Coordinate Modulation of Glycolytic Enzymes and OXPHOS by Imatinib in BCR-ABL Driven Chronic Myelogenous Leukemia Cells

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
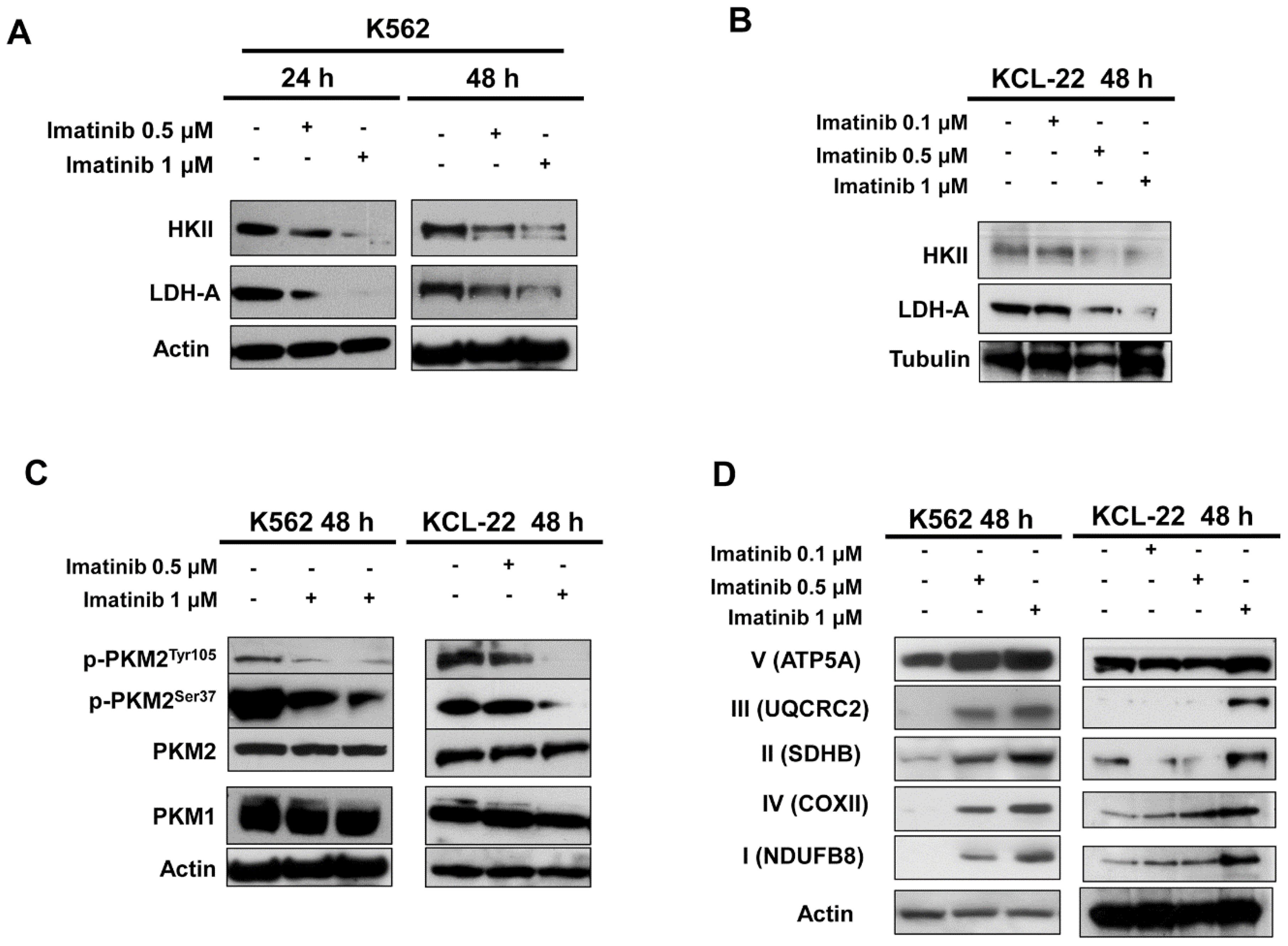
2. Results
3. Discussion
4. Materials and Methods
4.1. Cell Line and Treatment
4.2. Immunoblotting Analysis
4.3. Glucose Consumption, Lactate Secretion and Intracellular ATP
4.4. Oxygen Consumption and Extracellular Acidification Rates
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nowell, P.C.; Hungerford, D.A. A minute chromosome in human chronic granulocotic leukemia. Science 1960, 132, 1497–1501. [Google Scholar]
- Ren, R. Mechanisms of bcr-abl in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Melo, J.V.; Barnes, D.J. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat. Rev. Cancer 2007, 7, 441–453. [Google Scholar] [CrossRef]
- Cilloni, D.; Saglio, G. Molecular pathways: Bcr-abl. Clin. Cancer Res. 2012, 18, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Perrotti, D.; Caligiuri, M.A. Understanding the molecular basis of imatinib mesylate therapy in chronic myelogenous leukemia and the related mechanisms of resistance. Commentary re: A. N. Mohamed et al. The effect of imatinib mesylate on patients with philadelphia chromosome-positive chronic myeloid leukemia with secondary chromosomal aberrations. Clin. Cancer res., 9: 1333–1337, 2003. Clin. Cancer Res. 2003, 9, 1248–1252. [Google Scholar] [PubMed]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the bcr-abl tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Rea, D.; Etienne, G.; Nicolini, F.; Cony-Makhoul, P.; Johnson-Ansah, H.; Legros, L.; Huguet, F.; Tulliez, M.; Gardembas, M.; Bouabdallah, K.; et al. First-line imatinib mesylate in patients with newly diagnosed accelerated phase-chronic myeloid leukemia. Leukemia 2012, 26, 2254–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottschalk, S.; Anderson, N.; Hainz, C.; Eckhardt, S.G.; Serkova, N.J. Imatinib (sti571)-mediated changes in glucose metabolism in human leukemia bcr-abl-positive cells. Clin. Cancer Res. 2004, 10, 6661–6668. [Google Scholar] [CrossRef] [PubMed]
