Renin Activity in Heart Failure with Reduced Systolic Function—New Insights



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
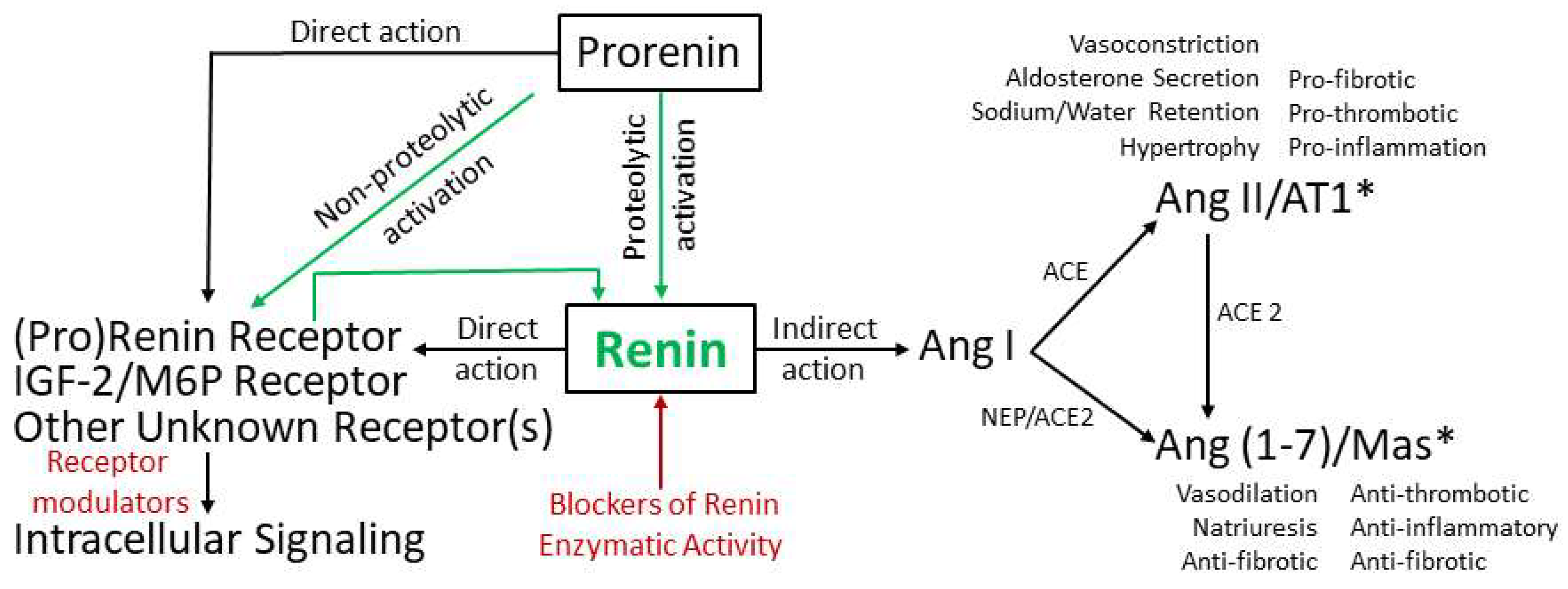
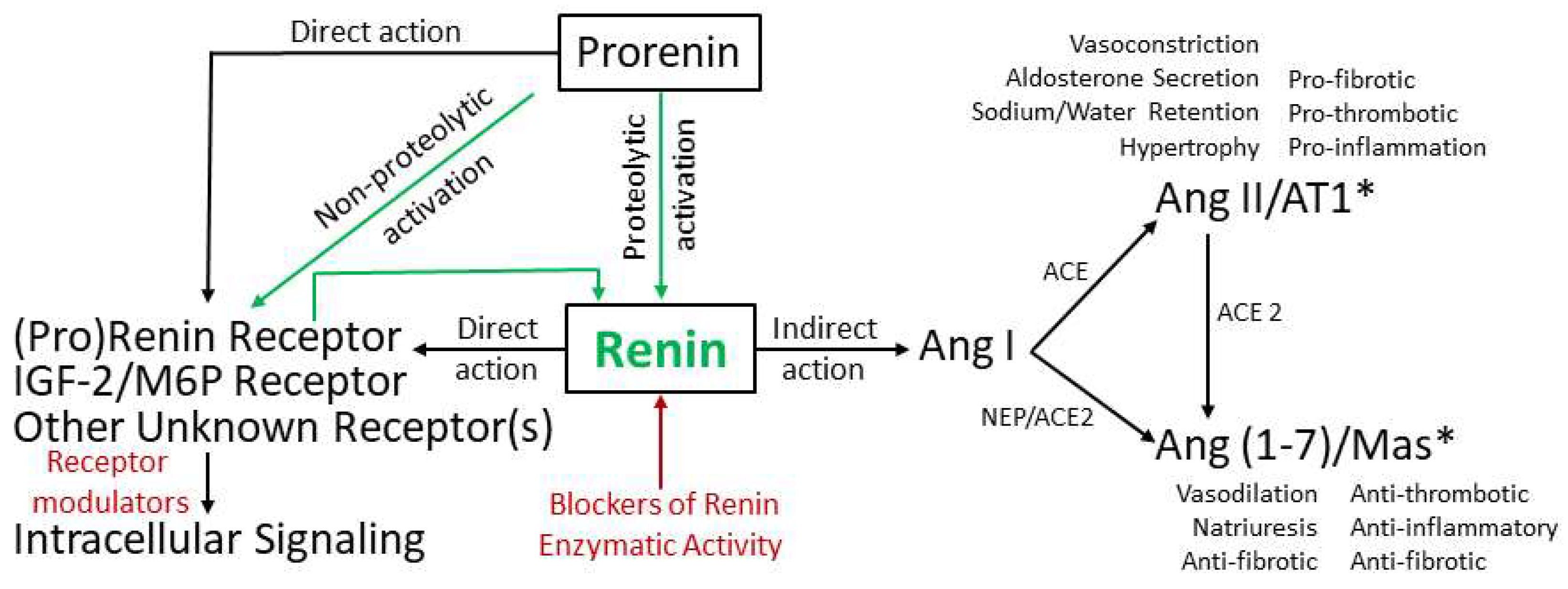
2. Pathways of Renin Generation and Function
3. Renin Activity as a Diagnostic and Prognostic Marker
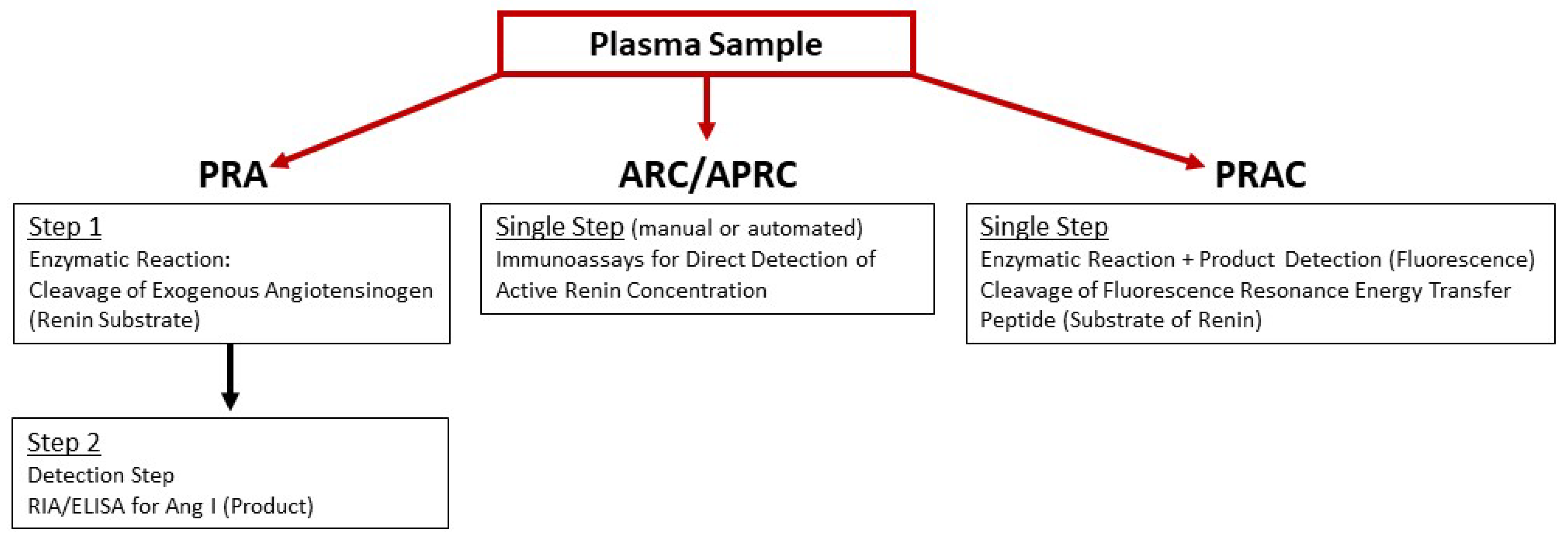
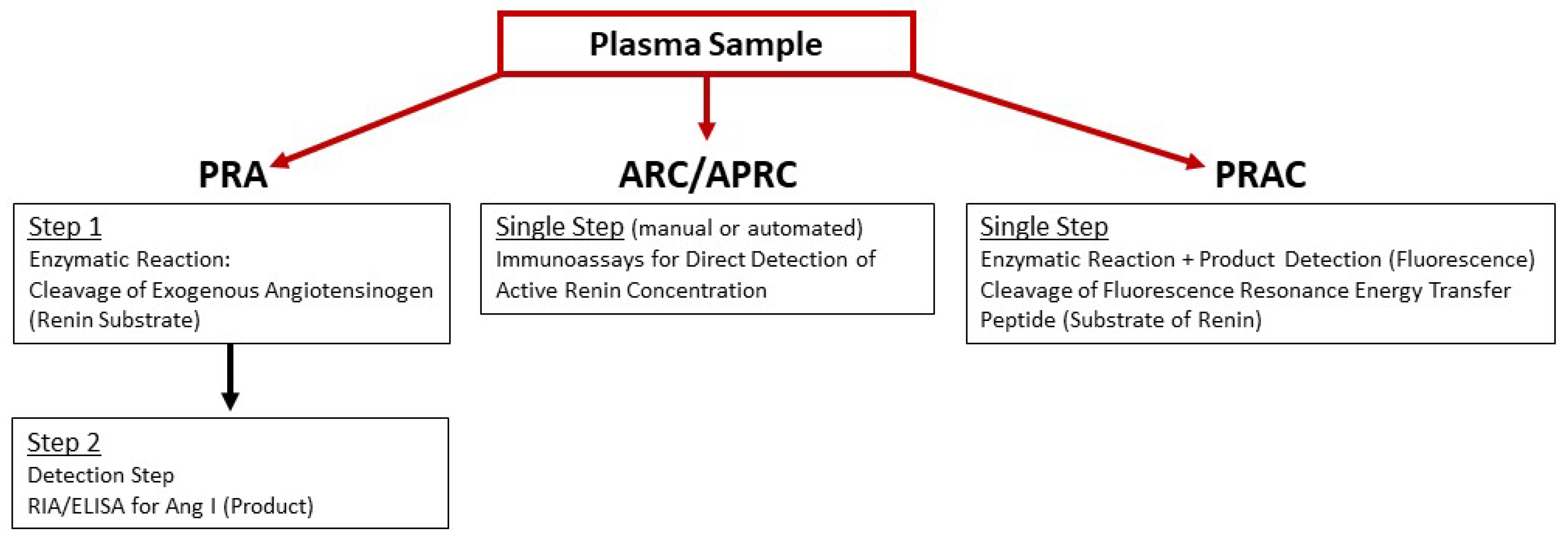
3.1. Assays of Plasma Renin Activity
3.1.1. Ang I Generation Assay
3.1.2. Immunoassay for Active Renin
3.1.3. Renin-Specific Substrate Cleavage Assay
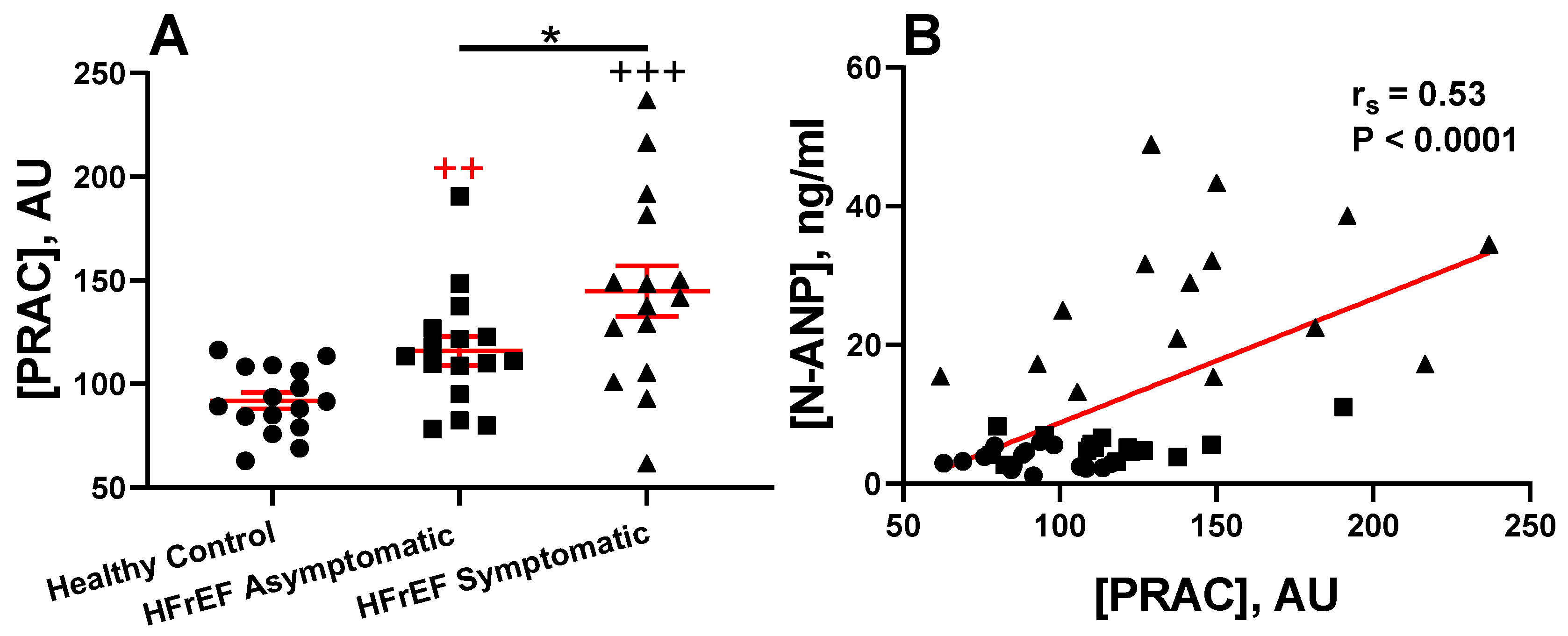
3.2. Prognostic Value of Plasma Renin Activity in HFrEF
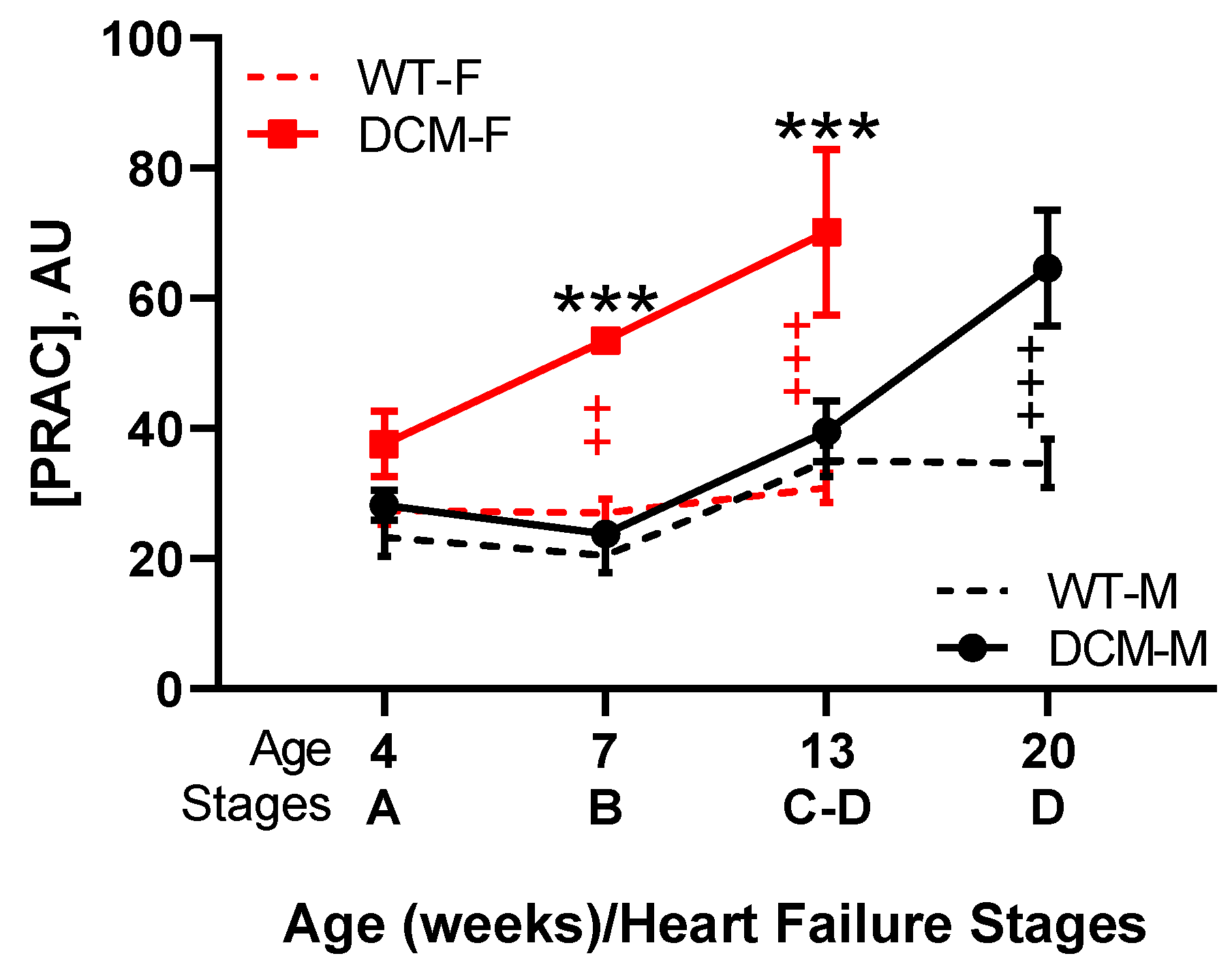
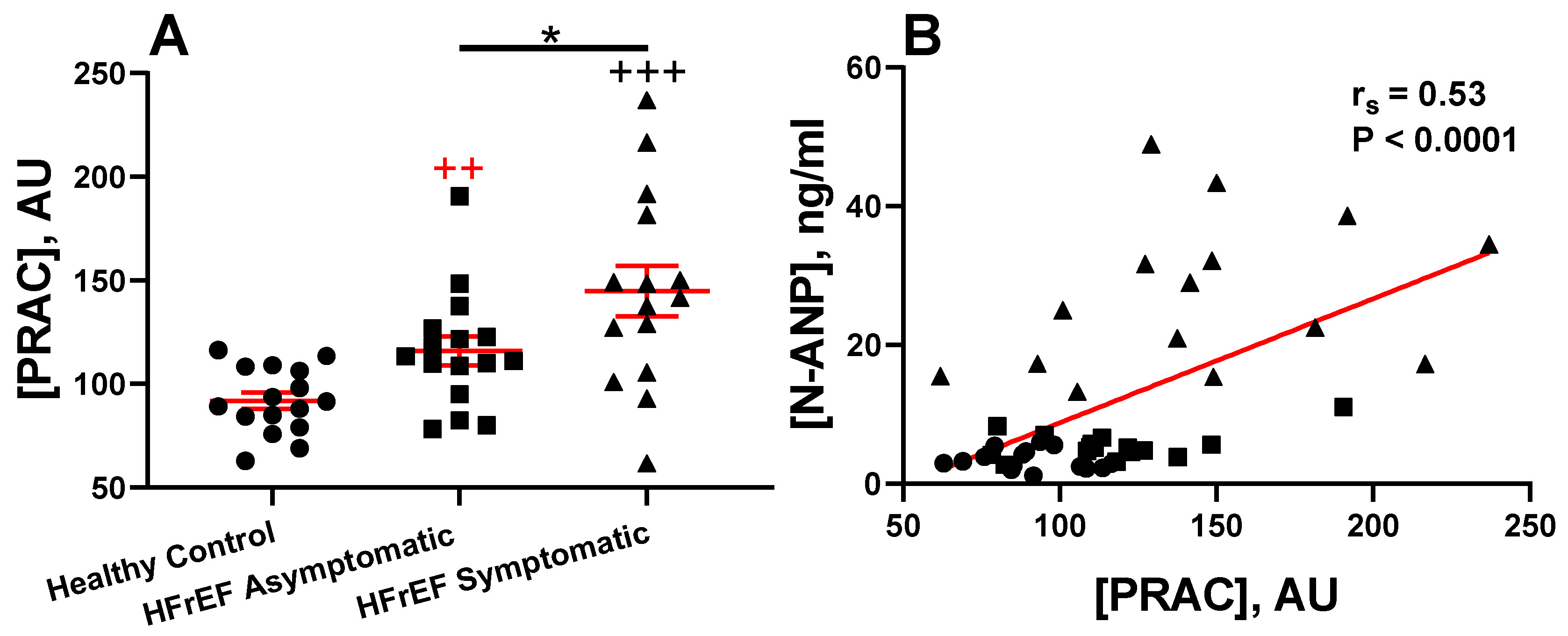
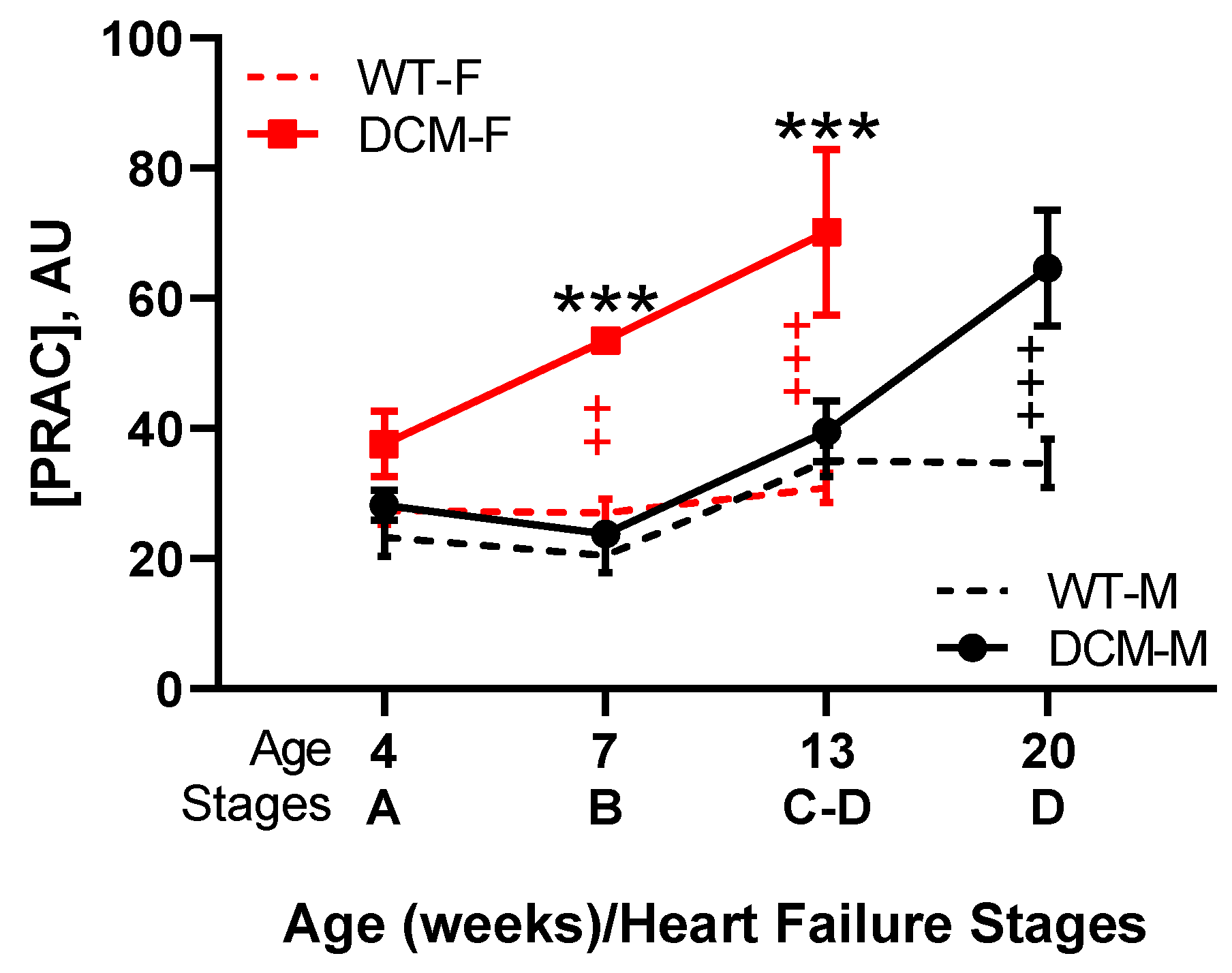
3.3. Renin Activity in Idopathic and Experimental (Genetic or Induced) Animal HFrEF Studies
4. Renin Activity as a Bio-Target for Pharmacological Intervention Using Direct Renin Inhibitors (DRI) in HFrEF
4.1. In Clinical HFrEF
4.2. In Animal Models of HFrEF
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HF | Heart Failure |
| rEF | Reduced Ejection Fraction |
| DCM | Dilated Cardiomyopathy |
| ACC | American College of Cardiology |
| AHA | American Heart Association |
| SNS | Sympathetic Nervous System |
| NP | Natriuretic Peptide |
| ANP | Atrial Natriuretic Peptide |
| BNP | B-Type Natriuretic Peptide |
| NT-proANP | N-terminal-pro ANP |
| NT-proBNP | N-terminal-pro hormone BNP |
| RAAS | Renin–Angiotensin–Aldosterone System |
| ACE | Angiotensin Converting Enzyme |
| ACE-I | Angiotensin Converting Enzyme Inhibitor |
| ACE2 | Angiotensin Converting Enzyme 2 |
| Ang I | Angiotensin I |
| Ang II | Angiotensin II |
| Ang (1–7) | Angiotensin (1–7) |
| ARB | Angiotensin II Receptor Blockers |
| AT1 | Angiotensin II Type 1 Receptor |
| AT2 | Angiotensin II Type 2 Receptor |
| Mas | G Protein-Coupled Receptor Mas |
| MRA | Mineralocorticoid Receptor Antagonist |
| ARNi | Angiotensin Receptor Neprilysin Inhibitors |
| DRI | Direct Renin Inhibitor |
| NEP | Neprilysin |
| JG | Juxtaglomerular |
| cAMP | Cyclic Adenosine Monophosphate |
| cGMP | Cyclic Guanosine Monophosphate |
| (P)RR | (Pro)Renin Receptor |
| MAPK | Mitogen-Regulated Protein Kinase |
| Erk1/2 | Extracellular Signal-Regulated Kinase 1 and 2 |
| TGF-β1 | Transforming Growth Factor-β1 |
| PAI-1 | Plasminogen Activator Inhibitor Type 1 |
| IGFII | Insulin-Like Growth Factor II |
| M6P | Mannose-6-Phosphate |
| PPARγ | Peroxisome Proliferator-Activated Receptor gamma |
| PRA | Plasma Renin Activity |
| ARC | Active Renin Concentration |
| APRC | Active Plasma Renin Concentration |
| PRAC | Plasma Renin Activity Concentration |
| RIA | Radioimmunoassay |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| EDTA | Ethylenediaminetetraacetic Acid |
| PMSF | Phenylmethylsulfonyl Fluoride |
| AU | Arbitrary Units |
| SBP | Systolic Blood Pressure |
| SOLVD | Studies of Left Ventricular Dysfunction |
| NYHA | New York Heart Association |
| ARIANA-CHF-RD | Additive Renin Inhibition with Aliskiren on Renal Blood Flow and Neurohormonal Activation in Patients with Chronic Heart Failure and Renal Dysfunction |
| ASTRONAUT | Six Months Efficacy and Safety of Aliskiren Therapy on Top of Standard Therapy, on Morbidity and Mortality in Patients with Acute Decompensated Heart Failure |
| ALOFT | Aliskiren Observation of Heart Failure Treatment |
| BUN | Blood Urea Nitrogen |
| SEM | Standard Error of the Mean |
| SC | Subcutaneous |
| dTg | Double Transgenic |
| CSQ | Calsequestrin |
References
- Japp, A.G.; Gulati, A.; Cook, S.A.; Cowie, M.R.; Prasad, S.K. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement from the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, E.; Di Palo, K.E.; Pina, I.L. Sex differences in heart failure. Clin. Cardiol. 2018, 41, 211–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, 1810–1852. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Matthay, M.A. Clinical practice. Acute pulmonary edema. N. Engl. J. Med. 2005, 353, 2788–2796. [Google Scholar] [CrossRef]
- Dzau, V.J.; Colucci, W.S.; Hollenberg, N.K.; Williams, G.H. Relation of the renin-angiotensin-aldosterone system to clinical state in congestive heart failure. Circulation 1981, 63, 645–651. [Google Scholar] [CrossRef]
- Levine, T.B.; Francis, G.S.; Goldsmith, S.R.; Simon, A.B.; Cohn, J.N. Activity of the sympathetic nervous system and renin-angiotensin system assessed by plasma hormone levels and their relation to hemodynamic abnormalities in congestive heart failure. Am. J. Cardiol. 1982, 49, 1659–1666. [Google Scholar] [CrossRef]
- Schrier, R.W.; Abraham, W.T. Hormones and hemodynamics in heart failure. N. Engl. J. Med. 1999, 341, 577–585. [Google Scholar] [CrossRef]
- Zucker, I.H.; Xiao, L.; Haack, K.K. The central renin-angiotensin system and sympathetic nerve activity in chronic heart failure. Clin. Sci. (Lond.) 2014, 126, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Hartupee, J.; Mann, D.L. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Karayannis, G.; Giamouzis, G.; Skoularigis, J.; Louridas, G.; Butler, J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J. Am. Coll. Cardiol. 2009, 54, 1747–1762. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.W.; Stopps, T.P.; Ford, S.E.; de Bold, A.J. Rapid ventricular pacing in the dog: Pathophysiologic studies of heart failure. Circulation 1986, 74, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Francis, G.S.; Benedict, C.; Johnstone, D.E.; Kirlin, P.C.; Nicklas, J.; Liang, C.S.; Kubo, S.H.; Rudin-Toretsky, E.; Yusuf, S. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Left Ventricular Dysfunction (SOLVD). Circulation 1990, 82, 1724–1729. [Google Scholar] [CrossRef] [PubMed]
- Kalra, P.R.; Anker, S.D.; Coats, A.J. Water and sodium regulation in chronic heart failure: The role of natriuretic peptides and vasopressin. Cardiovasc. Res. 2001, 51, 495–509. [Google Scholar] [CrossRef]
- Gladysheva, I.P.; Wang, D.; McNamee, R.A.; Houng, A.K.; Mohamad, A.A.; Fan, T.M.; Reed, G.L. Corin overexpression improves cardiac function, heart failure, and survival in mice with dilated cardiomyopathy. Hypertension 2013, 61, 327–332. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, Q. Corin in natriuretic peptide processing and hypertension. Curr. Hypertens. Rep. 2014, 16, 415. [Google Scholar] [CrossRef]
- Bayes-Genis, A.; Barallat, J.; Galan, A.; de Antonio, M.; Domingo, M.; Zamora, E.; Urrutia, A.; Lupon, J. Soluble neprilysin is predictive of cardiovascular death and heart failure hospitalization in heart failure patients. J. Am. Coll. Cardiol. 2015, 65, 657–665. [Google Scholar] [CrossRef]
- Huntley, B.K.; Sandberg, S.M.; Heublein, D.M.; Sangaralingham, S.J.; Burnett, J.C., Jr.; Ichiki, T. Pro-B-type natriuretic peptide-1-108 processing and degradation in human heart failure. Circ. Heart Fail. 2015, 8, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C., Jr.; Kao, P.C.; Hu, D.C.; Heser, D.W.; Heublein, D.; Granger, J.P.; Opgenorth, T.J.; Reeder, G.S. Atrial natriuretic peptide elevation in congestive heart failure in the human. Science 1986, 231, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Ibebuogu, U.N.; Gladysheva, I.P.; Houng, A.K.; Reed, G.L. Decompensated heart failure is associated with reduced corin levels and decreased cleavage of pro-atrial natriuretic peptide. Circ. Heart Fail. 2011, 4, 114–120. [Google Scholar] [CrossRef]
