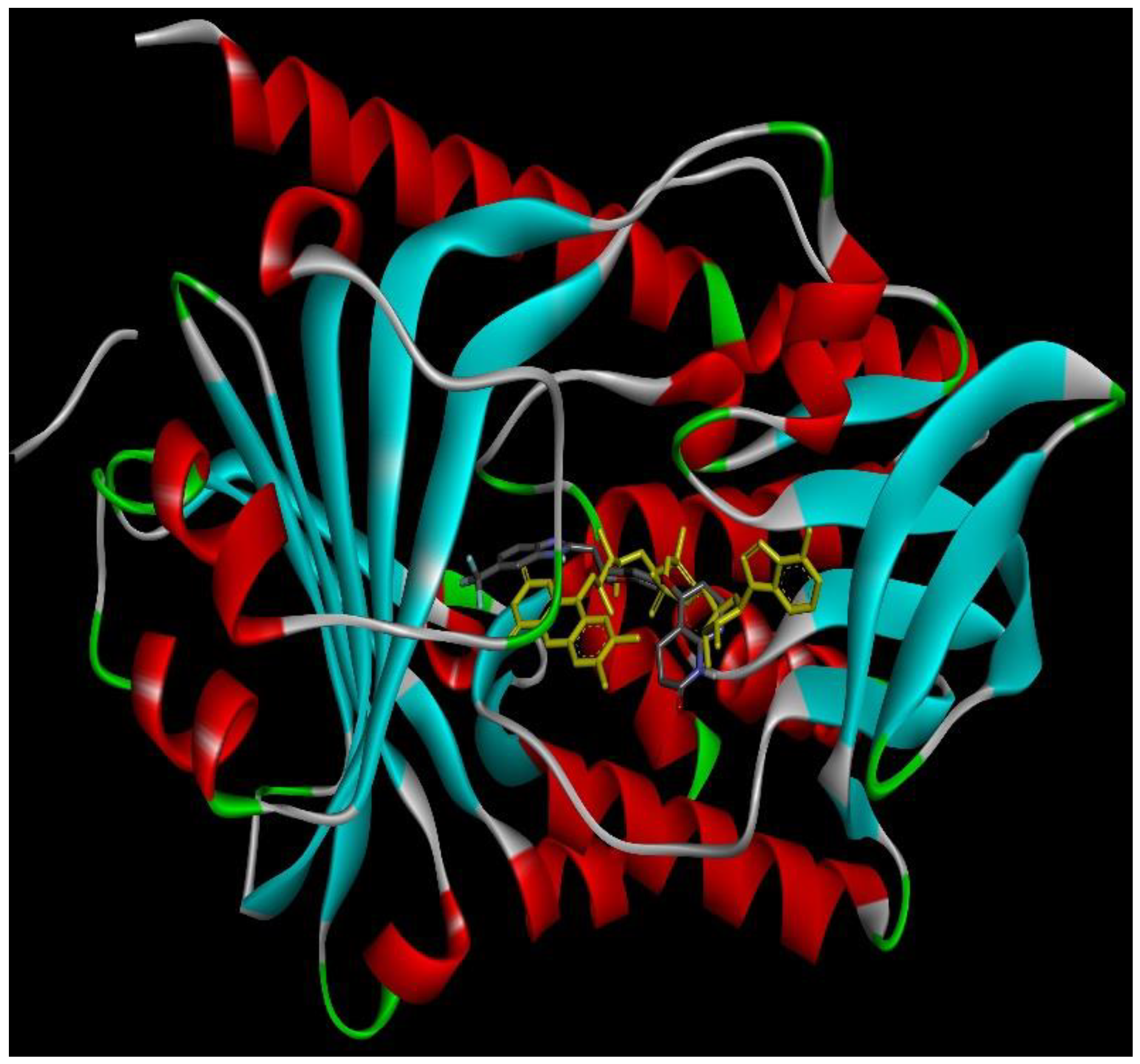
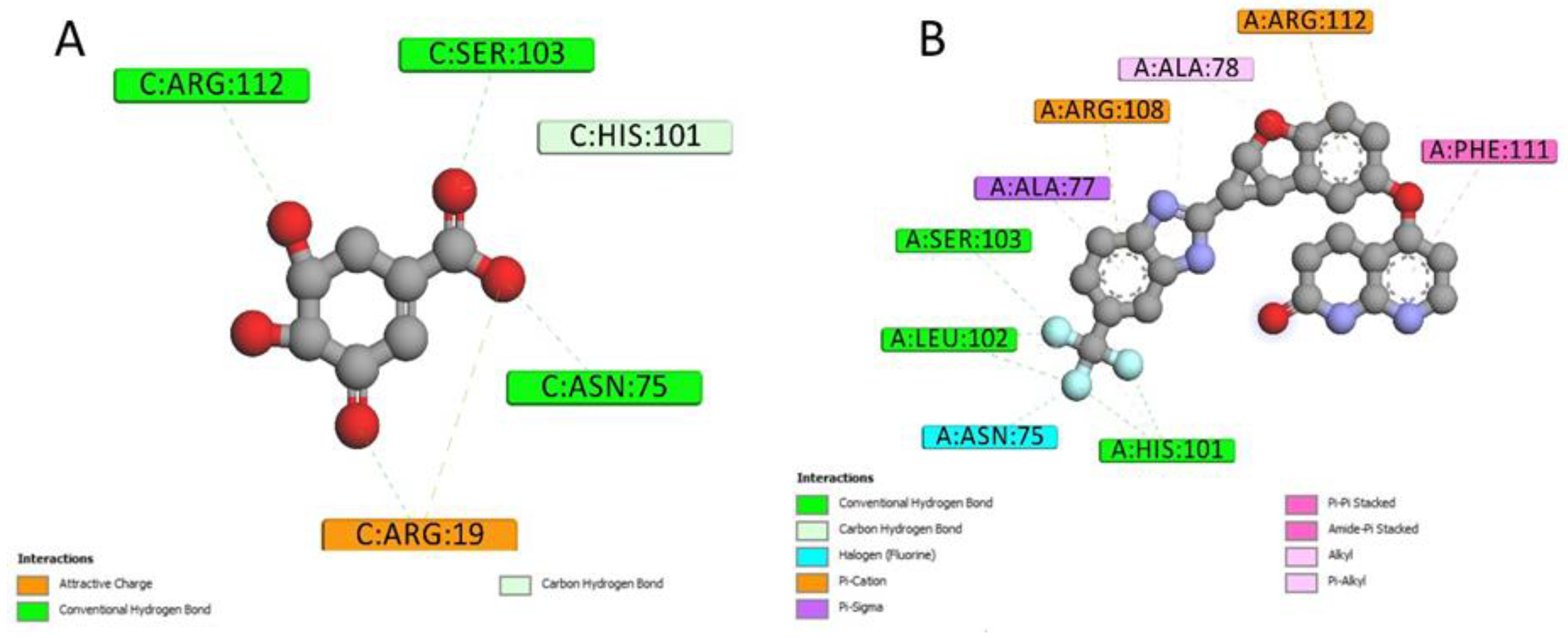
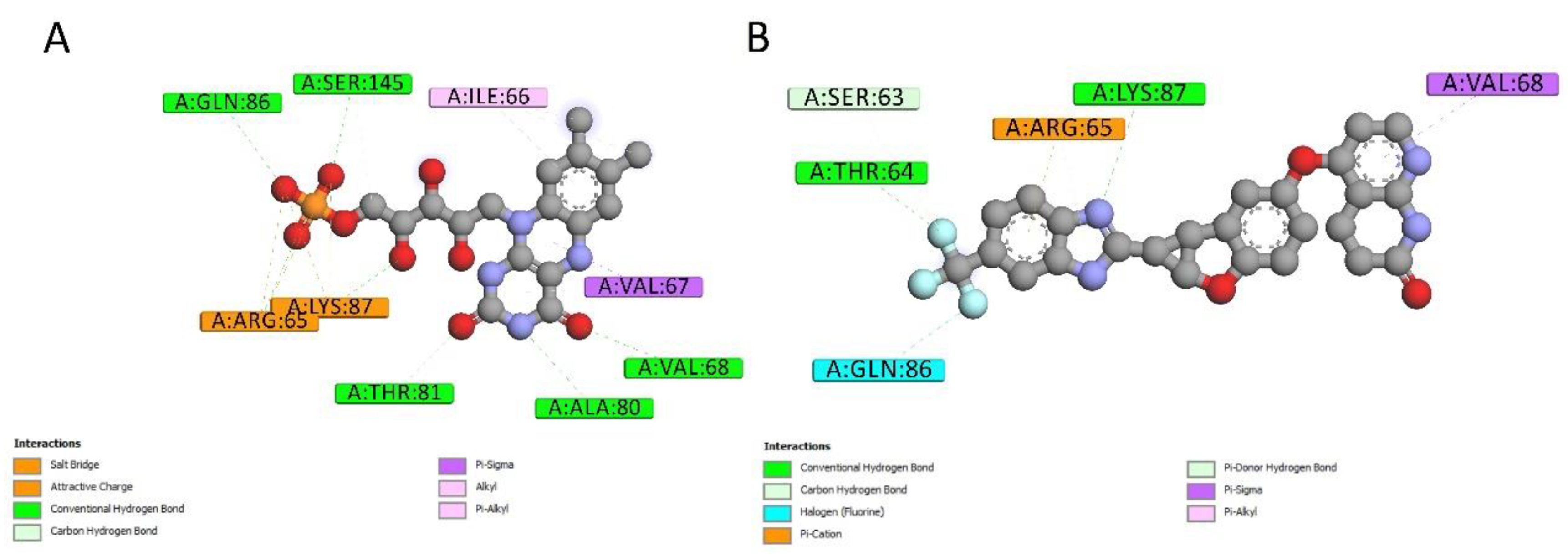
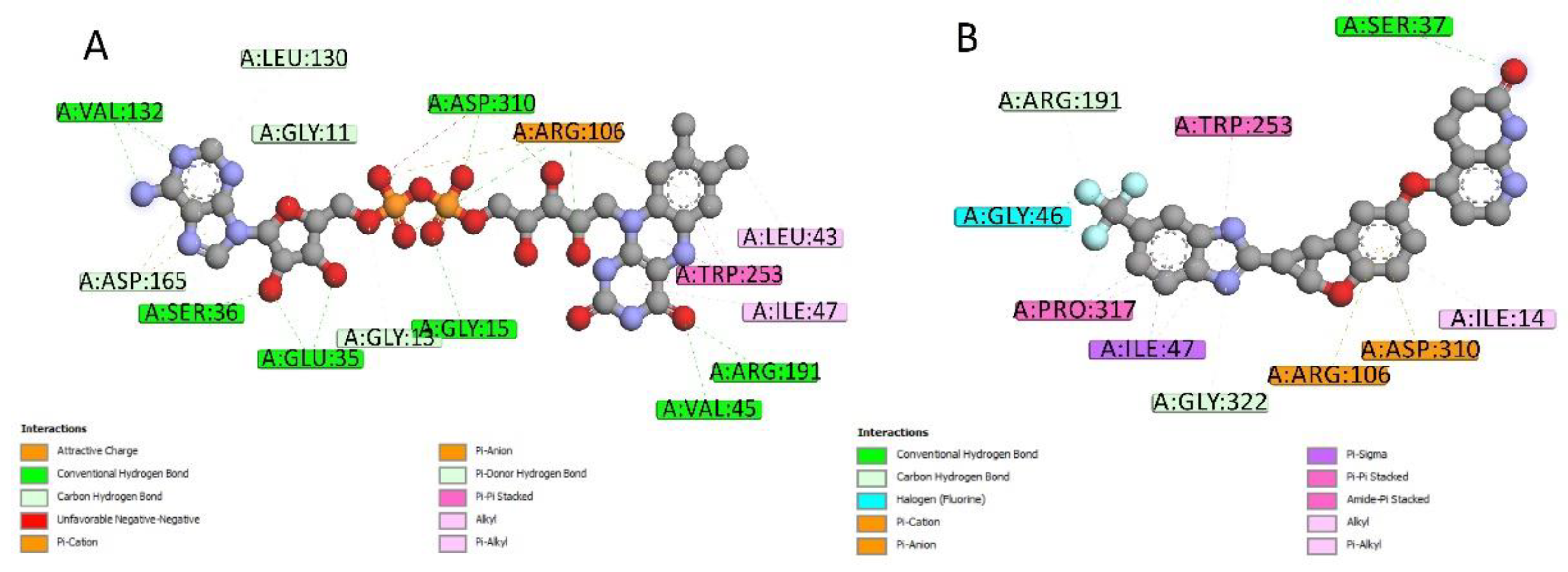
2.4. Docking Conformations and 2D Ligand–Protein Interactions
3. Discussion
4. Materials and Methods
4.1. Drug Library
4.2. Autodock Vina Screening
4.3. Linear Regression
4.4. Normalized Docking Score
4.5. Consensus Ranking of Small Molecules
4.6. 2D Interactions between Small Molecules and Proteins
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance. 2014. Available online: https://apps.who.int/iris/bitstream/handle/10665/112642/9789241564748_eng.pdf (accessed on 17 July 2019).
