Role of B-Cell Translocation Gene 1 in the Pathogenesis of Endometriosis

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Clinical Characteristics
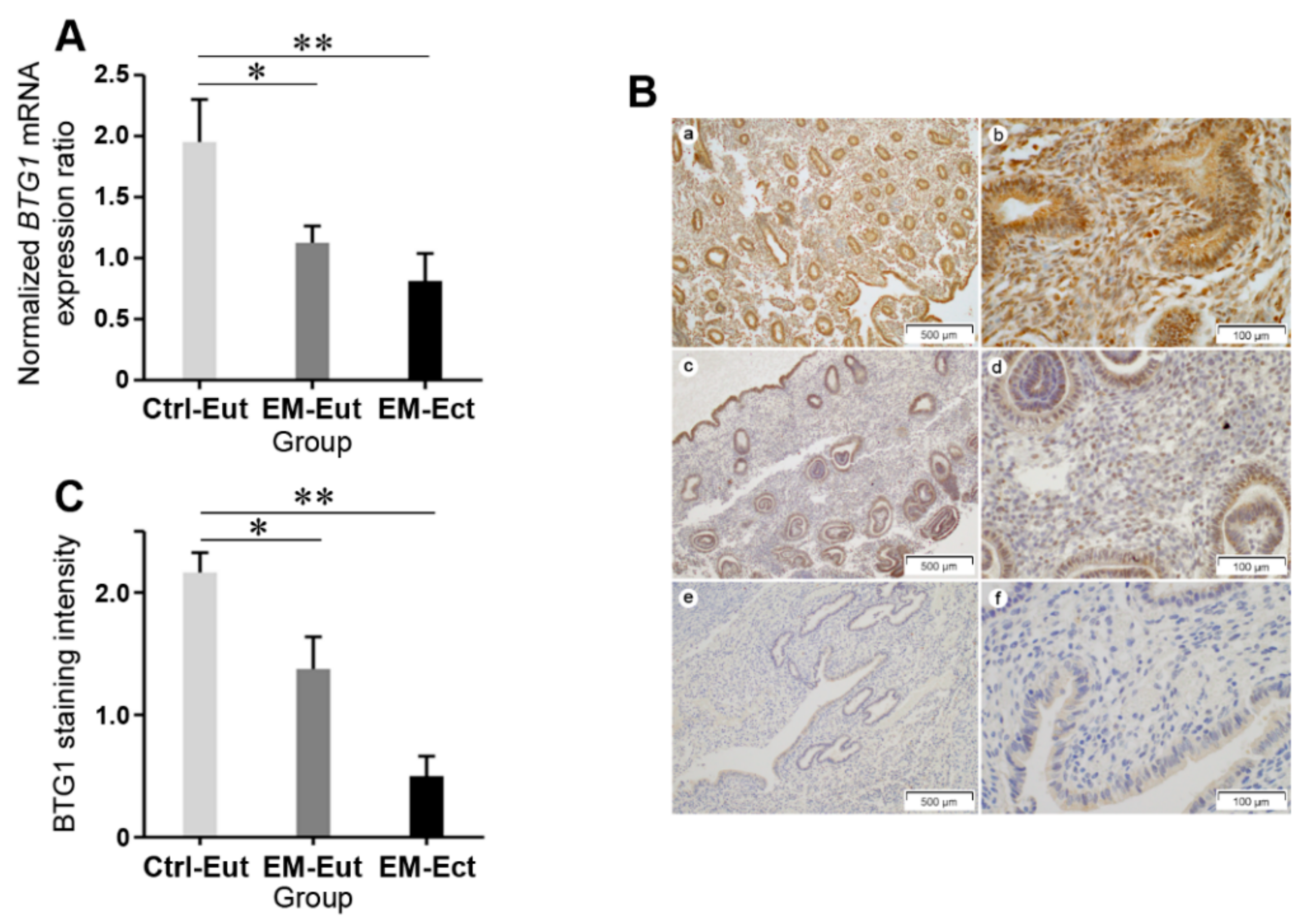
2.2. Expression of BTG1 mRNA and Protein in Eutopic and Ectopic Endometrium of Patients with and without Endometriosis
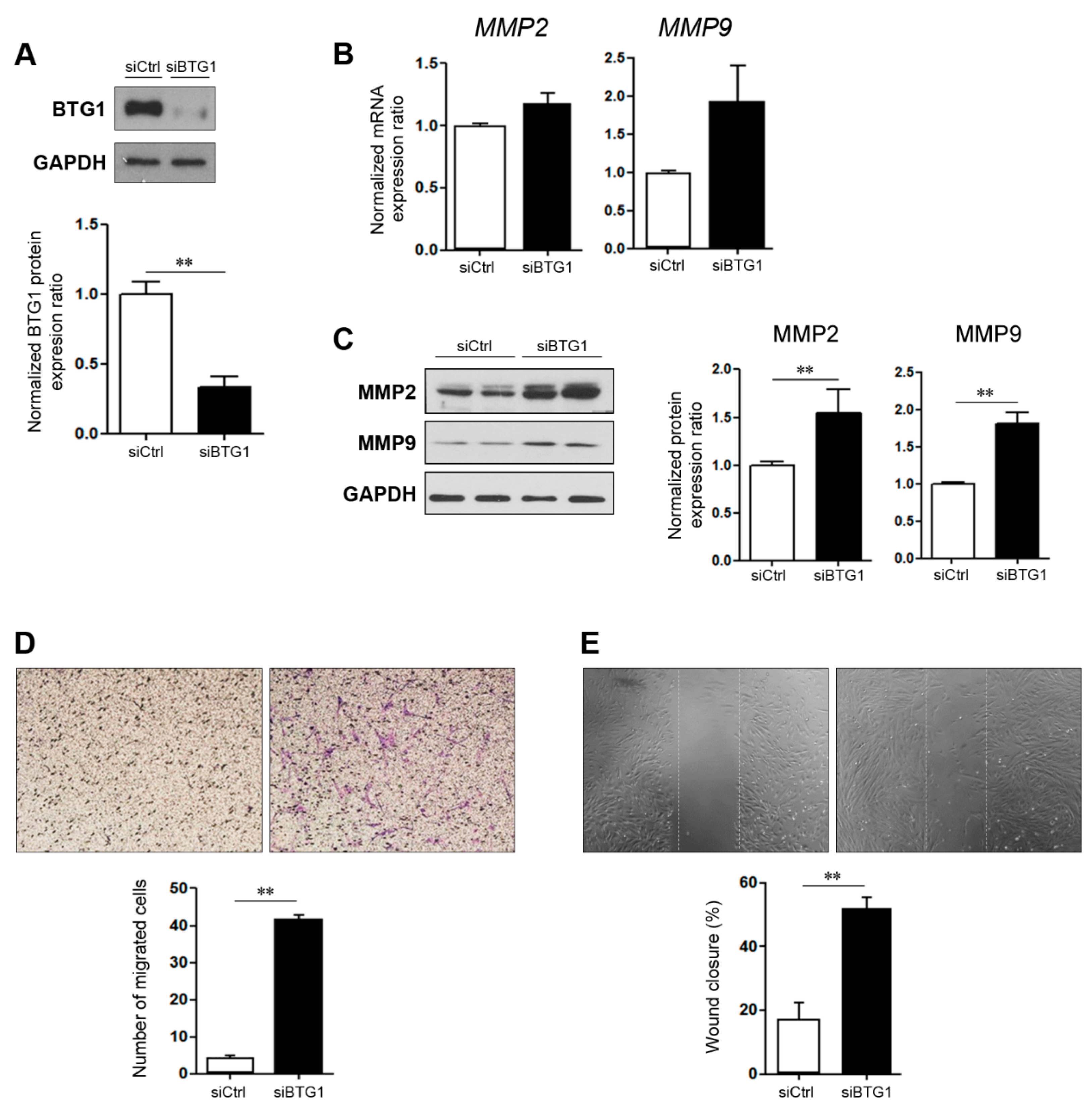
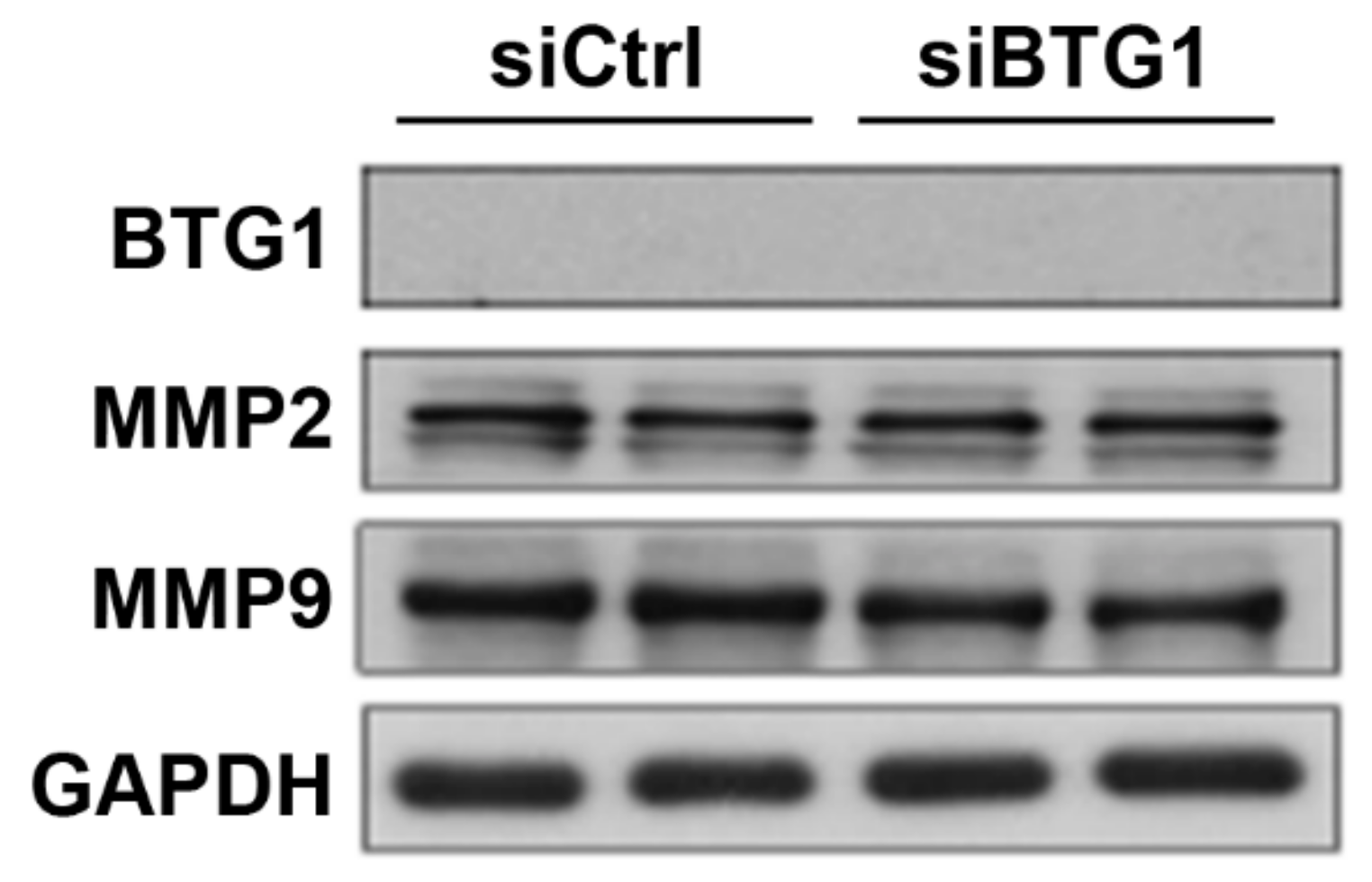
2.3. Effect of BTG1 Downregulation on Migration Potential of HESCs
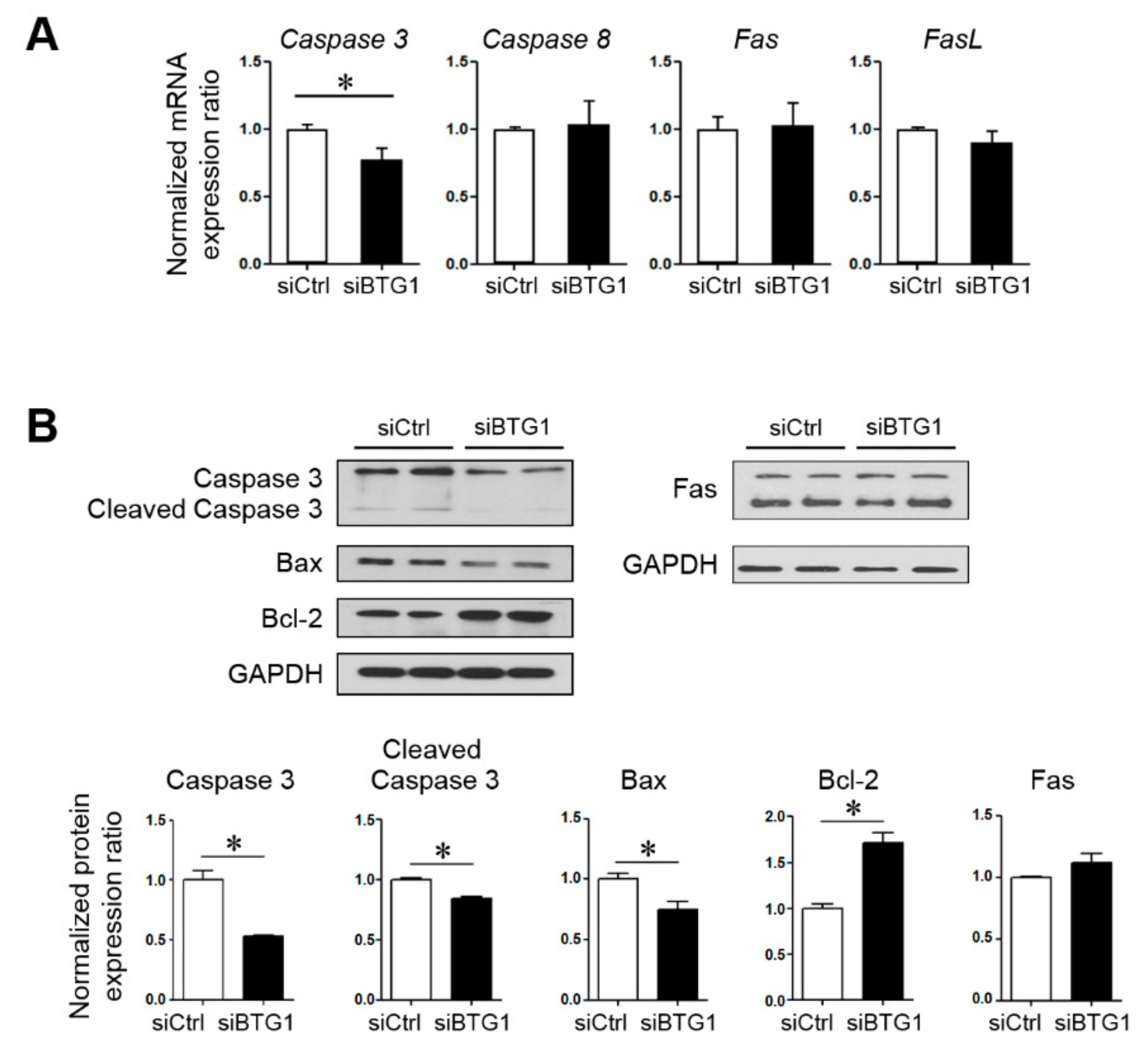
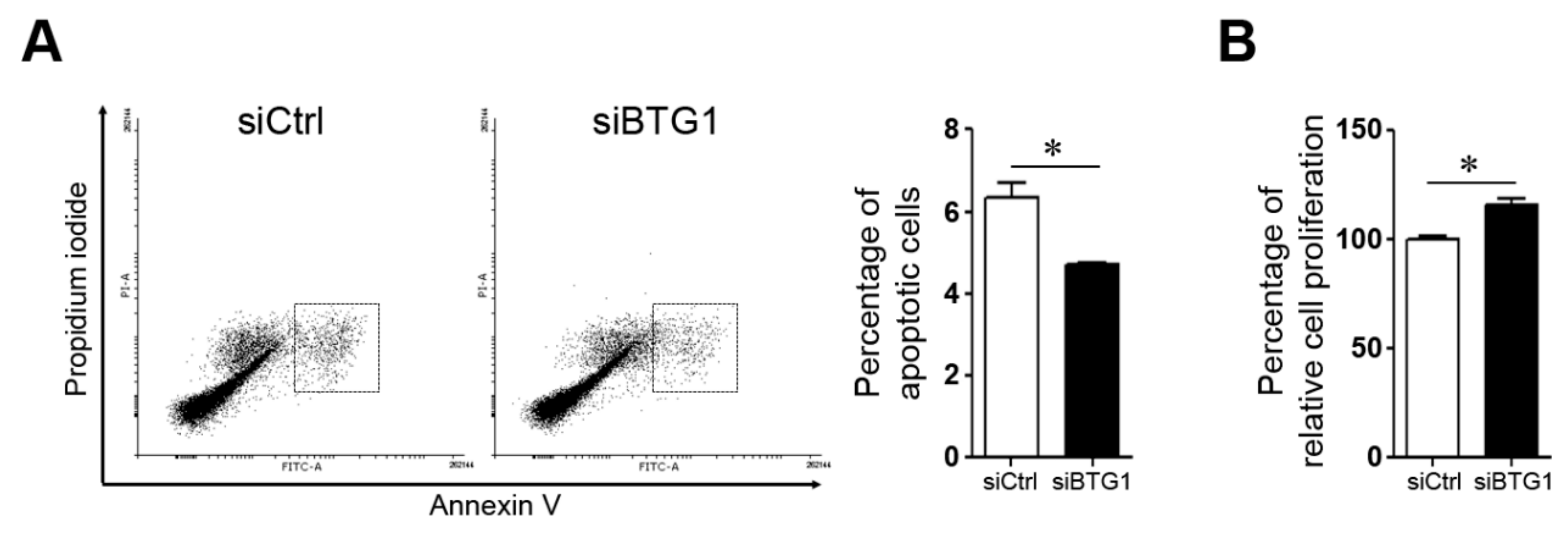
2.4. Effect of BTG1 Downregulation on Apoptotic Potential and Proliferative Activity of HESCs
3. Discussion
4. Materials and Methods
4.1. Patients and Tissue Samples
4.2. Culture of Primary Endometrial Stromal Cells and Ishikawa Cell Line
4.3. Cell Transfection
4.4. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.5. Immunohistochemistry
4.6. Protein Extraction and Western Blot Analysis
4.7. Migration and Wound Healing Assay
4.8. Flow Cytometry
4.9. MTT Assay
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zegers-Hochschild, F.; Adamson, G.D.; Dyer, S.; Racowsky, C.; de Mouzon, J.; Sokol, R.; Rienzi, L.; Sunde, A.; Schmidt, L.; Cooke, I.D.; et al. The International Glossary on Infertility and Fertility Care, 2017. Hum. Reprod. 2017, 32, 1786–1801. [Google Scholar] [CrossRef] [PubMed]
- Giudice, L.C.; Kao, L.C. Endometriosis. Lancet 2004, 364, 1789–1799. [Google Scholar] [CrossRef]