- Dries, D.L. Process matters: Emerging concepts underlying impaired natriuretic peptide system function in heart failure. Circ. Heart Fail. 2011, 4, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.S.; Ward, R.D.; Ramanathan, K.; Yu, X.; Gladysheva, I.P.; Reed, G.L. Possible Enzymatic Downregulation of the Natriuretic Peptide System in Patients with Reduced Systolic Function and Heart Failure: A Pilot Study. Biomed. Res. Int. 2018, 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Struthers, A.D.; Lang, C.C. Modulation of the renin-angiotensin-aldosterone system in heart failure. Curr. Atheroscler. Rep. 2014, 16, 403. [Google Scholar] [CrossRef] [PubMed]
- Skeggs, L.T., Jr.; Kahn, J.R.; Lentz, K.; Shumway, N.P. The preparation, purification, and amino acid sequence of a polypeptide renin substrate. J. Exp. Med. 1957, 106, 439–453. [Google Scholar] [CrossRef]
- Group, C.T.S. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N. Engl. J. Med. 1987, 316, 1429–1435. [Google Scholar] [CrossRef]
- Investigators, S.; Yusuf, S.; Pitt, B.; Davis, C.E.; Hood, W.B., Jr.; Cohn, J.N. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N. Engl. J. Med. 1992, 327, 685–691. [Google Scholar] [CrossRef]
- Packer, M.; Poole-Wilson, P.A.; Armstrong, P.W.; Cleland, J.G.; Horowitz, J.D.; Massie, B.M.; Ryden, L.; Thygesen, K.; Uretsky, B.F. Comparative effects of low and high doses of the angiotensin-converting enzyme inhibitor, lisinopril, on morbidity and mortality in chronic heart failure. ATLAS Study Group. Circulation 1999, 100, 2312–2318. [Google Scholar] [CrossRef]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Poole-Wilson, P.A.; Segal, R.; Martinez, F.A.; Dickstein, K.; Camm, A.J.; Konstam, M.A.; Riegger, G.; Klinger, G.H.; Neaton, J.; et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: Randomised trial--the Losartan Heart Failure Survival Study ELITE II. Lancet 2000, 355, 1582–1587. [Google Scholar] [CrossRef]
- Flather, M.D.; Yusuf, S.; Kober, L.; Pfeffer, M.; Hall, A.; Murray, G.; Torp-Pedersen, C.; Ball, S.; Pogue, J.; Moye, L.; et al. Long-term ACE-inhibitor therapy in patients with heart failure or left-ventricular dysfunction: A systematic overview of data from individual patients. ACE-Inhibitor Myocardial Infarction Collaborative Group. Lancet 2000, 355, 1575–1581. [Google Scholar] [CrossRef]
- Cohn, J.N.; Tognoni, G.; Valsartan Heart Failure Trial Investigators. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N. Engl. J. Med. 2001, 345, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Granger, C.B.; McMurray, J.J.; Yusuf, S.; Held, P.; Michelson, E.L.; Olofsson, B.; Ostergren, J.; Pfeffer, M.A.; Swedberg, K.; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: The CHARM-Alternative trial. Lancet 2003, 362, 772–776. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Swedberg, K.; Granger, C.B.; Held, P.; McMurray, J.J.; Michelson, E.L.; Olofsson, B.; Ostergren, J.; Yusuf, S.; Pocock, S.; et al. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: The CHARM-Overall programme. Lancet 2003, 362, 759–766. [Google Scholar] [CrossRef]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M.; et al. Survival Study, I. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2017, 136, e137–e161. [Google Scholar] [CrossRef]
- Orsborne, C.; Chaggar, P.S.; Shaw, S.M.; Williams, S.G. The renin-angiotensin-aldosterone system in heart failure for the non-specialist: The past, the present and the future. Postgrad. Med. J. 2017, 93, 29–37. [Google Scholar] [CrossRef]
- Gheorghiade, M.; Bohm, M.; Greene, S.J.; Fonarow, G.C.; Lewis, E.F.; Zannad, F.; Solomon, S.D.; Baschiera, F.; Botha, J.; Hua, T.A.; et al. Effect of aliskiren on postdischarge mortality and heart failure readmissions among patients hospitalized for heart failure: The ASTRONAUT randomized trial. JAMA 2013, 309, 1125–1135. [Google Scholar] [CrossRef]
- McMurray, J.J.; Krum, H.; Abraham, W.T.; Dickstein, K.; Kober, L.V.; Desai, A.S.; Solomon, S.D.; Greenlaw, N.; Ali, M.A.; Chiang, Y.; et al. Aliskiren, Enalapril, or Aliskiren and Enalapril in Heart Failure. N. Engl. J. Med. 2016, 374, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Luo, H.; Wang, S.; Zhang, C.; Hao, J.; Gao, C. Aliskiren for heart failure: A systematic review and meta-analysis of randomized controlled trials. Oncotarget 2017, 8, 88189–88198. [Google Scholar] [CrossRef] [PubMed]
- Wollert, K.C.; Drexler, H. The renin-angiotensin system and experimental heart failure. Cardiovasc. Res. 1999, 43, 838–849. [Google Scholar] [CrossRef]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical Renin-Angiotensin system in kidney physiology. Compr. Physiol. 2014, 4, 1201–1228. [Google Scholar] [CrossRef] [PubMed]
- Von Lueder, T.G.; Sangaralingham, S.J.; Wang, B.H.; Kompa, A.R.; Atar, D.; Burnett, J.C., Jr.; Krum, H. Renin-angiotensin blockade combined with natriuretic peptide system augmentation: Novel therapeutic concepts to combat heart failure. Circ. Heart Fail. 2013, 6, 594–605. [Google Scholar] [CrossRef]
- Tigerstedt, R.B.P.G. Niere und Kreislauf. Skand. Arch. Physiol. 1898, 8, 223–271. [Google Scholar] [CrossRef]
- Sielecki, A.R.; Hayakawa, K.; Fujinaga, M.; Murphy, M.E.; Fraser, M.; Muir, A.K.; Carilli, C.T.; Lewicki, J.A.; Baxter, J.D.; James, M.N. Structure of recombinant human renin, a target for cardiovascular-active drugs, at 2.5 A resolution. Science 1989, 243, 1346–1351. [Google Scholar] [CrossRef]
- Muller, D.N.; Luft, F.C. Direct renin inhibition with aliskiren in hypertension and target organ damage. Clin. J. Am. Soc. Nephrol. 2006, 1, 221–228. [Google Scholar] [CrossRef]
- Gould, A.B.; Green, D. Kinetics of the human renin and human substrate reaction. Cardiovasc. Res. 1971, 5, 86–89. [Google Scholar] [CrossRef]
- Cleland, S.J.; Reid, J.L. The renin-angiotensin system and the heart: A historical review. Heart 1996, 76 (Suppl. 3), 7–12. [Google Scholar] [CrossRef]
- Weber, K.T. Aldosterone in congestive heart failure. N. Engl. J. Med. 2001, 345, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Chappell, M.C.; Tallant, E.A.; Brosnihan, K.B.; Diz, D.I. Counterregulatory actions of angiotensin-(1-7). Hypertension 1997, 30 (3 Pt 2), 535–541. [Google Scholar] [CrossRef]
- Santos, R.A.; Campagnole-Santos, M.J.; Andrade, S.P. Angiotensin-(1-7): An update. Regul. Pept. 2000, 91, 45–62. [Google Scholar] [CrossRef]
- Tallant, E.A.; Ferrario, C.M.; Gallagher, P.E. Angiotensin-(1-7) inhibits growth of cardiac myocytes through activation of the mas receptor. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1560–H1566. [Google Scholar] [CrossRef] [PubMed]
- Fyhrquist, F.; Saijonmaa, O. Renin-angiotensin system revisited. J. Intern. Med. 2008, 264, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Shang, D.; Zhang, T.; Li, L.; Zhou, T.; Lu, W. Modeling of angiotensin II-angiotensin-(1-7) counterbalance in disease progression in spontaneously hypertensive rats treated with/without perindopril. Pharm. Res. 2012, 66, 177–184. [Google Scholar] [CrossRef]
- Santos, R.A.; Ferreira, A.J.; Verano-Braga, T.; Bader, M. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: New players of the renin-angiotensin system. J. Endocrinol. 2013, 216, R1–R17. [Google Scholar] [CrossRef]
- Gironacci, M.M.; Adamo, H.P.; Corradi, G.; Santos, R.A.; Ortiz, P.; Carretero, O.A. Angiotensin (1-7) induces MAS receptor internalization. Hypertension 2011, 58, 176–181. [Google Scholar] [CrossRef]
- Santos, R.A.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef]
- Carey, R.M. Update on the role of the AT2 receptor. Curr. Opin. Nephrol. Hypertens. 2005, 14, 67–71. [Google Scholar] [CrossRef]
- Carey, R.M. AT2 Receptors: Potential Therapeutic Targets for Hypertension. Am. J. Hypertens. 2017, 30, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Booz, G.W. Cardiac angiotensin AT2 receptor: What exactly does it do? Hypertension 2004, 43, 1162–1163. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.S.; Vinh, A.; McCarthy, C.A.; Gaspari, T.A.; Widdop, R.E. AT2 receptors: Functional relevance in cardiovascular disease. Pharmacol. Ther. 2008, 120, 292–316. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J. Renin inhibitors—Mechanisms of action. Aust. Prescr. 2009, 32, 132–135. [Google Scholar] [CrossRef]
