Selenium and Glutathione-Depleted Rats as a Sensitive Animal Model to Predict Drug-Induced Liver Injury in Humans

Abstract
1. Introduction
2. Results
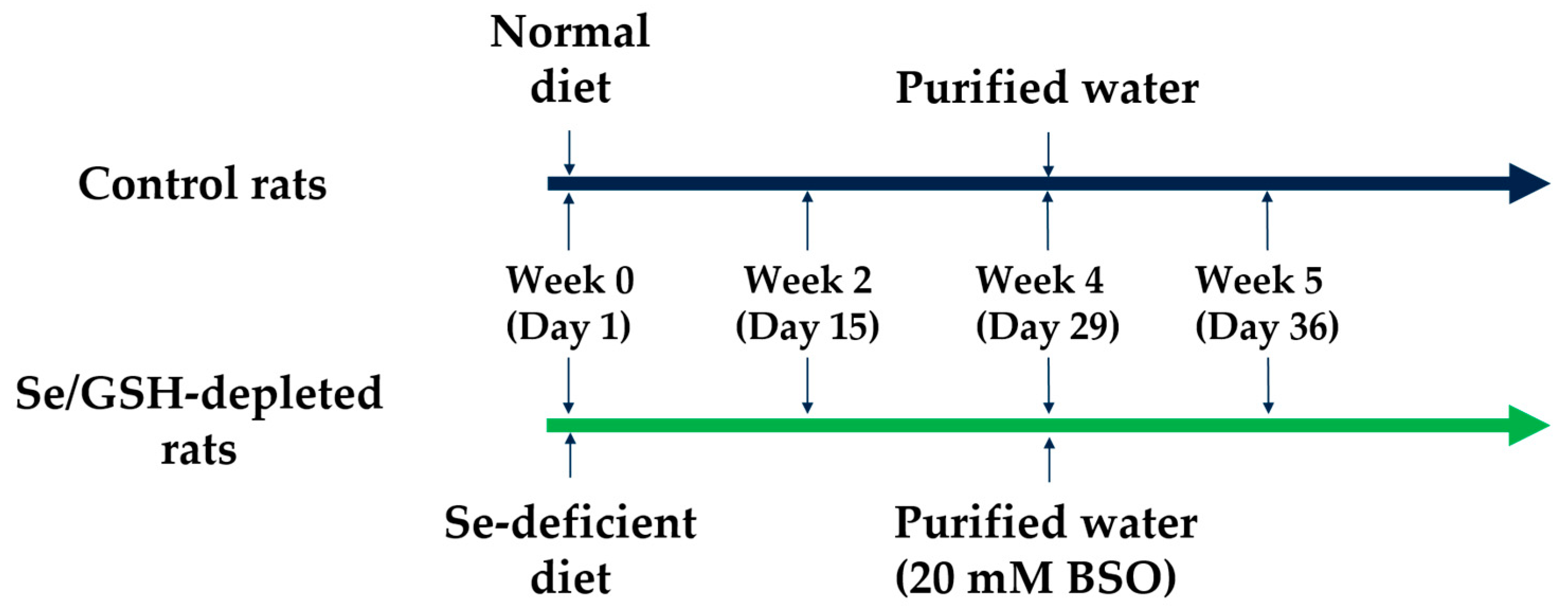
2.1. Preparation of Se/GSH-Depleted Rats
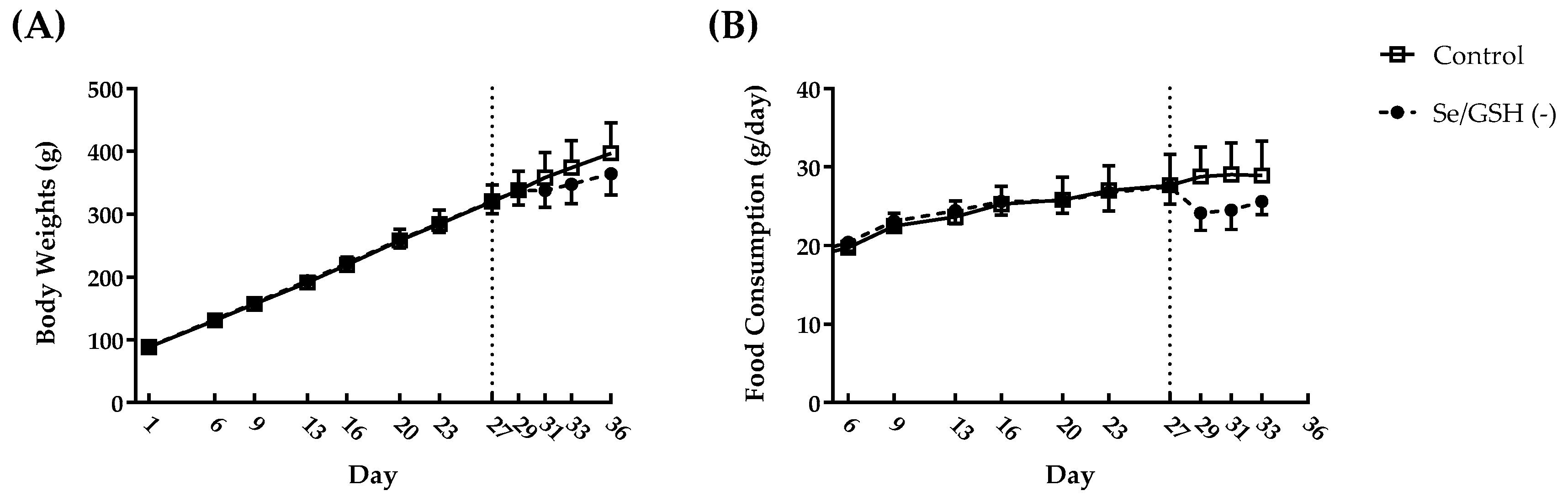
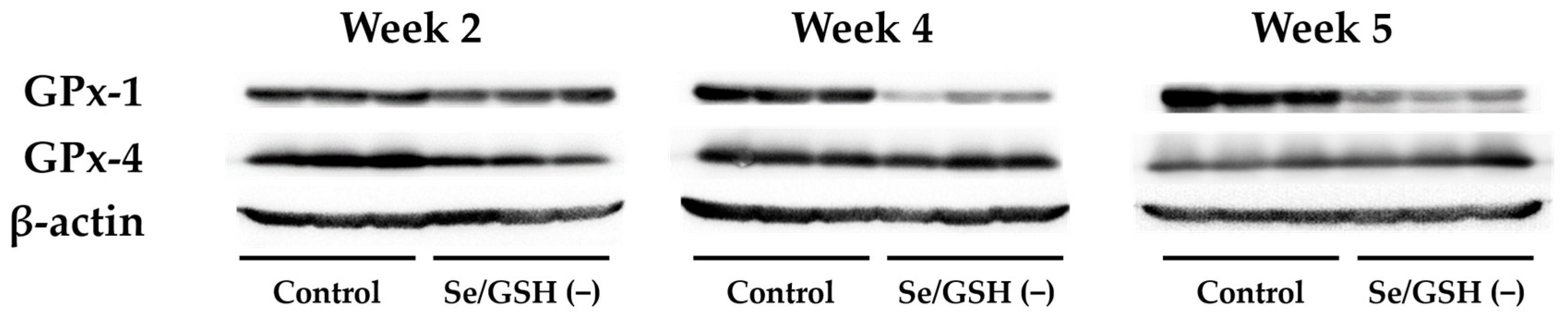
2.2. Profiling of Se/GSH-Depleted Rats
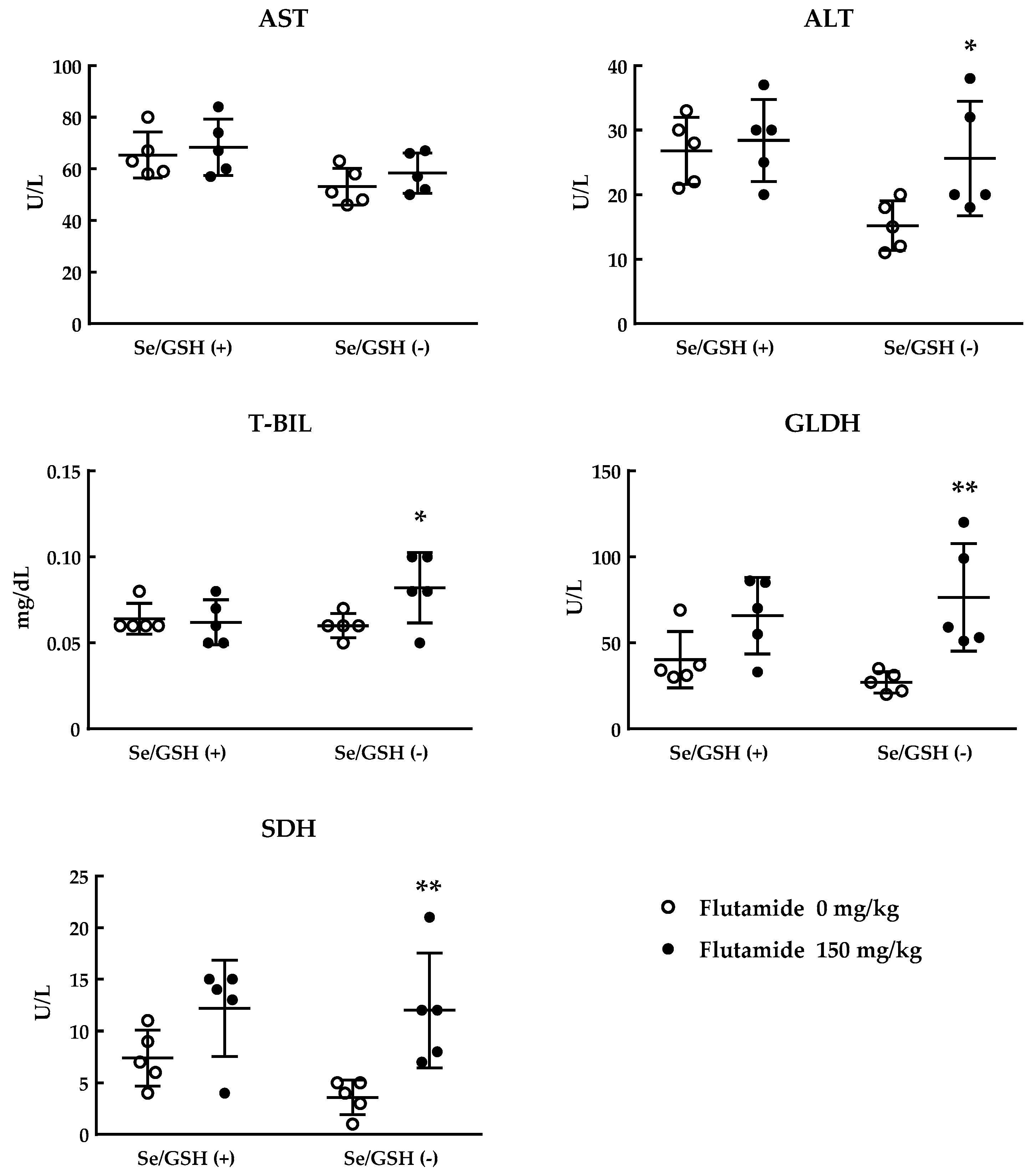
2.3. Toxicity Study of Flutamide Using Se/GSH-Depleted Rats
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Preparing Se/GSH-Depleted Rats
4.4. Toxicity Study of Flutamide Using Se/GSH-Depleted Rats
4.5. Sampling of Blood and Liver Samples
4.6. Measurements of Serum Se Levels
4.7. Measurements of Hepatic GSH and GSSG Levels
4.8. Measurements of Hepatic MDA Levels
4.9. Western Blotting
4.10. Measurements of Plasma Liver Function Parameters Levels
4.11. Histopathology
4.12. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, W.M. Drug-induced hepatotoxicity. N. Engl. J. Med. 2003, 349, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.; Schmid, E.F. Drug withdrawals and the lessons within. Curr. Opin. Drug Discov. Dev. 2006, 9, 38–46. [Google Scholar]
- Kieliszek, M.; Blazejak, S. Selenium: Significance, and outlook for supplementation. Nutrition 2013, 29, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.G.; Cheng, W.H.; McClung, J.P. Metabolic regulation and function of glutathione peroxidase-1. Annu. Rev. Nutr. 2007, 27, 41–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.K.; Weaver, J.D.; Zhang, S.; Lei, X.G. Knockout of sod1 promotes conversion of selenocysteine to dehydroalanine in murine hepatic gpx1 protein. Free Radic. Biol. Med. 2011, 51, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Patel, V.B.; Young, T.A.; Asayama, K.; Cunningham, C.C. Chronic ethanol consumption alters the glutathione/glutathione peroxidase-1 system and protein oxidation status in rat liver. Alcohol. Clin. Exp. Res. 2001, 25, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Esposito, L.A.; Kokoszka, J.E.; Waymire, K.G.; Cottrell, B.; MacGregor, G.R.; Wallace, D.C. Mitochondrial oxidative stress in mice lacking the glutathione peroxidase-1 gene. Free Radic. Biol. Med. 2000, 28, 754–766. [Google Scholar] [CrossRef]
