Inhibition of Sodium Glucose Cotransporters Improves Cardiac Performance

 and
and 
Abstract
1. Introduction
2. Cardiac Metabolism and Substrate Bioenergetics
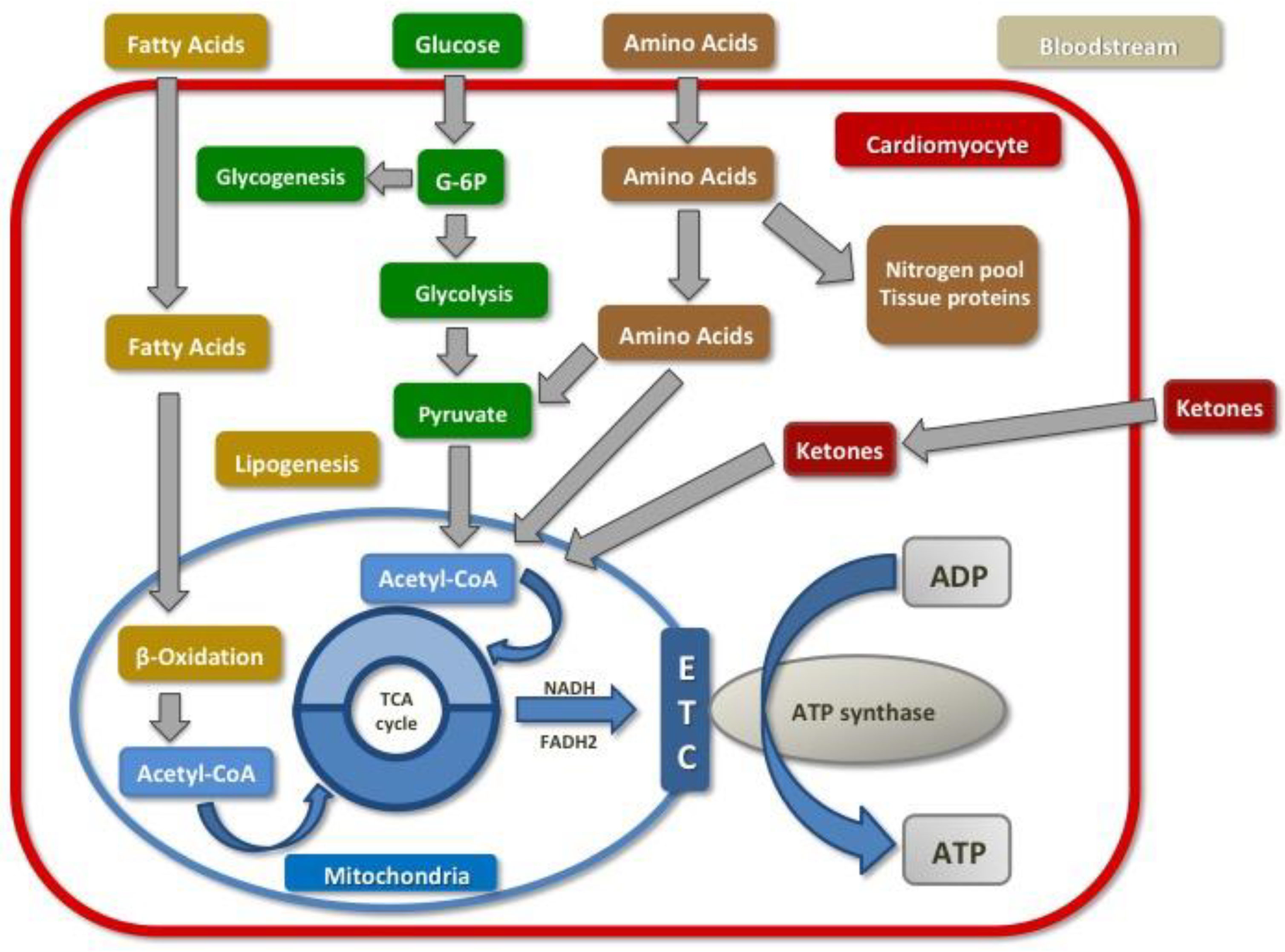
2.1. Normal Cardiac Metabolism
2.2. The Failing Heart and Diabetic Cardiomyopathy: Metabolic Alterations
2.3. Myocardial Schemia: Metabolic Alterations
3. The Role of SGLT1 Receptor in Ischemic Cardiomyopathy
3.1. SGLT-1 Receptors Overview
3.2. Impact of SGLT1 Inhibition on Ischemic Heart Disease
4. Inhibition of SGLT2 Receptors
4.1. SGLT2 Receptor Overview
4.2. Impact of SGLT2 Inhibition on Heart Failure and Diabetic Cardiomyopathy
4.3. SGLT2 Inhibitors and Clinical Outcomes
5. Conclusions
Funding
Conflicts of Interest
References
- Beer, M.; Seyfarth, T.; Sandstede, J.; Landschütz, W.; Lipke, C.; Köstler, H.; Von Kienlin, M.; Harre, K.; Hahn, D.; Neubauer, S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with31P-SLOOP magnetic resonance spectroscopy. J. Am. Coll. Cardiol. 2002, 40, 1267–1274. [Google Scholar] [CrossRef]
- Kolwicz, S.J.; Olson, D.; Marney, L.; Garcia-Menendez, L.; Synovec, R.; Tian, R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ. Res. 2012, 111, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-T.; Grayburn, P.; Karim, A.; Shimabukuro, M.; Higa, M.; Baetens, D.; Orci, L.; Unger, R.H. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 1784–1789. [Google Scholar] [CrossRef] [PubMed]
- Abubakar, I.I.; Tillmann, T.; Banerjee, A. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canvas. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2018, 380, 347–357. [Google Scholar] [CrossRef]
- Flores, E.; Santos-Gallego, C.G.; Diaz-Mejía, N.; Badimon, J.J. Do the SGLT-2 Inhibitors Offer More than Hypoglycemic Activity? Cardiovasc. Drugs Ther. 2018, 32, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Di Franco, A.; Cantini, G.; Tani, A.; Coppini, R.; Zecchi-Orlandini, S.; Raimondi, L.; Luconi, M.; Mannucci, E. Sodium-dependent glucose transporters (SGLT) in human ischemic heart: A new potential pharmacological target. Int. J. Cardiol. 2017, 243, 86–90. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Garcia-Ropero, A.; Mancini, D.; Pinney, S.P.; Contreras, J.P.; Fergus, I.; Abascal, V.; Moreno, P.; Atallah-Lajam, F.; Tamler, R.; et al. Rationale and Design of the EMPA-TROPISM Trial (ATRU-4): Are the “Cardiac Benefits” of Empagliflozin Independent of its Hypoglycemic Activity? Cardiovasc. Drugs Ther. 2019, 33, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Salvadó, L.; Barroso, E.; Vázquez-Carrera, M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int. J. Cardiol. 2013, 168, 3160–3172. [Google Scholar] [CrossRef]