- Opie, L.H.; Sack, M.N. Enhanced angiotensin II activity in heart failure: Reevaluation of the counterregulatory hypothesis of receptor subtypes. Circ. Res. 2001, 88, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.I. Can angiotensin II type 2 receptors have deleterious effects in cardiovascular disease? Implications for therapeutic blockade of the renin-angiotensin system. Circulation 2004, 109, 8–13. [Google Scholar] [CrossRef]
- Lemarie, C.A.; Schiffrin, E.L. The angiotensin II type 2 receptor in cardiovascular disease. J. Renin Angiotensin Aldosterone Syst. 2010, 11, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.D.; Morgan, J.P.; Yan, X. Genetically modified mouse models used for studying the role of the AT2 receptor in cardiac hypertrophy and heart failure. J. Biomed. Biotechnol. 2011, 2011, 141039. [Google Scholar] [CrossRef]
- Hobart, P.M.; Fogliano, M.; O’Connor, B.A.; Schaefer, I.M.; Chirgwin, J.M. Human renin gene: Structure and sequence analysis. Proc. Natl. Acad. Sci. USA 1984, 81, 5026–5030. [Google Scholar] [CrossRef]
- Khan, A.R.; James, M.N. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 1998, 7, 815–836. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Carrasquero, M.C.; Harrison-Bernard, L.M.; Kobori, H.; Ozawa, Y.; Hering-Smith, K.S.; Hamm, L.L.; Navar, L.G. Enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Hypertension 2004, 44, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Krop, M.; Lu, X.; Danser, A.H.; Meima, M.E. The (pro)renin receptor. A decade of research: What have we learned? Pflug. Arch. 2013, 465, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Castrop, H.; Hocherl, K.; Kurtz, A.; Schweda, F.; Todorov, V.; Wagner, C. Physiology of kidney renin. Physiol. Rev. 2010, 90, 607–673. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, A. Control of renin synthesis and secretion. Am. J. Hypertens. 2012, 25, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Heger, J.; Schluter, K.D. Renin and the IGFII/M6P receptor system in cardiac biology. Sci. World J. 2013, 2013, 260298. [Google Scholar] [CrossRef] [PubMed]
- Grunberger, C.; Obermayer, B.; Klar, J.; Kurtz, A.; Schweda, F. The calcium paradoxon of renin release: Calcium suppresses renin exocytosis by inhibition of calcium-dependent adenylate cyclases AC5 and AC6. Circ. Res. 2006, 99, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Beierwaltes, W.H. The role of calcium in the regulation of renin secretion. Am. J. Physiol. Ren. Physiol. 2010, 298, F1–F11. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, A.; Della Bruna, R.; Pfeilschifter, J.; Taugner, R.; Bauer, C. Atrial natriuretic peptide inhibits renin release from juxtaglomerular cells by a cGMP-mediated process. Proc. Natl. Acad. Sci. USA 1986, 83, 4769–4773. [Google Scholar] [CrossRef] [PubMed]
- Gambaryan, S.; Wagner, C.; Smolenski, A.; Walter, U.; Poller, W.; Haase, W.; Kurtz, A.; Lohmann, S.M. Endogenous or overexpressed cGMP-dependent protein kinases inhibit cAMP-dependent renin release from rat isolated perfused kidney, microdissected glomeruli, and isolated juxtaglomerular cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9003–9008. [Google Scholar] [CrossRef] [Green Version]
- Wagner, C.; Pfeifer, A.; Ruth, P.; Hofmann, F.; Kurtz, A. Role of cGMP-kinase II in the control of renin secretion and renin expression. J. Clin. Investig. 1998, 102, 1576–1582. [Google Scholar] [CrossRef]
- Fournier, D.; Luft, F.C.; Bader, M.; Ganten, D.; Andrade-Navarro, M.A. Emergence and evolution of the renin-angiotensin-aldosterone system. J. Mol. Med. (Berl.) 2012, 90, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.J. Renin: Friend or foe? Heart 2007, 93, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, J.N.; Weihprecht, H.; Schnermann, J.; Skott, O.; Briggs, J.P. Renin release from isolated juxtaglomerular apparatus depends on macula densa chloride transport. Am. J. Physiol. 1991, 260, F486–F493. [Google Scholar] [CrossRef] [PubMed]
- Bock, H.A.; Hermle, M.; Brunner, F.P.; Thiel, G. Pressure dependent modulation of renin release in isolated perfused glomeruli. Kidney Int. 1992, 41, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, R.M.; McGrath, H.E.; Pentz, E.S.; Gomez, R.A.; Barrett, P.Q. Biomechanical coupling in renin-releasing cells. J. Clin. Investig. 1997, 100, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Tatemichi, S.R.; Osmond, D.H. Factor XII in endogenous activation of inactive renin. Lancet 1978, 1, 1313. [Google Scholar] [CrossRef]
- Sealey, J.E.; Atlas, S.A.; Laragh, J.H.; Silverberg, M.; Kaplan, A.P. Initiation of plasma prorenin activation by Hageman factor-dependent conversion of plasma prekallikrein to kallikrein. Proc. Natl. Acad. Sci. USA 1979, 76, 5914–5918. [Google Scholar] [CrossRef] [PubMed]
- Rumpf, K.W.; Becker, K.; Kreusch, U.; Schmidt, S.; Vetter, R.; Scheler, F. Evidence for a role of plasma kallikrein in the activation of prorenin. Nature 1980, 283, 482–483. [Google Scholar] [CrossRef] [PubMed]
- Mantero, F.; Fallo, F.; Patrassi, G.; Sarandria, A.; Pedini, F.; Girolami, A. Effect of Captoril on Inactive Renin and Contact Phase of Coagulation System. Clin. Exp. Hypertens. 1982, 4, 2425–2434. [Google Scholar] [CrossRef]
- Patrassi, G.M.; Fallo, F.; Martinelli, S.; Mantero, F.; Boeri, G.; Girolami, A. The contact phase of blood coagulation and renin activation in essential hypertension before and after captopril. Eur. Heart J. 1984, 5, 561–567. [Google Scholar] [CrossRef]
- Reudelhuber, T.L.; Ramla, D.; Chiu, L.; Mercure, C.; Seidah, N.G. Proteolytic processing of human prorenin in renal and non-renal tissues. Kidney Int. 1994, 46, 1522–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derkx, F.H.; Schalekamp, M.P.; Schalekamp, M.A. Two-step prorenin-renin conversion. Isolation of an intermediary form of activated prorenin. J. Biol. Chem. 1987, 262, 2472–2477. [Google Scholar] [PubMed]
- Leckie, E.J. The reversible activation of inactive renin in human plasma: Role of acid and of plasma kallikrein and plasmin. Clin. Exp. Hypertens. A 1982, 4, 2133–2147. [Google Scholar] [CrossRef] [PubMed]
- Mercure, C.; Lacombe, M.J.; Khazaie, K.; Reudelhuber, T.L. Cathepsin B is not the processing enzyme for mouse prorenin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1212–R1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, G.; Delarue, F.; Burckle, C.; Bouzhir, L.; Giller, T.; Sraer, J.D. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J. Clin. Investig. 2002, 109, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Pratt, R.E.; Carleton, J.E.; Richie, J.P.; Heusser, C.; Dzau, V.J. Human renin biosynthesis and secretion in normal and ischemic kidneys. Proc. Natl. Acad. Sci. USA 1987, 84, 7837–7840. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, W.W.; Danser, A.H.J. Prorenin and the (pro)renin receptor: Binding kinetics, signalling and interaction with aliskiren. J. Renin-Angiotensin-Aldosterone Syst. 2008, 9, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G. Renin and prorenin receptor in hypertension: what’s new? Curr. Hypertens. Rep. 2011, 13, 79–85. [Google Scholar] [CrossRef]
- Schroten, N.F.; Gaillard, C.A.; van Veldhuisen, D.J.; Szymanski, M.K.; Hillege, H.L.; de Boer, R.A. New roles for renin and prorenin in heart failure and cardiorenal crosstalk. Heart Fail. Rev. 2012, 17, 191–201. [Google Scholar] [CrossRef]
- Binger, K.J.; Muller, D.N. Autophagy and the (Pro)renin Receptor. Front. Endocrinol. (Lausanne) 2013, 4, 155. [Google Scholar] [CrossRef] [Green Version]