- Agathocleous, M.; Harris, W.A. Metabolism in physiological cell proliferation and differentiation. Trends Cell Biol. 2013, 23, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Shibata, D.; Aaltonen, L.A. Genetic predisposition and somatic diversification in tumor development and progression. Adv. Cancer Res. 2001, 80, 83–114. [Google Scholar] [PubMed]
- Bulun, S.E. Endometriosis. N. Engl. J. Med. 2009, 360, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Huhtinen, K.; Desai, R.; Stahle, M.; Salminen, A.; Handelsman, D.J.; Perheentupa, A.; Poutanen, M. Endometrial and endometriotic concentrations of estrone and estradiol are determined by local metabolism rather than circulating levels. J. Clin. Endocrinol. Metab. 2012, 97, 4228–4235. [Google Scholar] [CrossRef] [PubMed]
- Vinci, G.; Arkwright, S.; Audebourg, A.; Radenen, B.; Chapron, C.; Borghese, B.; Dousset, B.; Mehats, C.; Vaiman, D.; Vacher-Lavenu, M.C.; et al. Correlation Between the Clinical Parameters and Tissue Phenotype in Patients Affected by Deep-Infiltrating Endometriosis. Reprod. Sci. 2016, 23, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Waanders, E.; Scheijen, B.; van der Meer, L.T.; van Reijmersdal, S.V.; van Emst, L.; Kroeze, Y.; Sonneveld, E.; Hoogerbrugge, P.M.; van Kessel, A.G.; van Leeuwen, F.N.; et al. The origin and nature of tightly clustered BTG1 deletions in precursor B-cell acute lymphoblastic leukemia support a model of multiclonal evolution. PLoS Genet. 2012, 8, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Berthet, C.; Guehenneux, F.; Revol, V.; Samarut, C.; Lukaszewicz, A.; Dehay, C.; Dumontet, C.; Magaud, J.P.; Rouault, J.P. Interaction of PRMT1 with BTG/TOB proteins in cell signalling: Molecular analysis and functional aspects. Genes Cells Devoted Mol. Cell. Mech. 2002, 7, 29–39. [Google Scholar] [CrossRef]