- Mari, M.; Morales, A.; Colell, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Klotz, L.O.; Sies, H. Defenses against peroxynitrite: Selenocompounds and flavonoids. Toxicol. Lett. 2003, 140–141, 125–132. [Google Scholar] [CrossRef]
- Sies, H.; Sharov, V.S.; Klotz, L.O.; Briviba, K. Glutathione peroxidase protects against peroxynitrite-mediated oxidations. A new function for selenoproteins as peroxynitrite reductase. J. Biol. Chem. 1997, 272, 27812–27817. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sies, H.; Lei, X.G. Opposite roles of selenium-dependent glutathione peroxidase-1 in superoxide generator diquat- and peroxynitrite-induced apoptosis and signaling. J. Biol. Chem. 2001, 276, 43004–43009. [Google Scholar] [CrossRef] [PubMed]
- Sunde, R.A.; Raines, A.M.; Barnes, K.M.; Evenson, J.K. Selenium status highly regulates selenoprotein mrna levels for only a subset of the selenoproteins in the selenoproteome. Biosci. Rep. 2009, 29, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Reed, D.J. Regulation of reductive processes by glutathione. Biochem. Pharmacol. 1986, 35, 7–13. [Google Scholar] [CrossRef]
- Lee, S.S.; Buters, J.T.; Pineau, T.; Fernandez-Salguero, P.; Gonzalez, F.J. Role of cyp2e1 in the hepatotoxicity of acetaminophen. J. Biol. Chem. 1996, 271, 12063–12067. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Murakami, M.; Shirai, M.; Tanaka, M.; Nakanishi, K. Metabolism-dependent hepatotoxicity of methimazole in mice depleted of glutathione. J. Appl. Toxicol. 1999, 19, 193–198. [Google Scholar] [CrossRef]
- Dahlin, D.C.; Miwa, G.T.; Lu, A.Y.; Nelson, S.D. N-acetyl-p-benzoquinone imine: A cytochrome p-450-mediated oxidation product of acetaminophen. Proc. Natl. Acad. Sci. USA 1984, 81, 1327–1331. [Google Scholar] [CrossRef]