- Wende, A.R.; Brahma, M.K.; McGinnis, G.R.; Young, M.E. Metabolic Origins of Heart Failure. JACC Basic Transl. Sci. 2017, 2, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac Metabolism in Heart Failure. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Noordali, H.; Loudon, B.L.; Frenneaux, M.P.; Madhani, M. Cardiac metabolism—A promising therapeutic target for heart failure. Pharmacol. Ther. 2018, 182, 95–114. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ropero, A.; Santos-Gallego, C.G.; Zafar, M.U.; Badimon, J.J. Metabolism of the failing heart and the impact of SGLT2 inhibitors. Expert Opin. Drug Metab. Toxicol. 2019, 15, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Maack, C.; Lehrke, M.; Backs, J.; Heinzel, F.R.; Hulot, J.-S.; Marx, N.; Paulus, W.J.; Rossignol, P.; Taegtmeyer, H.; Bauersachs, J.; et al. Heart failure and diabetes: Metabolic alterations and therapeutic interventions: A state-of-the-art review from the Translational Research Committee of the Heart Failure Association–European Society of Cardiology. Eur. Heart J. 2018, 39, 4243–4254. [Google Scholar] [CrossRef]
- Nikolajević Starčević, J.; Janić, M.; Šabovič, M. Molecular Mechanisms Responsible for Diastolic Dysfunction in Diabetes Mellitus Patients. Int. J. Mol. Sci. 2019, 20, 1197. [Google Scholar] [CrossRef]
- Pfeffer, M.; Braunwald, E. Ventricular Remodeling After Myocardial Infarction. Circulation 1990, 81, 1161–1172. [Google Scholar] [CrossRef]
- Gajarsa, J.J.; Kloner, R.A. Left ventricular remodeling in the post-infarction heart: A review of cellular, molecular mechanisms, and therapeutic modalities. Heart Fail. Rev. 2011, 16, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y. Myocardial repair/remodelling following infarction: Roles of local factors. Cardiovasc. Res. 2009, 81, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Vahl, T.P.; Goliasch, G.; Picatoste, B.; Arias, T.; Ishikawa, K.; Njerve, I.U.; Sanz, J.; Narula, J.; Sengupta, P.P.; et al. Sphingosine-1-Phosphate Receptor Agonist Fingolimod Increases Myocardial Salvage and Decreases Adverse Postinfarction Left Ventricular Remodeling in a Porcine Model of Ischemia/Reperfusion. Circulation 2016, 133, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Vilahur, G.; Juan-Babot, O.; Peña, E.; Oñate, B.; Casaní, L.; Badimon, L. Molecular and cellular mechanisms involved in cardiac remodeling after acute myocardial infarction. J. Mol. Cell. Cardiol. 2011, 50, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Dyck, J.R.B.; Lopaschuk, G.D. AMPK alterations in cardiac physiology and pathology: Enemy or ally? J. Physiol. 2006, 574, 95–112. [Google Scholar] [CrossRef]
- Goodyear, L.J.; Xing, Y.; Wolf, C.; Bali, D.; Perez-Atayde, A.R.; He, H.; Berul, C.I.; Tian, R.; Stapleton, D.; Ahmad, F.; et al. Increased α2 Subunit–Associated AMPK Activity and PRKAG2 Cardiomyopathy. Circulation 2005, 112, 3140–3148. [Google Scholar]
- Banerjee, S.K.; McGaffin, K.R.; Pastor-Soler, N.M.; Ahmad, F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc. Res. 2009, 84, 111–118. [Google Scholar] [CrossRef]
- Song, P.; Onishi, A.; Koepsell, H.; Vallon, V. Sodium glucose cotransporter SGLT1 as a therapeutic target in diabetes mellitus. Expert Opin. Ther. Targets 2016, 20, 1109–1125. [Google Scholar] [CrossRef]
- Elfeber, K.; Stümpel, F.; Gorboulev, V.; Mattig, S.; Deussen, A.; Kaissling, B.; Koepsell, H. Na+-D-glucose cotransporter in muscle capillaries increases glucose permeability. Biochem. Biophys. Res. Commun. 2004, 314, 301–305. [Google Scholar] [CrossRef]