- Doidge, R.; Mittal, S.; Aslam, A.; Winkler, G.S. The anti-proliferative activity of BTG/TOB proteins is mediated via the Caf1a (CNOT7) and Caf1b (CNOT8) deadenylase subunits of the Ccr4-not complex. PLoS ONE 2012, 7, e51331. [Google Scholar] [CrossRef]
- Rouault, J.P.; Rimokh, R.; Tessa, C.; Paranhos, G.; Ffrench, M.; Duret, L.; Garoccio, M.; Germain, D.; Samarut, J.; Magaud, J.P. BTG1, a member of a new family of antiproliferative genes. Embo J. 1992, 11, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Corjay, M.H.; Kearney, M.A.; Munzer, D.A.; Diamond, S.M.; Stoltenborg, J.K. Antiproliferative gene BTG1 is highly expressed in apoptotic cells in macrophage-rich areas of advanced lesions in Watanabe heritable hyperlipidemic rabbit and human. Lab. Investig. A J. Tech. Methods Pathol. 1998, 78, 847–858. [Google Scholar]
- Lee, H.; Cha, S.; Lee, M.S.; Cho, G.J.; Choi, W.S.; Suk, K. Role of antiproliferative B cell translocation gene-1 as an apoptotic sensitizer in activation-induced cell death of brain microglia. J. Immunol. 2003, 171, 5802–5811. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.G.; Wang, Y.D.; Cheng, Y.J.; Hu, W.N. BTG1 underexpression is an independent prognostic marker in esophageal squamous cell carcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 9707–9716. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.G.; Lu, Y.F.; Cheng, Y.J.; Hu, W.N. The expression of BTG1 is downregulated in NSCLC and possibly associated with tumor metastasis. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 2949–2957. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.G.; Wang, Y.D.; Cheng, Y.J.; Hu, W.N. The expression of BTG1 is downregulated in nasopharyngeal carcinoma and possibly associated with tumour metastasis. Mol. Biol. Rep. 2014, 41, 5979–5988. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Tao, T.; Xu, B.; Lu, K.; Zhang, L.; Jiang, L.; Chen, S.; Liu, D.; Zhang, X.; Cao, N.; et al. BTG1 potentiates apoptosis and suppresses proliferation in renal cell carcinoma by interacting with PRMT1. Oncol. Lett. 2015, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Zou, S.T.; Wan, J.M.; Li, W.; Li, X.L.; Zhu, W. BTG1 inhibits breast cancer cell growth through induction of cell cycle arrest and apoptosis. Oncol. Rep. 2013, 30, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Do, S.I.; Bae, G.E.; Kim, H.S. B-cell translocation gene 1 is downregulated by promoter methylation in ovarian carcinoma. J. Cancer 2017, 8, 2669–2675. [Google Scholar] [CrossRef] [PubMed]
- Aznavoorian, S.; Murphy, A.N.; Stetler-Stevenson, W.G.; Liotta, L.A. Molecular aspects of tumor cell invasion and metastasis. Cancer 1993, 71, 1368–1383. [Google Scholar] [CrossRef]
- Di Nezza, L.A.; Misajon, A.; Zhang, J.; Jobling, T.; Quinn, M.A.; Ostor, A.G.; Nie, G.; Lopata, A.; Salamonsen, L.A. Presence of active gelatinases in endometrial carcinoma and correlation of matrix metalloproteinase expression with increasing tumor grade and invasion. Cancer 2002, 94, 1466–1475. [Google Scholar] [CrossRef]