- Danser, A.H.; van Kats, J.P.; Admiraal, P.J.; Derkx, F.H.; Lamers, J.M.; Verdouw, P.D.; Saxena, P.R.; Schalekamp, M.A. Cardiac renin and angiotensins. Uptake from plasma versus in situ synthesis. Hypertension 1994, 24, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.L.; Sigmund, C.D. Minireview: Overview of the renin-angiotensin system--an endocrine and paracrine system. Endocrinology 2003, 144, 2179–2183. [Google Scholar] [CrossRef] [PubMed]
- Danser, A.H.; Deinum, J. Renin, prorenin and the putative (pro)renin receptor. J. Renin Angiotensin Aldosterone Syst. 2005, 6, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef] [PubMed]
- Nabi, A.H.; Kageshima, A.; Uddin, M.N.; Nakagawa, T.; Park, E.Y.; Suzuki, F. Binding properties of rat prorenin and renin to the recombinant rat renin/prorenin receptor prepared by a baculovirus expression system. Int J. Mol. Med. 2006, 18, 483–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawa, H.; Tokuchi, F.; Mochizuki, N.; Endo, Y.; Furuta, Y.; Shinohara, T.; Takada, A.; Kawaguchi, H.; Yasuda, H.; Nagashima, K. Expression of the angiotensinogen gene and localization of its protein in the human heart. Circulation 1992, 86, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Iwai, N.; Shimoike, H.; Kinoshita, M. Cardiac renin-angiotensin system in the hypertrophied heart. Circulation 1995, 92, 2690–2696. [Google Scholar] [CrossRef] [PubMed]
- Kleiger, R.E.; Miller, J.P.; Bigger, J.T., Jr.; Moss, A.J. Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Am. J. Cardiol. 1987, 59, 256–262. [Google Scholar] [CrossRef]
- Van Kesteren, C.A.; Danser, A.H.; Derkx, F.H.; Dekkers, D.H.; Lamers, J.M.; Saxena, P.R.; Schalekamp, M.A. Mannose 6-phosphate receptor-mediated internalization and activation of prorenin by cardiac cells. Hypertension 1997, 30, 1389–1396. [Google Scholar] [CrossRef]
- Ichihara, A.; Hayashi, M.; Kaneshiro, Y.; Suzuki, F.; Nakagawa, T.; Tada, Y.; Koura, Y.; Nishiyama, A.; Okada, H.; Uddin, M.N.; et al. Inhibition of diabetic nephropathy by a decoy peptide corresponding to the "handle" region for nonproteolytic activation of prorenin. J. Clin. Investig. 2004, 114, 1128–1135. [Google Scholar] [CrossRef]
- Ichihara, A.; Kaneshiro, Y.; Takemitsu, T.; Sakoda, M.; Suzuki, F.; Nakagawa, T.; Nishiyama, A.; Inagami, T.; Hayashi, M. Nonproteolytic activation of prorenin contributes to development of cardiac fibrosis in genetic hypertension. Hypertension 2006, 47, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Danser, A.H. Prorenin: Back into the arena. Hypertension 2006, 47, 824–826. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.A. Receptor-mediated actions of renin and prorenin. Kidney Int. 2006, 69, 13–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, G. Renin/prorenin receptors. Kidney Int. 2006, 69, 1503–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, G.; Danser, A.H. The (pro)renin receptor: Therapeutic consequences. Expert Opin. Investig. Drugs 2006, 15, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, A.; Suzuki, F.; Nakagawa, T.; Kaneshiro, Y.; Takemitsu, T.; Sakoda, M.; Nabi, A.H.; Nishiyama, A.; Sugaya, T.; Hayashi, M.; et al. Prorenin receptor blockade inhibits development of glomerulosclerosis in diabetic angiotensin II type 1a receptor-deficient mice. J. Am. Soc. Nephrol. 2006, 17, 1950–1961. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wongamorntham, S.; Kasting, J.; McQuillan, D.; Owens, R.T.; Yu, L.; Noble, N.A.; Border, W. Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney Int. 2006, 69, 105–113. [Google Scholar] [CrossRef]
- Cousin, C.; Bracquart, D.; Contrepas, A.; Corvol, P.; Muller, L.; Nguyen, G. Soluble form of the (pro)renin receptor generated by intracellular cleavage by furin is secreted in plasma. Hypertension 2009, 53, 1077–1082. [Google Scholar] [CrossRef]
- Yoshikawa, A.; Aizaki, Y.; Kusano, K.; Kishi, F.; Susumu, T.; Iida, S.; Ishiura, S.; Nishimura, S.; Shichiri, M.; Senbonmatsu, T. The (pro)renin receptor is cleaved by ADAM19 in the Golgi leading to its secretion into extracellular space. Hypertens. Res. 2011, 34, 599–605. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, N.; Isobe, S.; Ishigaki, S.; Suzuki, T.; Iwakura, T.; Ono, M.; Fujikura, T.; Tsuji, T.; Otsuka, A.; Ishii, Y.; et al. Plasma Soluble (Pro)renin Receptor Reflects Renal Damage. PLoS ONE 2016, 11, e0156165. [Google Scholar] [CrossRef]
- Gong, L.; Zhang, S.; Li, L.; Gao, X.; Wang, D.; Wu, D.; Wang, K.; Liu, Y. Elevated plasma soluble (pro)renin receptor levels are associated with left ventricular remodeling and renal function in chronic heart failure patients with reduced ejection fraction. Peptides 2019, 111, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Danser, A.H. The increase in renin during renin inhibition: Does it result in harmful effects by the (pro)renin receptor? Hypertens. Res. 2010, 33, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Trepiccione, F.; Gerber, S.D.; Grahammer, F.; Lopez-Cayuqueo, K.I.; Baudrie, V.; Paunescu, T.G.; Capen, D.E.; Picard, N.; Alexander, R.T.; Huber, T.B.; et al. Renal Atp6ap2/(Pro)renin Receptor Is Required for Normal Vacuolar H+-ATPase Function but Not for the Renin-Angiotensin System. J. Am. Soc. Nephrol. 2016, 27, 3320–3330. [Google Scholar] [CrossRef] [PubMed]
- Van den Eijnden, M.M.; Saris, J.J.; de Bruin, R.J.; de Wit, E.; Sluiter, W.; Reudelhuber, T.L.; Schalekamp, M.A.; Derkx, F.H.; Danser, A.H. Prorenin accumulation and activation in human endothelial cells: Importance of mannose 6-phosphate receptors. Arter. Thromb. Vasc. Biol. 2001, 21, 911–916. [Google Scholar] [CrossRef]
- Nguyen, G.; Contrepas, A. The (pro)renin receptors. J. Mol. Med. (Berl.) 2008, 86, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Saris, J.J.; Derkx, F.H.; De Bruin, R.J.; Dekkers, D.H.; Lamers, J.M.; Saxena, P.R.; Schalekamp, M.A.; Jan Danser, A.H. High-affinity prorenin binding to cardiac man-6-P/IGF-II receptors precedes proteolytic activation to renin. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1706–H1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batenburg, W.W.; Danser, A.H. (Pro)renin and its receptors: Pathophysiological implications. Clin. Sci. (Lond.) 2012, 123, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.D.; Chu, C.H.; Huang, E.J.; Lu, M.C.; Liu, J.Y.; Liu, C.J.; Hsu, H.H.; Lin, J.A.; Kuo, W.W.; Huang, C.Y. Roles of insulin-like growth factor II in cardiomyoblast apoptosis and in hypertensive rat heart with abdominal aorta ligation. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E306–E314. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Kuo, W.W.; Yeh, Y.L.; Ho, T.J.; Lin, J.Y.; Lin, D.Y.; Chu, C.H.; Tsai, F.J.; Tsai, C.H.; Huang, C.Y. ANG II promotes IGF-IIR expression and cardiomyocyte apoptosis by inhibiting HSF1 via JNK activation and SIRT1 degradation. Cell Death Differ. 2014, 21, 1262–1274. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Kuo, C.H.; Pai, P.Y.; Ho, T.J.; Lin, Y.M.; Chen, R.J.; Tsai, F.J.; Vijaya Padma, V.; Kuo, W.W.; Huang, C.Y. Inhibition of HSF2 SUMOylation via MEL18 upregulates IGF-IIR and leads to hypertension-induced cardiac hypertrophy. Int. J. Cardiol. 2018, 257, 283–290. [Google Scholar] [CrossRef]
- Huang, C.Y.; Lee, F.L.; Peng, S.F.; Lin, K.H.; Chen, R.J.; Ho, T.J.; Tsai, F.J.; Padma, V.V.; Kuo, W.W.; Huang, C.Y. HSF1 phosphorylation by ERK/GSK3 suppresses RNF126 to sustain IGF-IIR expression for hypertension-induced cardiomyocyte hypertrophy. J. Cell Physiol. 2018, 233, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, J.; Huang, J.; Zhang, X.; Zhao, H.; Cui, C.; Li, Y.; Hu, S. Elevation of IGF-2 receptor and the possible underlying implications in end-stage heart failure patients before and after heart transplantation. J. Cell Mol. Med. 2012, 16, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, S.; Heger, J.; Schreckenberg, R.; Wenzel, S.; Euler, G.; Arens, C.; Bader, M.; Rosenkranz, S.; Caglayan, E.; Schluter, K.D. Controlling cardiomyocyte length: The role of renin and PPAR-{gamma}. Cardiovasc. Res. 2011, 89, 344–352. [Google Scholar] [CrossRef] [PubMed]