- World Health Organization. Race against time to develop new antibiotics. Bull. World Health Organ. 2011, 89, 88–89. Available online: https://www.scielosp.org/article/bwho/2011.v89n2/88-89/en/ (accessed on 17 July 2019). [CrossRef] [PubMed]
- World Health Organization. Antimicrobial resistance. Fact Sheet. 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 17 July 2019).
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, J.A.; Brody, S.L.; Kollef, M.H. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 2007, 67, 351–368. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, S.E.; Faught, K.A.; Vethamuthu, J.; Lawson, M.L. Beneficial effects of raloxifene and tamoxifen in the treatment of pubertal gynecomastia. J. Pediatr. 2004, 145, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Ho Sui, S.J.; Lo, R.; Fernandes, A.R.; Caulfield, M.D.; Lerman, J.A.; Xie, L.; Bourne, P.E.; Baillie, D.L.; Brinkman, F.S. Raloxifene attenuates Pseudomonas aeruginosa pyocyanin production and virulence. Int. J. Antimicrob. Agents 2012, 40, 246–251. [Google Scholar] [CrossRef]
- Mavrodi, D.V.; Bonsall, R.F.; Delaney, S.M.; Soule, M.J.; Phillips, G.; Thomashow, L.S. Functional analysis of genes for biosynthesis of pyocyanin and phenazine-1-carboxamide from Pseudomonas aeruginosa PAO1. J. Bacteriol. 2001, 183, 6454–6465. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G.R.; Lehar, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef]
- Xie, L.; Li, J.; Xie, L.; Bourne, P.E. Drug discovery using chemical systems biology: Identification of the protein-ligand binding network to explain the side effects of CETP inhibitors. PLoS Comput. Biol. 2009, 5, e1000387. [Google Scholar] [CrossRef]
- Reiling, S.; Kelleher, A.; Matsumoto, M.M.; Robinson, G.; Asojo, O.A. Structure of type II dehydroquinase from Pseudomonas aeruginosa. Acta Cryst. Sect. F Struct. Biol. Commun. 2014, 70, 1485–1491. [Google Scholar] [CrossRef]
- Tang, Z.; Yuan, X.; Du, R.; Cheung, S.H.; Zhang, G.; Wei, J.; Zhao, Y.; Feng, Y.; Peng, H.; Zhang, Y.; et al. BGB-283, a Novel RAF Kinase and EGFR Inhibitor, Displays Potent Antitumor Activity in BRAF-Mutated Colorectal Cancers. Mol. Cancer 2015, 14, 2187–2197. [Google Scholar] [CrossRef]
- BIOvIA, D.S. Discovery Stuio Visualizer Client. San Diego, Dassault Systemes. 2019. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/ (accessed on 17 July 2019).
- Zuo, M.; Li, Y.; Wang, H.; Zhou, J.; Li, H.; Liu, H.; Xin, H.; Zhang, S.; Chen, X. The antitumor activity of meisoindigo against human colorectal cancer HT-29 cells in vitro and in vivo. J. Chemother. 2008, 20, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Imperi, F.; Leoni, L.; Visca, P. Antivirulence activity of azithromycin in Pseudomonas aeruginosa. Front. Microbiol. 2014, 5, 178. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.; Vira, D.; Medikonda, R.; Kumar, N. Extensively and pan-drug resistant Pseudomonas aeruginosa keratitis: Clinical features, risk factors, and outcome. Graefes. Arch. Clin. Exp. Ophthalmol. 2016, 254, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Aka, S.T.; Haji, S.H. Sub-MIC of antibiotics induced biofilm formation of Pseudomonas aeruginosa in the presence of chlorhexidine. Braz. J. Microbiol. 2015, 46, 149–154. [Google Scholar] [CrossRef] [PubMed]