- Gibson, J.D.; Pumford, N.R.; Samokyszyn, V.M.; Hinson, J.A. Mechanism of acetaminophen-induced hepatotoxicity: Covalent binding versus oxidative stress. Chem. Res. Toxicol. 1996, 9, 580–585. [Google Scholar] [CrossRef]
- Hinson, J.A.; Reid, A.B.; McCullough, S.S.; James, L.P. Acetaminophen-induced hepatotoxicity: Role of metabolic activation, reactive oxygen/nitrogen species, and mitochondrial permeability transition. Drug Metab. Rev. 2004, 36, 805–822. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. Iv. Protective role of glutathione. J. Pharmacol. Exp. Ther. 1973, 187, 211–217. [Google Scholar]
- Drew, R.; Miners, J.O. The effects of buthionine sulphoximine (bso) on glutathione depletion and xenobiotic biotransformation. Biochem. Pharmacol. 1984, 33, 2989–2994. [Google Scholar] [CrossRef]
- Nishiya, T.; Mori, K.; Hattori, C.; Kai, K.; Kataoka, H.; Masubuchi, N.; Jindo, T.; Manabe, S. The crucial protective role of glutathione against tienilic acid hepatotoxicity in rats. Toxicol. Appl. Pharmacol. 2008, 232, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Atsumi, R.; Itokawa, K.; Iwasaki, M.; Aoki, T.; Ono, C.; Izumi, T.; Sudo, K.; Okazaki, O. Metabolism-dependent hepatotoxicity of amodiaquine in glutathione-depleted mice. Arch. Toxicol. 2009, 83, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Russmann, S.; Kullak-Ublick, G.A.; Grattagliano, I. Current concepts of mechanisms in drug-induced hepatotoxicity. Curr. Med. Chem. 2009, 16, 3041–3053. [Google Scholar] [CrossRef] [PubMed]
- Pirmohamed, M.; Madden, S.; Park, B.K. Idiosyncratic drug reactions. Metabolic bioactivation as a pathogenic mechanism. Clin. Pharmacokinet. 1996, 31, 215–230. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, T.M.; Watkins, P.B. The application of metabonomics to predict drug-induced liver injury. Clin. Pharmacol. Ther. 2010, 88, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Sagisaka, H.; Arakawa, S.; Shibaya, Y.; Watanabe, M.; Igarashi, I.; Tanaka, K.; Totsuka, S.; Takasaki, W.; Manabe, S. A novel model of continuous depletion of glutathione in mice treated with l-buthionine (s,r)-sulfoximine. J. Toxicol. Sci. 2003, 28, 455–469. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Higashi, T.; Furukawa, M.; Hikita, K.; Naruse, A.; Tateishi, N.; Sakamoto, Y. Re-evaluation of protein-bound glutathione in rat liver. J. Biochem. 1985, 98, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, K.W.; Williams, F.M.; Mutch, E.; Wright, P.; James, O.F.; Rawlins, M.D. The effect of alcoholic cirrhosis on the activities of microsomal aldrin epoxidase, 7-ethoxycoumarin o-de-ethylase and epoxide hydrolase, and on the concentrations of reduced glutathione in human liver. Br. J. Clin. Pharmacol. 1983, 15, 667–672. [Google Scholar] [CrossRef]
- Mertens, K.; Vercruysse, A.; Rahmani, R.; Kaufman, S.; Waterschoot, S.; Rogiers, V. Interspecies differences in glutathione-dependent detoxication of hydroperoxides in short-term cultures of hepatocytes. Toxicol. In Vitro. 1996, 10, 473–478. [Google Scholar] [CrossRef]
- Grover, P.L.; Sims, P. Conjugations with glutathione. Distribution of glutathione s-aryltransferase in vertebrate species. Biochem. J. 1964, 90, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Shalini, S.; Bansal, M.P. Role of selenium in regulation of spermatogenesis: Involvement of activator protein 1. BioFactors 2005, 23, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, Y.; Ikemoto, I.; Kishimoto, K.; Wada, T.; Yamazaki, H.; Ohishi, Y.; Kiyota, H.; Furuta, N.; Suzuki, H.; Ueda, M. Flutamide-induced hepatic dysfunction in relation to steady-state plasma concentrations of flutamide and its metabolites. Mol. Cell. Biochem. 2003, 252, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Osculati, A.; Castiglioni, C. Fatal liver complications with flutamide. Lancet 2006, 367, 1140–1141. [Google Scholar] [CrossRef]