- El-Shewy, H.M.; Lee, M.H.; Obeid, L.M.; Jaffa, A.A.; Luttrell, L.M. The insulin-like growth factor type 1 and insulin-like growth factor type 2/mannose-6-phosphate receptors independently regulate ERK1/2 activity in HEK293 cells. J. Biol. Chem. 2007, 282, 26150–26157. [Google Scholar] [CrossRef] [PubMed]
- Todorov, V.T.; Desch, M.; Schmitt-Nilson, N.; Todorova, A.; Kurtz, A. Peroxisome proliferator-activated receptor-gamma is involved in the control of renin gene expression. Hypertension 2007, 50, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Wylie, A.A.; Pulford, D.J.; McVie-Wylie, A.J.; Waterland, R.A.; Evans, H.K.; Chen, Y.T.; Nolan, C.M.; Orton, T.C.; Jirtle, R.L. Tissue-specific inactivation of murine M6P/IGF2R. Am. J. Pathol. 2003, 162, 321–328. [Google Scholar] [CrossRef]
- Muller, D.N.; Derer, W.; Dechend, R. Aliskiren--mode of action and preclinical data. J. Mol. Med. (Berl.) 2008, 86, 659–662. [Google Scholar] [CrossRef]
- Teisman, A.C.; van Veldhuisen, D.J.; Boomsma, F.; de Kam, P.J.; Tjeerdsma, G.; Pinto, Y.M.; de Zeeuw, D.; van Gilst, W.H. Chronic beta-blocker treatment in patients with advanced heart failure. Effects on neurohormones. Int. J. Cardiol. 2000, 73, 7–12; discussion 13–14. [Google Scholar] [CrossRef]
- Bodyak, N.; Ayus, J.C.; Achinger, S.; Shivalingappa, V.; Ke, Q.; Chen, Y.S.; Rigor, D.L.; Stillman, I.; Tamez, H.; Kroeger, P.E.; et al. Activated vitamin D attenuates left ventricular abnormalities induced by dietary sodium in Dahl salt-sensitive animals. Proc. Natl. Acad. Sci. USA 2007, 104, 16810–16815. [Google Scholar] [CrossRef] [Green Version]
- Schroten, N.F.; Ruifrok, W.P.; Kleijn, L.; Dokter, M.M.; Sillje, H.H.; Lambers Heerspink, H.J.; Bakker, S.J.; Kema, I.P.; van Gilst, W.H.; van Veldhuisen, D.J.; et al. Short-term vitamin D3 supplementation lowers plasma renin activity in patients with stable chronic heart failure: An open-label, blinded end point, randomized prospective trial (VitD-CHF trial). Am. Heart J. 2013, 166, 357–364.e2. [Google Scholar] [CrossRef]
- Bellabarba, G.; Davila, D.F.; Torres, A.; Donis, J.H.; Gonzalez, J.C.; Figueroa, O.; Vasquez, C.J.; Faddoul, M.; Khoury, A. Plasma renin activity in chagasic patients with and without congestive heart failure. Int. J. Cardiol. 1994, 47, 5–11. [Google Scholar] [CrossRef]
- Koch, J.; Pedersen, H.D.; Jensen, A.L.; Flagstad, A.; Poulsen, K. Activation of the renin-angiotensin system in dogs with asymptomatic and symptomatic dilated cardiomyopathy. Res. Vet. Sci. 1995, 59, 172–175. [Google Scholar] [CrossRef]
- Latini, R.; Masson, S.; Anand, I.; Salio, M.; Hester, A.; Judd, D.; Barlera, S.; Maggioni, A.P.; Tognoni, G.; Cohn, J.N.; et al. The comparative prognostic value of plasma neurohormones at baseline in patients with heart failure enrolled in Val-HeFT. Eur. Heart J. 2004, 25, 292–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentz, R.J.; Stevens, S.R.; DeVore, A.D.; Lala, A.; Vader, J.M.; AbouEzzeddine, O.F.; Khazanie, P.; Redfield, M.M.; Stevenson, L.W.; O’Connor, C.M.; et al. Decongestion strategies and renin-angiotensin-aldosterone system activation in acute heart failure. JACC Heart Fail. 2015, 3, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Sullivan, R.; Fan, T.M.; Wang, D.; Sun, Y.; Reed, G.L.; Gladysheva, I.P. Enhanced heart failure, mortality and renin activation in female mice with experimental dilated cardiomyopathy. PLoS ONE 2017, 12, e0189315. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, L.A.; Kolka, M.A.; Francesconi, R.; Gonzalez, R.R. Circadian variations in plasma renin activity, catecholamines and aldosterone during exercise in women. Eur. J. Appl. Physiol. Occup. Physiol. 1989, 58, 756–764. [Google Scholar] [CrossRef]
- Sealey, J.E. Plasma renin activity and plasma prorenin assays. Clin. Chem. 1991, 37 (10 Pt 2), 1811–1819. [Google Scholar]
- Pitarresi, T.M.; Rubattu, S.; Heinrikson, R.; Sealey, J.E. Reversible cryoactivation of recombinant human prorenin. J. Biol. Chem. 1992, 267, 11753–11759. [Google Scholar]
- Campbell, D.J. Critical review of prorenin and (pro)renin receptor research. Hypertension 2008, 51, 1259–1264. [Google Scholar] [CrossRef]
- Cartledge, S.; Lawson, N. Aldosterone and renin measurements. Ann. Clin. Biochem. 2000, 37, 262–278. [Google Scholar] [CrossRef]
- Campbell, D.J.; Nussberger, J.; Stowasser, M.; Danser, A.H.; Morganti, A.; Frandsen, E.; Menard, J. Activity assays and immunoassays for plasma Renin and prorenin: Information provided and precautions necessary for accurate measurement. Clin. Chem. 2009, 55, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Hartman, D.; Sagnella, G.A.; Chesters, C.A.; Macgregor, G.A. Direct renin assay and plasma renin activity assay compared. Clin. Chem. 2004, 50, 2159–2161. [Google Scholar] [CrossRef] [PubMed]
- Tsutamoto, T.; Sakai, H.; Tanaka, T.; Fujii, M.; Yamamoto, T.; Wada, A.; Ohnishi, M.; Horie, M. Comparison of active renin concentration and plasma renin activity as a prognostic predictor in patients with heart failure. Circ. J. 2007, 71, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Wedatilake, Y.N.; Scanlon, M.J.; Barnes, S.C. The clinical utility of two renin mass methods to detect primary hyperaldosteronism compared with renin activity. Ann. Clin. Biochem. 2011, 48, 256–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, R.; Ono, Y.; Tezuka, Y.; Kudo, M.; Yamamoto, S.; Arai, T.; Gomez-Sanchez, C.E.; Sasano, H.; Ito, S.; Satoh, F. Rapid Screening of Primary Aldosteronism by a Novel Chemiluminescent Immunoassay. Hypertension 2017, 70, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Pavo, N.; Goliasch, G.; Wurm, R.; Novak, J.; Strunk, G.; Gyongyosi, M.; Poglitsch, M.; Saemann, M.D.; Hulsmann, M. Low- and High-renin Heart Failure Phenotypes with Clinical Implications. Clin. Chem. 2018, 64, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.T.; Chung, C.C.; Holzman, T.F.; Krafft, G.A. A continuous fluorescence assay of renin activity. Anal. Biochem. 1993, 210, 351–359. [Google Scholar] [CrossRef]
- Paschalidou, K.; Neumann, U.; Gerhartz, B.; Tzougraki, C. Highly sensitive intramolecularly quenched fluorogenic substrates for renin based on the combination of L-2-amino-3-(7-methoxy-4-coumaryl)propionic acid with 2,4-dinitrophenyl groups at various positions. Biochem. J. 2004, 382 (Pt 3), 1031–1038. [Google Scholar] [CrossRef] [Green Version]
- Schmiedt, C.W.; Hurley, K.A.; Tong, X.; Rakhmanova, V.A.; Po, C.L.; Hurley, D.J. Measurement of plasma renin concentration in cats by use of a fluorescence resonance energy transfer peptide substrate of renin. Am. J. Vet. Res. 2009, 70, 1315–1322. [Google Scholar] [CrossRef]
- Schmiedt, C.W.; Nelson, S.A.; Brainard, B.M.; Brown, C.A.; Vandenplas, M.; Hurley, D.J. Bilateral renal ischemia as a model of acute kidney injury in cats. Res. Vet. Sci. 2012, 93, 950–959. [Google Scholar] [CrossRef]
- Prysyazhna, O.; Rudyk, O.; Eaton, P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat. Med. 2012, 18, 286–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakoki, M.; Pochynyuk, O.M.; Hathaway, C.M.; Tomita, H.; Hagaman, J.R.; Kim, H.S.; Zaika, O.L.; Mamenko, M.; Kayashima, Y.; Matsuki, K.; et al. Primary aldosteronism and impaired natriuresis in mice underexpressing TGFbeta1. Proc. Natl. Acad. Sci. USA 2013, 110, 5600–5605. [Google Scholar] [CrossRef] [PubMed]