- Poulsen, S.; Fenton, R.; Rieg, T. Sodium-glucose cotransport. Curr. Opin. Nephrol. Hypertens. 2015, 24, 463–469. [Google Scholar] [CrossRef]
- Young, L.; Coven, D.; Russell, R. Cellular and molecular regulation of cardiac glucose transport. J. Nucl. Cardiol. 2000, 7, 267–276. [Google Scholar] [CrossRef]
- Szablewski, L. Glucose transporters in healthy heart and in cardiac disease. Int. J. Cardiol. 2017, 230, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.K.; Wang, D.W.; Alzamora, R.; Huang, X.N.; Pastor-Soler, N.M.; Hallows, K.R.; McGaffin, K.R.; Ahmad, F. SGLT1, a novel cardiac glucose transporter, mediates increased glucose uptake in PRKAG2 cardiomyopathy. J. Mol. Cell. Cardiol. 2010, 49, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Ramratnam, M.; Sharma, R.K.; D’Auria, S.; Lee, S.J.; Wang, D.; Huang, X.Y.N.; Ahmad, F. Transgenic Knockdown of Cardiac Sodium/Glucose Cotransporter 1 (SGLT1) Attenuates PRKAG2 Cardiomyopathy, Whereas Transgenic Overexpression of Cardiac SGLT1 Causes Pathologic Hypertrophy and Dysfunction in Mice. J. Am. Heart Assoc. 2014, 3, e000899. [Google Scholar] [CrossRef] [PubMed]
- Jakubiak, M.; Agrawal, V.; D’Auria, S.; Li, Z.; Ramratnam, M.; Sincoular, A.; Sharma, R.K.; Music, M.L.; Gifford, L.; Huang, X.N.; et al. Cardiac Sodium-Glucose Co-Transporter 1 (SGLT1) is a Novel Mediator of Ischemia/Reperfusion Injury. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef]
- Garcia-Ropero, A.; Santos-Gallego, C.G.; Badimon, J.J. SGLT receptors and myocardial ischaemia-reperfusion injury: Inhibition of SGLT-1, SGLT-2, or both? Cardiovasc. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Connelly, K.A.; Zhang, Y.; Desjardins, J.-F.; Thai, K.; Gilbert, R.E. Dual inhibition of sodium–glucose linked cotransporters 1 and 2 exacerbates cardiac dysfunction following experimental myocardial infarction. Cardiovasc. Diabetol. 2018, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, Y.; Nagoshi, T.; Yoshino, T.; Tanaka, T.D.; Ito, K.; Harada, T.; Takahashi, H.; Ikegami, M.; Anzawa, R.; Yoshimura, M. Expression of SGLT1 in Human Hearts and Impairment of Cardiac Glucose Uptake by Phlorizin during Ischemia-Reperfusion Injury in Mice. PLoS ONE 2015, 10, e0130605. [Google Scholar] [CrossRef] [PubMed]
- Clar, C.; Gill, J.A.; Court, R.; Waugh, N. Systematic review of SGLT2 receptor inhibitors in dual or triple therapy in type 2 diabetes. BMJ Open 2012, 2, e001007. [Google Scholar] [CrossRef]
- Garcia-Ropero, A.; Badimon, J.J.; Santos-Gallego, C.G. The pharmacokinetics and pharmacodynamics of SGLT2 inhibitors for type 2 diabetes mellitus: The latest developments. Expert Opin. Drug Metab. Toxicol. 2018, 14, 1287–1302. [Google Scholar] [CrossRef]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Browning and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-induced Obese Mice. EBioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G. Editorial: Cardiovascular Disease and Diabetes. Front. Endocrinol. 2019, 10, 314. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Node, K. Exploration of the clinical benefits of sodium glucose co-transporter 2 inhibitors in diabetic patients with concomitant heart failure. Cardiovasc. Diabetol. 2018, 17, 74. [Google Scholar] [CrossRef] [PubMed]