- Thole, Z.; Manso, G.; Salgueiro, E.; Revuelta, P.; Hidalgo, A. Hepatotoxicity induced by antiandrogens: A review of the literature. Urol. Int. 2004, 73, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Wysowski, D.K.; Fourcroy, J.L. Flutamide hepatotoxicity. J. Urol. 1996, 155, 209–212. [Google Scholar] [CrossRef]
- Fau, D.; Eugene, D.; Berson, A.; Letteron, P.; Fromenty, B.; Fisch, C.; Pessayre, D. Toxicity of the antiandrogen flutamide in isolated rat hepatocytes. J. Pharmacol. Exp. Ther. 1994, 269, 954–962. [Google Scholar]
- Kang, P.; Dalvie, D.; Smith, E.; Zhou, S.; Deese, A. Identification of a novel glutathione conjugate of flutamide in incubations with human liver microsomes. Drug Metab. Dispos. 2007, 35, 1081–1088. [Google Scholar] [CrossRef]
- Kang, P.; Dalvie, D.; Smith, E.; Zhou, S.; Deese, A.; Nieman, J.A. Bioactivation of flutamide metabolites by human liver microsomes. Drug Metab. Dispos. 2008, 36, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- Kostrubsky, S.E.; Strom, S.C.; Ellis, E.; Nelson, S.D.; Mutlib, A.E. Transport, metabolism, and hepatotoxicity of flutamide, drug-drug interaction with acetaminophen involving phase i and phase ii metabolites. Chem. Res. Toxicol. 2007, 20, 1503–1512. [Google Scholar] [CrossRef]
- Ohbuchi, M.; Miyata, M.; Nagai, D.; Shimada, M.; Yoshinari, K.; Yamazoe, Y. Role of enzymatic n-hydroxylation and reduction in flutamide metabolite-induced liver toxicity. Drug Metab. Dispos. 2009, 37, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Friry-Santini, C.; Rouquie, D.; Kennel, P.; Tinwell, H.; Benahmed, M.; Bars, R. Correlation between protein accumulation profiles and conventional toxicological findings using a model antiandrogenic compound, flutamide. Toxicol. Sci. 2007, 97, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Coe, K.J.; Nelson, S.D.; Ulrich, R.G.; He, Y.; Dai, X.; Cheng, O.; Caguyong, M.; Roberts, C.J.; Slatter, J.G. Profiling the hepatic effects of flutamide in rats: A microarray comparison with classical aryl hydrocarbon receptor ligands and atypical cyp1a inducers. Drug Metab. Dispos. 