- Takada, S.; Kinugawa, S.; Hirabayashi, K.; Suga, T.; Yokota, T.; Takahashi, M.; Fukushima, A.; Homma, T.; Ono, T.; Sobirin, M.A.; et al. Angiotensin II receptor blocker improves the lowered exercise capacity and impaired mitochondrial function of the skeletal muscle in type 2 diabetic mice. J. Appl. Physiol. (1985) 2013, 114, 844–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Gladysheva, I.P.; Fan, T.H.; Sullivan, R.; Houng, A.K.; Reed, G.L. Atrial natriuretic peptide affects cardiac remodeling, function, heart failure, and survival in a mouse model of dilated cardiomyopathy. Hypertension 2014, 63, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.D.; Mehta, R.M.; Tripathi, R.; Gladysheva, I.P.; Reed, G.L. Abstract 16743: Targeting renin activity in heart failure: precision therapy with aliskiren improves systolic function and prolongs survival in female experimental dilated cardiomyopathy. Circulation 2018, 138 (Suppl. 1). [Google Scholar]
- Vergaro, G.; Emdin, M.; Iervasi, A.; Zyw, L.; Gabutti, A.; Poletti, R.; Mammini, C.; Giannoni, A.; Fontana, M.; Passino, C. Prognostic value of plasma renin activity in heart failure. Am. J. Cardiol. 2011, 108, 246–251. [Google Scholar] [CrossRef]
- Poletti, R.; Vergaro, G.; Zyw, L.; Prontera, C.; Passino, C.; Emdin, M. Prognostic value of plasma renin activity in heart failure patients with chronic kidney disease. Int. J. Cardiol. 2013, 167, 711–715. [Google Scholar] [CrossRef]
- Sim, J.J.; Shi, J.; Al-Moomen, R.; Behayaa, H.; Kalantar-Zadeh, K.; Jacobsen, S.J. Plasma renin activity and its association with ischemic heart disease, congestive heart failure, and cerebrovascular disease in a large hypertensive cohort. J. Clin. Hypertens. (Greenwich) 2014, 16, 805–813. [Google Scholar] [CrossRef]
- Bhandari, S.K.; Batech, M.; Shi, J.; Jacobsen, S.J.; Sim, J.J. Plasma renin activity and risk of cardiovascular and mortality outcomes among individuals with elevated and nonelevated blood pressure. Kidney Res. Clin. Pract. 2016, 35, 219–228. [Google Scholar] [CrossRef]
- Nijst, P.; Verbrugge, F.H.; Martens, P.; Bertrand, P.B.; Dupont, M.; Francis, G.S.; Tang, W.W.; Mullens, W. Plasma renin activity in patients with heart failure and reduced ejection fraction on optimal medical therapy. J. Renin Angiotensin Aldosterone Syst. 2017, 18, 1470320317729919. [Google Scholar] [CrossRef]
- Osterziel, K.J.; Dietz, R.; Schmid, W.; Mikulaschek, K.; Manthey, J.; Kubler, W. ACE inhibition improves vagal reactivity in patients with heart failure. Am. Heart J. 1990, 120, 1120–1129. [Google Scholar] [CrossRef]
- Borgarelli, M.; Tarducci, A.; Tidholm, A.; Haggstrom, J. Canine idiopathic dilated cardiomyopathy. Part II: Pathophysiology and therapy. Vet. J. 2001, 162, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Monnet, E.; Orton, E.C.; Salman, M.; Boon, J. Idiopathic dilated cardiomyopathy in dogs: Survival and prognostic indicators. J. Vet. Intern. Med. 1995, 9, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Tidholm, A.; Haggstrom, J.; Borgarelli, M.; Tarducci, A. Canine idiopathic dilated cardiomyopathy. Part I: Aetiology, clinical characteristics, epidemiology and pathology. Vet. J. 2001, 162, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Tidholm, A. Survival in dogs with dilated cardiomyopathy and congestive heart failure treated with digoxin, furosemide and propranolol: A retrospective study of 62 dogs. J. Vet. Cardiol. 2006, 8, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Sisson, D.D.; Thomas, W.P. Myocardial Diseases. In Textbook of Veterinary Internal Medicine, 4th ed.; Ettinger, S.J., Ed.; W. B. Saunders: Philadelphia, PA, USA, 1995; pp. 995–1005. [Google Scholar]
- Tidholm, A.; Jonsson, L. A retrospective study of canine dilated cardiomyopathy (189 cases). J. Am. Anim. Hosp. Assoc. 1997, 33, 544–550. [Google Scholar] [CrossRef]
- Vollmar, A.C. The prevalence of cardiomyopathy in the Irish wolfhound: A clinical study of 500 dogs. J. Am. Anim. Hosp. Assoc. 2000, 36, 125–132. [Google Scholar] [CrossRef]
- Tidholm, A.; Haggstrom, J.; Hansson, K. Effects of dilated cardiomyopathy on the renin-angiotensin-aldosterone system, atrial natriuretic peptide activity, and thyroid hormone concentrations in dogs. Am. J. Vet. Res. 2001, 62, 961–967. [Google Scholar] [CrossRef]
- Tolwani, R.J.; Waggie, K.S.; Green, S.L.; Tolwani, A.J.; Lyons, D.M.; Schatzberg, A.F. Dilative cardiomyopathy leading to congestive heart failure in a male squirrel monkey (Saimiri sciureus). J. Med. Primatol. 2000, 29, 42–45. [Google Scholar] [CrossRef]
- Shannon, R.P.; Simon, M.A.; Mathier, M.A.; Geng, Y.J.; Mankad, S.; Lackner, A.A. Dilated cardiomyopathy associated with simian AIDS in nonhuman primates. Circulation 2000, 101, 185–193. [Google Scholar] [CrossRef]
- Sleeper, M.M.; Doane, C.J.; Langner, P.H.; Curtis, S.; Avila, K.; Lee, D.R. Successful treatment of idiopathic dilated cardiomyopathy in an adult chimpanzee (Pan troglodytes). Comp. Med. 2005, 55, 80–84. [Google Scholar] [PubMed]
- Rajendra, R.S.; Brady, A.G.; Parks, V.L.; Massey, C.V.; Gibson, S.V.; Abee, C.R. The normal and abnormal owl monkey (Aotus sp.) heart: Looking at cardiomyopathy changes with echocardiography and electrocardiography. J. Med. Primatol. 2010, 39, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Felkai, A.; Vogelnest, L.; McNabb, S.; Allan, G.; Sangster, C. Dilated cardiomyopathy in a De Brazza’s monkey (Cercopithecus neglectus). J. Med. Primatol. 2014, 43, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Houser, S.R.; Margulies, K.B.; Murphy, A.M.; Spinale, F.G.; Francis, G.S.; Prabhu, S.D.; Rockman, H.A.; Kass, D.A.; Molkentin, J.D.; Sussman, M.A.; et al. Animal models of heart failure: A scientific statement from the American Heart Association. Circ. Res. 2012, 111, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Becher, P.M.; Jugdutt, B.I.; Baugh, J.; Schmack, B. Experimental Heart Failure Models and Their Pathophysiological Characterization. Biomed. Res. Int. 2016, 2016, 2538263. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Gross, K.W. Transcriptional regulation of renin: An update. Hypertension 2005, 45, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sigmund, C.D.; Gross, K.W. Structure, expression, and regulation of the murine renin genes. Hypertension 1991, 18, 446–457. [Google Scholar] [CrossRef]
- Holm, I.; Ollo, R.; Panthier, J.J.; Rougeon, F. Evolution of aspartyl proteases by gene duplication: The mouse renin gene is organized in two homologous clusters of four exons. EMBO J. 1984, 3, 557–562. [Google Scholar] [CrossRef]
- Hansen, P.B.; Yang, T.; Huang, Y.; Mizel, D.; Briggs, J.; Schnermann, J. Plasma renin in mice with one or two renin genes. Acta Physiol. Scand. 2004, 181, 431–437. [Google Scholar] [CrossRef]
- Moreno, C.; Hoffman, M.; Stodola, T.J.; Didier, D.N.; Lazar, J.; Geurts, A.M.; North, P.E.; Jacob, H.J.; Greene, A.S. Creation and characterization of a renin knockout rat. Hypertension 2011, 57, 614–619. [Google Scholar] [CrossRef]
- Clark, A.F.; Sharp, M.G.; Morley, S.D.; Fleming, S.; Peters, J.; Mullins, J.J. Renin-1 is essential for normal renal juxtaglomerular cell granulation and macula densa morphology. J. Biol. Chem. 1997, 272, 18185–18190. [Google Scholar] [CrossRef] [PubMed]
- Sharp, M.G.; Fettes, D.; Brooker, G.; Clark, A.F.; Peters, J.; Fleming, S.; Mullins, J.J. Targeted inactivation of the Ren-2 gene in mice. Hypertension 1996, 28, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Ganten, D.; Wagner, J.; Zeh, K.; Bader, M.; Michel, J.B.; Paul, M.; Zimmermann, F.; Ruf, P.; Hilgenfeldt, U.; Ganten, U.; et al. Species specificity of renin kinetics in transgenic rats harboring the human renin and angiotensinogen genes. Proc. Natl. Acad. Sci. USA 1992, 89, 7806–7810. [Google Scholar] [CrossRef] [PubMed]