- Greene, S.J.; Butler, J. Primary Prevention of Heart Failure in Patients with Type 2 Diabetes Mellitus. Circulation 2019, 139, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.; Requen Ibañez, J.; San Antonio, R.; Picatoste, B.; Watanabe, S.; Ishikawa, K.; Flores, E.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.; et al. Empagliflozin Ameliorates Adverse LV Remodeling in a Non-Diabetic Model of Heart Failure Mediated via a Switch in Myocardial Metabolism That Enhances Energetics. J. Am. Coll. Cardiol. 2019, 73, 1931–1944. [Google Scholar] [CrossRef] [PubMed]
- Yurista, S.R.; Silljé, H.H.; Oberdorf-Maass, S.U.; Schouten, E.; Pavez Giani, M.G.; Hillebrands, J.; van Goor, H.; van Veldhuisen, D.J.; de Boer, R.A.; Westenbrink, B.D. Sodium–glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur. J. Heart Fail. 2019. [Google Scholar] [CrossRef]
- Baker, H.E.; Kiel, A.M.; Luebbe, S.T.; Simon, B.R.; Earl, C.C.; Regmi, A.; Roell, W.C.; Mather, K.J.; Tune, J.D.; Goodwill, A.G. Inhibition of sodium–glucose cotransporter-2 preserves cardiac function during regional myocardial ischemia independent of alterations in myocardial substrate utilization. Basic Res. Cardiol. 2019, 114, 25. [Google Scholar] [CrossRef]
- Takasu, T.; Takakura, S. Effect of ipragliflozin, an SGLT2 inhibitor, on cardiac histopathological changes in a non-diabetic rat model of cardiomyopathy. Life Sci. 2019, 230, 19–27. [Google Scholar] [CrossRef]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Ibanez, J.A.R.; Antonio, R.S.; Ishikawa, K.; Watanabe, S.; Picatoste Botija, M.B.; Salvo, A.J.S.; Hajjar, R.; Fuster, V.; Badimon, J. Empagliflozin Induces a Myocardial Metabolic Shift from Glucose Consumption to Ketone Metabolism That Mitigates Adverse Cardiac Remodeling and Improves Myocardial Contractility. J. Am. Coll. Cardiol. 2018, 71, A674. [Google Scholar] [CrossRef]
- SantosGallego, C.G.; Requena-Ibanez, J.A.; San Antonio, R.; Ishikawa, K.; Picatoste, B.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.; Fuster, V.; Badimon, J.J. Abstract 17367: Infusion of the Ketone Body β-Hydroxybutyrate Improves Left Ventricular Systolic Function in an Animal Model of Heart Failure with Reduced Ejection Fraction. Circulation 2018, 138, A17367. [Google Scholar]
- Prasad, V.; Lorenz, J.; Miller, M.; Vairamani, K.; Nieman, M.; Wang, Y.; Shull, G. Loss of NHE1 activity leads to reduced oxidative stress in heart and mitigates high-fat diet-induced myocardial stress. J. Mol. Cell. Cardiol. 2013, 65, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Baartscheer, A.; Schumacher, C.A.; Wüst, R.C.; Fiolet, J.W.; Stienen, G.J.; Coronel, R.; Zuurbier, C.J. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 2017, 60, 568. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Chang, N.C.; Lin, S.Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 2017, 104, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Durak, A.; Olgar, Y.; Degirmenci, S.; Akkus, E.; Tuncay, E.; Turan, B. A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc. Diabetol. 2018, 17, 144. [Google Scholar] [CrossRef]