2006, 34, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Nagai, D.; Ichimura, E.; Goda, R.; Tomura, A.; Doi, M.; Nishikawa, K. Metabolism and hepatic toxicity of flutamide in cytochrome p450 1a2 knockout sv129 mice. J. Gastroenterol. 2006, 41, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Akai, S.; Hosomi, H.; Tsuneyama, K.; Nakajima, M.; Yokoi, T. Drug-induced hepatotoxicity test using gamma-glutamylcysteine synthetase knockdown rat. Toxicol. Lett. 2009, 189, 159–165. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Group | Week 2 | Week 4 | Week 5 |
|---|---|---|---|---|
| Serum Se level (μg/L) | Control | 509.0 ± 34.8 | 550.0 ± 34.8 | 607.0 ± 69.7 |
| Se/GSH (−) | 355.0 ± 4.6 ** | 396.0 ± 56.0 * | 412.0 ± 79.7 * | |
| Hepatic GSH level (μmol/g liver) | Control | 2.90 ± 0.10 | 3.03 ± 0.06 | 3.53 ± 0.35 |
| Se/GSH (−) | 5.37 ± 0.97 * | 5.80 ± 0.89 ** | 1.43 ± 0.47 ** | |
| Hepatic GSSG level (μmol/g liver) | Control | 0.95 ± 0.15 | 1.44 ± 0.56 | 1.47 ± 0.22 |
| Se/GSH (−) | 0.16 ± 0.04 ** | 0.11 ± 0.03 * | 0.61 ± 0.33 * | |
| GSSG/GSH ratio | Control | 0.33 ± 0.01 | 0.47 ± 0.17 | 0.41 ± 0.17 |
| Se/GSH (−) | 0.03 ± 0.01 ** | 0.02 ± 0.002 * | 0.41 ± 0.10 | |
| Hepatic MDA level (×10−2 μmol/g liver) | Control | 13.7 ± 0.3 | 12.6 ± 0.4 | 12.2 ± 0.5 |
| Se/GSH (−) | 13.4 ± 0.1 | 14.4 ± 0.4 ** | 14.3 ± 0.4 ** | |
| Plasma AST level (U/L) | Control | 75.7 ± 1.5 | 57.3 ± 6.5 | 59.0 ± 7.5 |
| Se/GSH (−) | 65.7 ± 8.6 | 60.0 ± 57.3 | 55.7 ± 1.5 | |
| Plasma ALT level (U/L) | Control | 25.7 ± 4.0 | 18.3 ± 2.5 | 22.3 ± 7.4 |
| Se/GSH (−) | 21.0 ± 3.0 | 21.0 ± 1.7 | 17.3 ± 3.2 | |
| Plasma T-BIL level (mg/dL) | Control | 0.047 ± 0.006 | 0.053 ± 0.021 | 0.053 ± 0.025 |
| Se/GSH (−) | 0.043 ± 0.006 | 0.050 ± 0.010 | 0.053 ± 0.021 | |