- El-Mowafy, S.A.; Abd El Galil, K.H.; El-Messery, S.M.; Shaaban, M.I. Aspirin is an efficient inhibitor of quorum sensing, virulence and toxins in Pseudomonas aeruginosa. Microb. Pathog. 2014, 74, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Starkey, M.; Lepine, F.; Maura, D.; Bandyopadhaya, A.; Lesic, B.; He, J.; Kitao, T.; Righi, V.; Milot, S.; Tzika, A.; et al. Identification of anti-virulence compounds that disrupt quorum-sensing regulated acute and persistent pathogenicity. PLoS Pathog. 2014, 10, e1004321. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.X.; Xu, Z.H.; Zhang, Y.Q.; Tian, J.; Weng, L.X.; Wang, L.H. A new quorum-sensing inhibitor attenuates virulence and decreases antibiotic resistance in Pseudomonas aeruginosa. J. Microbiol. 2012, 50, 987–993. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, C.T.; Miller, L.C.; Siryaporn, A.; Drescher, K.; Semmelhack, M.F.; Bassler, B.L. A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc. Natl. Acad. Sci. USA 2013, 110, 17981–17986. [Google Scholar] [CrossRef]
- Jakobsen, T.H.; Bragason, S.K.; Phipps, R.K.; Christensen, L.D.; van Gennip, M.; Alhede, M.; Skindersoe, M.; Larsen, T.O.; Hoiby, N.; Bjarnsholt, T.; et al. Food as a source for quorum sensing inhibitors: Iberin from horseradish revealed as a quorum sensing inhibitor of Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2012, 78, 2410–2421. [Google Scholar] [CrossRef]
- Smyth, A.R.; Cifelli, P.M.; Ortori, C.A.; Righetti, K.; Lewis, S.; Erskine, P.; Holland, E.D.; Givskov, M.; Williams, P.; Camara, M.; et al. Garlic as an inhibitor of Pseudomonas aeruginosa quorum sensing in cystic fibrosis—A pilot randomized controlled trial. Pediatr. Pulmonol. 2010, 45, 356–362. [Google Scholar]
- Kadirvel, M.; Fanimarvasti, F.; Forbes, S.; McBain, A.; Gardiner, J.M.; Brown, G.D.; Freeman, S. Inhibition of quorum sensing and biofilm formation in Vibrio harveyi by 4-fluoro-DPD; a novel potent inhibitor of signalling. Chem. Commun. 2014, 50, 5000–5002. [Google Scholar] [CrossRef] [PubMed]
- Cevizci, R.; Duzlu, M.; Dundar, Y.; Noyanalpan, N.; Sultan, N.; Tutar, H.; Bayazit, Y.A. Preliminary results of a novel quorum sensing inhibitor against pneumococcal infection and biofilm formation with special interest to otitis media and cochlear implantation. Eur. Arch. Otorhinolaryngol. 2015, 272, 1389–1393. [Google Scholar] [CrossRef] [PubMed]
- Ilangovan, A.; Fletcher, M.; Rampioni, G.; Pustelny, C.; Rumbaugh, K.; Heeb, S.; Camara, M.; Truman, A.; Chhabra, S.R.; Emsley, J.; et al. Structural basis for native agonist and synthetic inhibitor recognition by the Pseudomonas aeruginosa quorum sensing regulator PqsR (MvfR). PLoS Pathog. 2013, 9, e1003508. [Google Scholar] [CrossRef] [PubMed]
- Majik, M.S.; Naik, D.; Bhat, C.; Tilve, S.; Tilvi, S.; D’Souza, L. Synthesis of (R)-norbgugaine and its potential as quorum sensing inhibitor against Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett. 2013, 23, 2353–2356. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MCE ID | # of Heavy Atoms | Drug Name | Ranking of Z-Score | Bioactivities | ||
|---|---|---|---|---|---|---|
| aroQ | phzG | phzS | ||||
| HY-18957 | 35 | Lifirafenib | 1 | 9 | 39 | Raf Kinase and EGFR inhibitor |
| HY-13860 | 21 | Meisoindigo | 43 | 24 | 14 | derivative of Indigo naturalis |
| HY-111050 | 28 | JNJ-38877618 | 49 | 49 | 17 | Met kinase inhibitor |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, L.; Xie, L. Pathway-Centric Structure-Based Multi-Target Compound Screening for Anti-Virulence Drug Repurposing. Int. J. Mol. Sci. 2019, 20, 3504. https://doi.org/10.3390/ijms20143504
Xie L, Xie L. Pathway-Centric Structure-Based Multi-Target Compound Screening for Anti-Virulence Drug Repurposing. International Journal of Molecular Sciences. 2019; 20(14):3504. https://doi.org/10.3390/ijms20143504
Chicago/Turabian StyleXie, Li, and Lei Xie. 2019. "Pathway-Centric Structure-Based Multi-Target Compound Screening for Anti-Virulence Drug Repurposing" International Journal of Molecular Sciences 20, no. 14: 3504. https://doi.org/10.3390/ijms20143504
APA StyleXie, L., & Xie, L. (2019). Pathway-Centric Structure-Based Multi-Target Compound Screening for Anti-Virulence Drug Repurposing. International Journal of Molecular Sciences, 20(14), 3504. https://doi.org/10.3390/ijms20143504