- Kang, S.; Verma, S.; Teng, G.; Belke, D.; Svystonyuk, D.; Guzzardi, D.; Park, D.; Turnbull, J.; Malik, G.; Fedak, P. Direct effect of empagliflozin on extracellular matrix remodeling in human cardiac fibroblasts: Novel translational clues to EMPA-REG OUTCOME. Can. J. Cardiol. 2017, 33, S169. [Google Scholar] [CrossRef]
- Danne, T.; Biester, T.; Kordonouri, O. Combined SGLT1 and SGLT2 Inhibitors and Their Role in Diabetes Care. Diabetes Technol. Ther. 2018, 20, S269–S277. [Google Scholar] [CrossRef]
- ACCORD Study Group; Miller, M.; Genuth, S.; Ismail-Beigi, F.; Buse, J.; Goff, J.; Probstfield, J.; Cushman, W.; Ginsberg, H.; Bigger, J.; et al. Long-term effects of intensive glucose lowering on cardiovascular outcomes. N. Engl. J. Med. 2011, 364, 818–828. [Google Scholar]
- Cherney, D.Z.I.; Perkins, B.A.; Soleymanlou, N.; Har, R.; Fagan, N.; Johansen, O.E.; Woerle, H.J.; von Eynatten, M.; Broedl, U.C. The effect of empagliflozin on arterial stiffness and heart rate variability in subjects with uncomplicated type 1 diabetes mellitus. Cardiovasc. Diabetol. 2014, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J. Empagliflozin, calcium, and SGLT1/2 receptor affinity: Another piece of the puzzle. ESC Heart Fail. 2018, 5, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, J.; Wagemann, O.; Lücht, C.M.; Trum, M.; Hammer, K.P.; Sag, C.M.; Lebek, S.; Tarnowski, D.; Reinders, J.; Perbellini, F.; et al. Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. 2018, 5, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-I.; Chen, Y.-C.; Lin, Y.-K.; Chung, C.-C.; Lu, Y.-Y.; Kao, Y.-H.; Chen, Y.-J. Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and Reverses Cardiac Remodeling in Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 1680. [Google Scholar] [CrossRef] [PubMed]
- Bethel, M.A.; McMurray, J.V. Class Effect for Sodium Glucose-Cotransporter-2 Inhibitors in Cardiovascular Outcomes. Circulation 2018, 137, 1218–1220. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; de Zeeuw, D.; Mahaffey, K.W.; Fulcher, G.; Erondu, N.; Shaw, W.; Barrett, T.D.; Weidner-Wells, M.; Deng, H.; Matthews, D.R.; et al. Canagliflozin and renal outcomes in type 2 diabetes: Results from the CANVAS Program randomised clinical trials. Lancet Diabetes Endocrinol. 2018, 6, 691–704. [Google Scholar] [CrossRef]


{kind=link}
| SGLT2 Inhibitor | Bioavailability | Time-to Peak/Half-Life | Excretion | Initial (Max) Dose | Clinical Trials | CV Outcomes | Renal Outcome | Effects | Main Adverse Effects |
|---|---|---|---|---|---|---|---|---|---|
| Empagliflozin [5] | ~75% | 1.5 h/13 h | 55% Renal 40% Fecal | 10 mg (25 mg) | EMPA-REG OUTCOME 10 or 25 mg for 3.1 years | Reduced CV death (RRR of 38%) Reduced hospitalization for HF (RRR 35%) Reduced death from any cause (RRR 32%) | Reduced progression of kidney disease (RRR 44% of doubled creatinine) | Pancreatic β-cell function improvement Weight loss Natriuretic effect Decreased SBP Decreased Acid uric Small increase in HDL-c and LDL-c | GTI Hypoglycemia Infrequent |