- Shaco-Levy, R.; Sharabi, S.; Benharroch, D.; Piura, B.; Sion-Vardy, N. Matrix metalloproteinases 2 and 9, E-cadherin, and beta-catenin expression in endometriosis, low-grade endometrial carcinoma and non-neoplastic eutopic endometrium. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 139, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Talbi, S.; Hamilton, A.E.; Vo, K.C.; Tulac, S.; Overgaard, M.T.; Dosiou, C.; Le Shay, N.; Nezhat, C.N.; Kempson, R.; Lessey, B.A.; et al. Molecular phenotyping of human endometrium distinguishes menstrual cycle phases and underlying biological processes in normo-ovulatory women. Endocrinology 2006, 147, 1097–1121. [Google Scholar] [CrossRef] [PubMed]
- Weigel, M.T.; Kramer, J.; Schem, C.; Wenners, A.; Alkatout, I.; Jonat, W.; Maass, N.; Mundhenke, C. Differential expression of MMP-2, MMP-9 and PCNA in endometriosis and endometrial carcinoma. Eur. J. Obstet. Gynecol. Reprod. Biol. 2012, 160, 74–78. [Google Scholar] [CrossRef]
- Liu, H.; Wang, J.; Wang, H.; Tang, N.; Li, Y.; Zhang, Y.; Hao, T. Correlation between matrix metalloproteinase-9 and endometriosis. Int. J. Clin. Exp. Pathol. 2015, 8, 13399–13404. [Google Scholar]
- Collette, T.; Maheux, R.; Mailloux, J.; Akoum, A. Increased expression of matrix metalloproteinase-9 in the eutopic endometrial tissue of women with endometriosis. Hum. Reprod. 2006, 21, 3059–3067. [Google Scholar] [CrossRef]
- Emonard, H.; Grimaud, J.A. Matrix metalloproteinases. A review. Cell. Mol. Biol. 1990, 36, 131–153. [Google Scholar] [PubMed]