- Fukamizu, A.; Sugimura, K.; Takimoto, E.; Sugiyama, F.; Seo, M.S.; Takahashi, S.; Hatae, T.; Kajiwara, N.; Yagami, K.; Murakami, K. Chimeric renin-angiotensin system demonstrates sustained increase in blood pressure of transgenic mice carrying both human renin and human angiotensinogen genes. J. Biol. Chem. 1993, 268, 11617–11621. [Google Scholar] [PubMed]
- Merrill, D.C.; Thompson, M.W.; Carney, C.L.; Granwehr, B.P.; Schlager, G.; Robillard, J.E.; Sigmund, C.D. Chronic hypertension and altered baroreflex responses in transgenic mice containing the human renin and human angiotensinogen genes. J. Clin. Investig. 1996, 97, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, F. Development of genetically engineered mice with hypertension and hypotension. Exp. Anim. 1997, 46, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.L.; Bianco, R.A.; Sakai, K.; Keen, H.L.; Ryan, M.J.; Sigmund, C.D. Transgenic mice for studies of the renin-angiotensin system in hypertension. Acta Physiol. Scand. 2004, 181, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.E.; Sigmund, C.D. Genetic basis of hypertension: Revisiting angiotensinogen. Hypertension 2006, 48, 14–20. [Google Scholar] [CrossRef]
- Spinale, F.G.; de Gasparo, M.; Whitebread, S.; Hebbar, L.; Clair, M.J.; Melton, D.M.; Krombach, R.S.; Mukherjee, R.; Iannini, J.P.; O, S.J. Modulation of the renin-angiotensin pathway through enzyme inhibition and specific receptor blockade in pacing-induced heart failure: I. Effects on left ventricular performance and neurohormonal systems. Circulation 1997, 96, 2385–2396. [Google Scholar] [CrossRef]
- Fitzpatrick, M.A.; Rademaker, M.T.; Frampton, C.M.; Charles, C.J.; Yandle, T.G.; Espiner, E.A.; Ikram, H. Hemodynamic and hormonal effects of renin inhibition in ovine heart failure. Am. J. Physiol. 1990, 258 (6 Pt 2), H1625–H1631. [Google Scholar] [CrossRef]
- Kanamori, T.; Wada, A.; Tsutamoto, T.; Kinoshita, M. Possible regulation of renin release by ANP in dogs with heart failure. Am. J. Physiol. 1995, 268 (6 Pt 2), H2281–H2287. [Google Scholar] [CrossRef]
- Holycross, B.J.; Summers, B.M.; Dunn, R.B.; McCune, S.A. Plasma renin activity in heart failure-prone SHHF/Mcc-facp rats. Am. J. Physiol. 1997, 273 (1 Pt 2), H228–H233. [Google Scholar] [CrossRef]
- Yamada, C.; Kuwahara, K.; Yamazaki, M.; Nakagawa, Y.; Nishikimi, T.; Kinoshita, H.; Kuwabara, Y.; Minami, T.; Yamada, Y.; Shibata, J.; et al. The renin-angiotensin system promotes arrhythmogenic substrates and lethal arrhythmias in mice with non-ischaemic cardiomyopathy. Cardiovasc. Res. 2016, 109, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.R.; Suzuki, Y.J.; Wang, W.; Kobayashi, Y.M.; Ramesh, V.; Franzini-Armstrong, C.; Cleemann, L.; Morad, M. Regulation of Ca2+ signaling in transgenic mouse cardiac myocytes overexpressing calsequestrin. J. Clin. Investig. 1998, 101, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.C.; Rapacciuolo, A.; Koch, W.J.; Kobayashi, Y.; Jones, L.R.; Rockman, H.A. Defective beta-adrenergic receptor signaling precedes the development of dilated cardiomyopathy in transgenic mice with calsequestrin overexpression. J. Biol. Chem. 1999, 274, 22251–22256. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nishimura, S.; Yamamoto, T.; Kajimoto, Y.; Kusumoto, K.; Kanagawa, R.; Ikeda, S.; Nishimoto, T. TAK-272 (imarikiren), a novel renin inhibitor, improves cardiac remodeling and mortality in a murine heart failure model. PLoS ONE 2018, 13, e0202176. [Google Scholar] [CrossRef] [PubMed]
- Fentzke, R.C.; Korcarz, C.E.; Lang, R.M.; Lin, H.; Leiden, J.M. Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J. Clin. Investig. 1998, 101, 2415–2426. [Google Scholar] [CrossRef]
- Spencer, K.T.; Collins, K.; Korcarz, C.; Fentzke, R.; Lang, R.M.; Leiden, J.M. Effects of exercise training on LV performance and mortality in a murine model of dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H210–H215. [Google Scholar] [CrossRef]
- Tripathi, R.; Wang, D.; Sullivan, R.; Fan, T.H.; Gladysheva, I.P.; Reed, G.L. Depressed Corin Levels Indicate Early Systolic Dysfunction Before Increases of Atrial Natriuretic Peptide/B-Type Natriuretic Peptide and Heart Failure Development. Hypertension 2016, 67, 362–367. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Baessler, A.; Schunkert, H. Renin angiotensin system and gender differences in the cardiovascular system. Cardiovasc. Res. 2002, 53, 672–677. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, M.A.; Wirth, J.R.; Scott, J.L.; Eudaly, J.; Collins, E.L.; Gilkeson, G.S. Early Ovariectomy Results in Reduced Numbers of CD11c+/CD11b+ Spleen Cells and Impacts Disease Expression in Murine Lupus. Front. Immunol. 2016, 7, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradman, A.H.; Kad, R. Renin inhibition in hypertension. J. Am. Coll. Cardiol. 2008, 51, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Corminboeuf, O.; Bezencon, O.; Remen, L.; Grisostomi, C.; Richard-Bildstein, S.; Bur, D.; Prade, L.; Strickner, P.; Hess, P.; Fischli, W.; et al. Piperidine-based renin inhibitors: Upper chain optimization. Bioorg. Med. Chem. Lett. 2010, 20, 6291–6296. [Google Scholar] [CrossRef] [PubMed]
- Imaeda, Y.; Tokuhara, H.; Fukase, Y.; Kanagawa, R.; Kajimoto, Y.; Kusumoto, K.; Kondo, M.; Snell, G.; Behnke, C.A.; Kuroita, T. Discovery of TAK-272: A Novel, Potent, and Orally Active Renin Inhibitor. ACS Med. Chem. Lett. 2016, 7, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Wal, P.; Wal, A.; Rai, A.K.; Dixit, A. Aliskiren: An orally active renin inhibitor. J. Pharm. Bioallied. Sci. 2011, 3, 189–193. [Google Scholar] [CrossRef]
- Seed, A.; Gardner, R.; McMurray, J.; Hillier, C.; Murdoch, D.; MacFadyen, R.; Bobillier, A.; Mann, J.; McDonagh, T. Neurohumoral effects of the new orally active renin inhibitor, aliskiren, in chronic heart failure. Eur. J. Heart Fail. 2007, 9, 1120–1127. [Google Scholar] [CrossRef]
- Solomon, S.D.; Shin, S.H.; Shah, A.; Skali, H.; Desai, A.; Kober, L.; Maggioni, A.P.; Rouleau, J.L.; Kelly, R.Y.; Hester, A.; et al. Effect of the direct renin inhibitor aliskiren on left ventricular remodelling following myocardial infarction with systolic dysfunction. Eur. Heart J. 2011, 32, 1227–1234. [Google Scholar] [CrossRef]
- Schroten, N.F.; Damman, K.; Hemmelder, M.H.; Voors, A.A.; Navis, G.; Gaillard, C.A.; van Veldhuisen, D.J.; Van Gilst, W.H.; Hillege, H.L. Effect of additive renin inhibition with aliskiren on renal blood flow in patients with Chronic Heart Failure and Renal Dysfunction (Additive Renin Inhibition with Aliskiren on renal blood flow and Neurohormonal Activation in patients with Chronic Heart Failure and Renal Dysfunction). Am. Heart J. 2015, 169, 693–701.e3. [Google Scholar] [CrossRef]
- Krum, H.; McMurray, J.J.; Abraham, W.T.; Dickstein, K.; Kober, L.; Desai, A.S.; Solomon, S.D.; Chiang, Y.; Gimpelewicz, C.; Reimund, B.; et al. The Aliskiren Trial to Minimize OutcomeS in Patients with HEart failure trial (ATMOSPHERE): Revised statistical analysis plan and baseline characteristics. Eur. J. Heart Fail. 2015, 17, 1075–1083. [Google Scholar] [CrossRef]
- Azizi, M.; Guyene, T.T.; Chatellier, G.; Wargon, M.; Menard, J. Additive effects of losartan and enalapril on blood pressure and plasma active renin. Hypertension 1997, 29, 634–640. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Cheema, B.; Cleveland, E.; Sankar, K.; Subacius, H.; Fonarow, G.C.; Solomon, S.D.; Lewis, E.F.; Greene, S.J.; Maggioni, A.P.; et al. Plasma renin activity, response to aliskiren, and clinical outcomes in patients hospitalized for heart failure: The ASTRONAUT trial. Eur. J. Heart Fail. 2018, 20, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Chen, Q. Efficacy of aliskiren supplementation for heart failure: A meta-analysis of randomized controlled trials. Herz 2018. [Google Scholar] [CrossRef] [PubMed]