| Canagliflozin [6] | ~65% | 1–2 h/13 h | 41.5% Fecal 33% Renal | 100 mg (300 mg) | CANVAS 100 or 300 mg for 3.6 years | Reduced hospitalization for HF (RRR 33%) Reduced death from any cause (RRR 13%) | Reduced progression of kidney disease (RRR 27%, albuminuria progression) | Weight loss Natriuretic effect Decreased SBP Decreased Acid uric Small increase in HDL-c and LDL-c | GTI Hypoglycemia (more common than dapagliflozin/ empagliflozin) May ↑ lower extremities amputations |
| Dapagliflozin [7] | ~78% | 1–1.5 h/13 h | 75% Renal 21% Fecal | 5 mg (10 mg) | DECLARE-TIMI38 10 mg for 4.2 years DAPA-HF 5 or 10 mg for 3 years | No yet available | May reduce progression of kidney disease (scarce data) | Weight loss Natriuretic effect Decreased SBP Decreased Acid uric Small increase in HDL-c and LDL-c | GTI Hypoglycemia Headache Diarrhea May increase breast and bladder cancer rates |
| Ertugliflozin [37] | ~70–90% | 0.5–1.5 h/11–17 h | 50% Renal 41% Fecal | 5 mg (15 mg) | VERTIS-CV 5 or 15 mg for 6.1 years | No yet available | Not yet available | Weight loss Natriuretic effect Decreased SBP Decreased Acid uric Small increase in HDL-c and LDL-c | GTI Hypoglycemia |
| Sotagliflozin (dual SGLT 1 and 2 inhibitor) [41] | - | 3 h/13.5–20.7 h | Mostly Renal | 200 mg (400 mg) | inTANDEM 400 mg for 24 weeks | Not yet available | Not yet available | Under investigation | GTI Nausea/Diarrhea |
| Characteristic | SGLT1 | SGLT2 |
|---|---|---|
| Capacity | Low | High |
| Affinity | High | Low |
| Function | Dietary absorption glucose and galactose (GIT) Renal reabsorption glucose | Renal reabsorption glucose |
| Renal location | S3 of PCT | S1 and S2 of PCT |
| Renal glucose reabsorption | 10% | 90% |
| Ratio Na-Glucose cotransport | 2:1 | 1:1 |
| Gene encoding | SCL5A1 | SLC5A2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Ropero, Á.; Vargas-Delgado, A.P.; Santos-Gallego, C.G.; Badimon, J.J. Inhibition of Sodium Glucose Cotransporters Improves Cardiac Performance. Int. J. Mol. Sci. 2019, 20, 3289. https://doi.org/10.3390/ijms20133289
García-Ropero Á, Vargas-Delgado AP, Santos-Gallego CG, Badimon JJ. Inhibition of Sodium Glucose Cotransporters Improves Cardiac Performance. International Journal of Molecular Sciences. 2019; 20(13):3289. https://doi.org/10.3390/ijms20133289
Chicago/Turabian StyleGarcía-Ropero, Álvaro, Ariana P. Vargas-Delgado, Carlos G. Santos-Gallego, and Juan J. Badimon. 2019. "Inhibition of Sodium Glucose Cotransporters Improves Cardiac Performance" International Journal of Molecular Sciences 20, no. 13: 3289. https://doi.org/10.3390/ijms20133289
APA StyleGarcía-Ropero, Á., Vargas-Delgado, A. P., Santos-Gallego, C. G., & Badimon, J. J. (2019). Inhibition of Sodium Glucose Cotransporters Improves Cardiac Performance. International Journal of Molecular Sciences, 20(13), 3289. https://doi.org/10.3390/ijms20133289