- Nahta, R.; Esteva, F.J. Bcl-2 antisense oligonucleotides: A potential novel strategy for the treatment of breast cancer. Semin. Oncol. 2003, 30, 143–149. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Witzig, T.E.; Adjei, A.A. Targeting apoptosis pathways in cancer therapy. CA A Cancer J. Clin. 2005, 55, 178–194. [Google Scholar] [CrossRef]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326 Pt 1, 1–16. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Kymionis, G.D.; Dimitrakakis, C.E.; Konstadoulakis, M.M.; Arzimanoglou, I.; Leandros, E.; Chalkiadakis, G.; Keramopoulos, A.; Michalas, S. Can expression of apoptosis genes, bcl-2 and bax, predict survival and responsiveness to chemotherapy in node-negative breast cancer patients? J. Surg. Res. 2001, 99, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Canis, M.; Donnez, J.G.; Guzick, D.S.; Halme, J.K.; Rock, J.A.; Schenken, R.S.; Vernon, M.W. Revised American Society for Reproductive Medicine classification of endometriosis: 1996. Fertil. Steril. 1997, 67, 817–821. [Google Scholar]
- Bourdel, N.; Alves, J.; Pickering, G.; Ramilo, I.; Roman, H.; Canis, M. Systematic review of endometriosis pain assessment: How to choose a scale? Hum. Reprod. Update 2015, 21, 136–152. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Mutlu, L.; Zhou, Y.; Taylor, H.S. Aromatase inhibitor regulates let-7 expression and let-7f-induced cell migration in endometrial cells from women with endometriosis. Fertil. Steril. 2016, 106, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Ahn, Y.S.; Choi, Y.S.; Seo, S.K.; Nam, A.; Kim, H.Y.; Kim, J.H.; Park, K.H.; Cho, D.J.; Lee, B.S. Endometrial osteopontin mRNA expression and plasma osteopontin levels are increased in patients with endometriosis. Am. J. Reprod. Immunol. 