- Gradman, A.H.; Vivas, Y. New drugs for hypertension: What do they offer? Curr. Hypertens. Rep. 2006, 8, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Oh, B.H.; Mitchell, J.; Herron, J.R.; Chung, J.; Khan, M.; Keefe, D.L. Aliskiren, an oral renin inhibitor, provides dose-dependent efficacy and sustained 24-hour blood pressure control in patients with hypertension. J. Am. Coll. Cardiol. 2007, 49, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Azizi, M.; Webb, R.; Nussberger, J.; Hollenberg, N.K. Renin inhibition with aliskiren: Where are we now, and where are we going? J. Hypertens. 2006, 24, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Nussberger, J.; Wuerzner, G.; Jensen, C.; Brunner, H.R. Angiotensin II suppression in humans by the orally active renin inhibitor Aliskiren (SPP100): Comparison with enalapril. Hypertension 2002, 39, E1–E8. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Pitt, B.; Latini, R.; Maggioni, A.P.; Solomon, S.D.; Keefe, D.L.; Ford, J.; Verma, A.; Lewsey, J.; Aliskiren Observation of Heart Failure Treatment. Effects of the oral direct renin inhibitor aliskiren in patients with symptomatic heart failure. Circ. Heart Fail. 2008, 1, 17–24. [Google Scholar] [CrossRef]
- Pitt, B.; Latini, R.; Maggioni, A.P.; Solomon, S.D.; Smith, B.A.; Wright, M.; Prescott, M.F.; McMurray, J.J. Neurohumoral effects of aliskiren in patients with symptomatic heart failure receiving a mineralocorticoid receptor antagonist: The Aliskiren Observation of Heart Failure Treatment study. Eur. J. Heart Fail. 2011, 13, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Tamargo, J.; Lopez-Sendon, J. Novel therapeutic targets for the treatment of heart failure. Nat. Rev. Drug Discov. 2011, 10, 536–555. [Google Scholar] [CrossRef]
- Wang, W. Chronic administration of aldosterone depresses baroreceptor reflex function in the dog. Hypertension 1994, 24, 571–575. [Google Scholar] [CrossRef]
- Barr, C.S.; Lang, C.C.; Hanson, J.; Arnott, M.; Kennedy, N.; Struthers, A.D. Effects of adding spironolactone to an angiotensin-converting enzyme inhibitor in chronic congestive heart failure secondary to coronary artery disease. Am. J. Cardiol. 1995, 76, 1259–1265. [Google Scholar] [CrossRef]
- Upadhya, B.; Brubaker, P.H.; Morgan, T.M.; Eggebeen, J.D.; Jao, G.T.; Stewart, K.P.; Kitzman, D.W. The effect of Aliskiren on exercise capacity in older patients with heart failure and preserved ejection fraction: A randomized, placebo-controlled, double-blind trial. Am. Heart J. 2018, 201, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metra, M.; Teerlink, J.R. Heart failure. Lancet 2017, 390, 1981–1995. [Google Scholar] [CrossRef]
- Fedele, F.; Mancone, M.; Adamo, F.; Severino, P. Heart Failure With Preserved, Mid-Range, and Reduced Ejection Fraction: The Misleading Definition of the New Guidelines. Cardiol. Rev. 2017, 25, 4–5. [Google Scholar] [CrossRef]
- Seferovic, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Gal, T.B.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart failure in cardiomyopathies: A position paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef] [PubMed]
- Connelly, K.A.; Advani, A.; Advani, S.; Zhang, Y.; Thai, K.; Thomas, S.; Krum, H.; Kelly, D.J.; Gilbert, R.E. Combination angiotensin converting enzyme and direct renin inhibition in heart failure following experimental myocardial infarction. Cardiovasc. Ther. 2013, 31, 84–91. [Google Scholar] [CrossRef]
- Thomas, C.M.; Yong, Q.C.; Seqqat, R.; Chandel, N.; Feldman, D.L.; Baker, K.M.; Kumar, R. Direct renin inhibition prevents cardiac dysfunction in a diabetic mouse model: Comparison with an angiotensin receptor antagonist and angiotensin-converting enzyme inhibitor. Clin. Sci. (Lond.) 2013, 124, 529–541. [Google Scholar] [CrossRef]
- Westermann, D.; Riad, A.; Lettau, O.; Roks, A.; Savvatis, K.; Becher, P.M.; Escher, F.; Jan Danser, A.H.; Schultheiss, H.P.; Tschope, C. Renin inhibition improves cardiac function and remodeling after myocardial infarction independent of blood pressure. Hypertension 2008, 52, 1068–1075. [Google Scholar] [CrossRef]
- Rashikh, A.; Ahmad, S.J.; Pillai, K.K.; Kohli, K.; Najmi, A.K. Aliskiren attenuates myocardial apoptosis and oxidative stress in chronic murine model of cardiomyopathy. Biomed. Pharm. 2012, 66, 138–143. [Google Scholar] [CrossRef]
- Moniwa, N.; Varagic, J.; Ahmad, S.; VonCannon, J.L.; Simington, S.W.; Wang, H.; Groban, L.; Brosnihan, K.B.; Nagata, S.; Kato, J.; et al. Hemodynamic and hormonal changes to dual renin-angiotensin system inhibition in experimental hypertension. Hypertension 2013, 61, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Li, X.; Wu, W.; He, H.; Li, Y.; Gao, L.; Gan, L.; Wang, M.; Ou, S.; Liu, J. [Aliskiren inhibits angiotensin II/angiotensin 1-7(Ang II/Ang1-7) signal pathway in rats with diabetic nephropathy]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2018, 34, 891–895. [Google Scholar] [PubMed]
- Antlanger, M.; Bernhofer, S.; Kovarik, J.J.; Kopecky, C.; Kaltenecker, C.C.; Domenig, O.; Poglitsch, M.; Saemann, M.D. Effects of direct renin inhibition versus angiotensin II receptor blockade on angiotensin profiles in non-diabetic chronic kidney disease. Ann. Med. 2017, 49, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J.; Zhang, Y.; Kelly, D.J.; Gilbert, R.E.; McCarthy, D.J.; Shi, W.; Smyth, G.K. Aliskiren increases bradykinin and tissue kallikrein mRNA levels in the heart. Clin. Exp. Pharm. Physiol. 2011, 38, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Koid, S.S.; Ziogas, J.; Campbell, D.J. Aliskiren reduces myocardial ischemia-reperfusion injury by a bradykinin B2 receptor- and angiotensin AT2 receptor-mediated mechanism. Hypertension 2014, 63, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, M.; Gohda, T.; Hagiwara, S.; Tanimoto, M.; Horikoshi, S.; Funabiki, K.; Tomino, Y. Effect of the Direct Renin Inhibitor Aliskiren on Urinary Albumin Excretion in Spontaneous Type 2 Diabetic KK-A (y) Mouse. Int. J. Nephrol. 2013, 2013, 519130. [Google Scholar] [CrossRef] [PubMed]
- Poss, J.; Werner, C.; Lorenz, D.; Gensch, C.; Bohm, M.; Laufs, U. The renin inhibitor aliskiren upregulates pro-angiogenic cells and reduces atherogenesis in mice. Basic Res. Cardiol. 2010, 105, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Seto, S.W.; Krishna, S.M.; Moran, C.S.; Liu, D.; Golledge, J. Aliskiren limits abdominal aortic aneurysm, ventricular hypertrophy and atherosclerosis in an apolipoprotein-E-deficient mouse model. Clin. Sci. (Lond.) 2014, 127, 123–134. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sullivan, R.D.; Mehta, R.M.; Tripathi, R.; Reed, G.L.; Gladysheva, I.P. Renin Activity in Heart Failure with Reduced Systolic Function—New Insights. Int. J. Mol. Sci. 2019, 20, 3182. https://doi.org/10.3390/ijms20133182
Sullivan RD, Mehta RM, Tripathi R, Reed GL, Gladysheva IP. Renin Activity in Heart Failure with Reduced Systolic Function—New Insights. International Journal of Molecular Sciences. 2019; 20(13):3182. https://doi.org/10.3390/ijms20133182
Chicago/Turabian StyleSullivan, Ryan D., Radhika M. Mehta, Ranjana Tripathi, Guy L. Reed, and Inna P. Gladysheva. 2019. "Renin Activity in Heart Failure with Reduced Systolic Function—New Insights" International Journal of Molecular Sciences 20, no. 13: 3182. https://doi.org/10.3390/ijms20133182
APA StyleSullivan, R. D., Mehta, R. M., Tripathi, R., Reed, G. L., & Gladysheva, I. P. (2019). Renin Activity in Heart Failure with Reduced Systolic Function—New Insights. International Journal of Molecular Sciences, 20(13), 3182. https://doi.org/10.3390/ijms20133182