| Plasma GLDH level (U/L) | Control | 18.3 ± 1.2 | 16.7 ± 2.3 | 15.7 ± 2.9 |
| Se/GSH (−) | 14.7 ± 2.9 | 17.0 ± 4.4 | 15.0 ± 2.6 | |
| Plasma SDH level (U/L) | Control | 1.17 ± 0.76 | 2.17 ± 1.44 | 4.33 ± 1.53 |
| Se/GSH (−) | 1.67 ± 1.15 | 2.17 ± 2.47 | 4.00 ± 2.00 |
| Dose Level of Flutamide (mg/kg/day) | Se/GSH (+) | Se/GSH (−) | ||
|---|---|---|---|---|
| 0 | 150 | 0 | 150 | |
| Hypertrophy, hepatocyte, centrilobular | −: 5 | ±: 3 | −: 5 | ±: 2 |
| +: 2 | +: 3 | |||
| Infiltration, inflammatory cell, periportal | −: 5 | −: 5 | −: 5 | −: 4 |
| ±: 1 | ||||
| Single cell necrosis, hepatocyte, centrilobular | −: 5 | −: 5 | −: 5 | −: 4 |
| ±: 1 | ||||
| Necrosis, hepatocyte, focal, subcapsular | −: 5 | −: 5 | −: 5 | −: 5 |
| +: 1 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goda, K.; Muta, K.; Yasui, Y.; Oshida, S.-i.; Kitatani, K.; Takekoshi, S. Selenium and Glutathione-Depleted Rats as a Sensitive Animal Model to Predict Drug-Induced Liver Injury in Humans. Int. J. Mol. Sci. 2019, 20, 3141. https://doi.org/10.3390/ijms20133141
Goda K, Muta K, Yasui Y, Oshida S-i, Kitatani K, Takekoshi S. Selenium and Glutathione-Depleted Rats as a Sensitive Animal Model to Predict Drug-Induced Liver Injury in Humans. International Journal of Molecular Sciences. 2019; 20(13):3141. https://doi.org/10.3390/ijms20133141
Chicago/Turabian StyleGoda, Keisuke, Kyotaka Muta, Yuzo Yasui, Shin-ichi Oshida, Kanae Kitatani, and Susumu Takekoshi. 2019. "Selenium and Glutathione-Depleted Rats as a Sensitive Animal Model to Predict Drug-Induced Liver Injury in Humans" International Journal of Molecular Sciences 20, no. 13: 3141. https://doi.org/10.3390/ijms20133141
APA StyleGoda, K., Muta, K., Yasui, Y., Oshida, S.-i., Kitatani, K., & Takekoshi, S. (2019). Selenium and Glutathione-Depleted Rats as a Sensitive Animal Model to Predict Drug-Induced Liver Injury in Humans. International Journal of Molecular Sciences, 20(13), 3141. https://doi.org/10.3390/ijms20133141