2009, 61, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.; Carver, J.G.; Ridley, A.J.; Mardon, H.J. Implantation of the human embryo requires Rac1-dependent endometrial stromal cell migration. Proc. Natl. Acad. Sci. USA 2008, 105, 16189–16194. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Endometriosis Group | Control Group | p-Value |
|---|---|---|---|
| Number of patients | 30 | 22 | |
| Age (years) | 34.93 ± 1.33 | 37.41 ± 1.78 | 0.349 |
| Gravidity (frequency) | 0.90 ± 0.24 | 1.91 ± 0.38 | 0.034 * |
| Parity (frequency) | 0.47 ± 0.16 | 1.23 ± 0.22 | 0.006 * |
| Body mass index (kg/m2) | 20.67 ± 0.34 | 21.76 ± 0.53 | 0.092 |
| Cancer antigen 125 (U/mL) | 84.06 ± 25.83 | 18.60 ± 3.17 | 0.002 * |
| Visual analogue scale | 6.57 ± 0.84 | 1.55 ± 0.98 | <0.001 * |
| Phase of Endometrium | Group | p-Value | |
|---|---|---|---|
| Endometriosis | Control | ||
| Proliferative phase | 1.23 ± 0.21 (n = 14) | 1.75 ± 0.50 (n = 8) | 0.570 |
| Secretory phase | 1.03 ± 0.20 (n = 15) | 1.88 ± 0.60 (n = 10) | 0.091 |
| Gene | Sequence | |
|---|---|---|
| BTG1 | Forward | CAA GGG ATC GGG TTA CCG TTG T |
| Reverse | AGC CAT CCT CTC CAA TTC TGT AGG | |
| Caspase 3 | Forward | GGA AGC GAA TCA ATG GAC TCT GG |
| Reverse | GCA TCG ACA TCT GTA CCA GAC C | |
| Caspase 8 | Forward | CCA GAG ACT CCA GGA AAA GAG A |
| Reverse | GAT AGA GCA TGA CCC TGT AGG C | |
| Fas | Forward | AGC TTG GTC TAG AGT GAA AA |
| Reverse | GAG GCA GAA TCA TGA GAT AT | |
| FasL | Forward | CAG CTC TTC CAC CTG CAG AAG G |
| Reverse | AGA TTC CTC AAA ATT GAT CAG AGA GAG | |
| MMP2 | Forward | ACC GCG ACA AGA AGT ATG GC |
| Reverse | CCA CTT GCG GTC ATC ATC GT | |
| MMP9 | Forward | CGA TGA CGA GTT GTG GTC CC |
| Reverse | TCG TAG TTG GCC GTG GTA CT | |
| GAPDH | Forward | ACC ACA GTC CAT GCC ATC AC |
| Reverse | TCC ACC ACC CTG TTG CTG TA | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.S.; Choi, Y.S.; Park, J.H.; Yun, J.; Kim, S.; Lee, J.H.; Yun, B.H.; Park, J.H.; Seo, S.K.; Cho, S.; et al. Role of B-Cell Translocation Gene 1 in the Pathogenesis of Endometriosis. Int. J. Mol. Sci. 2019, 20, 3372. https://doi.org/10.3390/ijms20133372
Kim JS, Choi YS, Park JH, Yun J, Kim S, Lee JH, Yun BH, Park JH, Seo SK, Cho S, et al. Role of B-Cell Translocation Gene 1 in the Pathogenesis of Endometriosis. International Journal of Molecular Sciences. 2019; 20(13):3372. https://doi.org/10.3390/ijms20133372
Chicago/Turabian StyleKim, Jeong Sook, Young Sik Choi, Ji Hyun Park, Jisun Yun, Soohyun Kim, Jae Hoon Lee, Bo Hyon Yun, Joo Hyun Park, Seok Kyo Seo, SiHyun Cho, and et al. 2019. "Role of B-Cell Translocation Gene 1 in the Pathogenesis of Endometriosis" International Journal of Molecular Sciences 20, no. 13: 3372. https://doi.org/10.3390/ijms20133372
APA StyleKim, J. S., Choi, Y. S., Park, J. H., Yun, J., Kim, S., Lee, J. H., Yun, B. H., Park, J. H., Seo, S. K., Cho, S., Kim, H.-S., & Lee, B. S. (2019). Role of B-Cell Translocation Gene 1 in the Pathogenesis of Endometriosis. International Journal of Molecular Sciences, 20(13), 3372. https://doi.org/10.3390/ijms20133372