- Kominsky, D.J.; Klawitter, J.; Brown, J.L.; Boros, L.G.; Melo, J.V.; Eckhardt, S.G.; Serkova, N.J. Abnormalities in glucose uptake and metabolism in imatinib-resistant human bcr-abl-positive cells. Clin. Cancer Res. 2009, 15, 3442–3450. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Chen, J. Post-translational modifications and the warburg effect. Oncogene 2014, 33, 4279–4285. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Kang, S.; Vander Heiden, M.G.; Chung, T.W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine phosphorylation inhibits pkm2 to promote the warburg effect and tumor growth. Sci. Signal 2009, 2, ra73. [Google Scholar] [CrossRef] [PubMed]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. Pkm2, cancer metabolism, and the road ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The m2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase m2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. Erk1/2-dependent phosphorylation and nuclear translocation of pkm2 promotes the warburg effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Prakasam, G.; Iqbal, M.A.; Bamezai, R.N.K.; Mazurek, S. Posttranslational modifications of pyruvate kinase m2: Tweaks that benefit cancer. Front Oncol. 2018, 8, 22. [Google Scholar] [CrossRef]
- De Rosa, V.; Iommelli, F.; Monti, M.; Fonti, R.; Votta, G.; Stoppelli, M.P.; Del Vecchio, S. Reversal of warburg effect and reactivation of oxidative phosphorylation by differential inhibition of egfr signaling pathways in non-small cell lung cancer. Clin. Cancer Res. 2015, 21, 5110–5120. [Google Scholar] [CrossRef]
- Alvarez-Calderon, F.; Gregory, M.A.; Pham-Danis, C.; DeRyckere, D.; Stevens, B.M.; Zaberezhnyy, V.; Hill, A.A.; Gemta, L.; Kumar, A.; Kumar, V.; et al. Tyrosine kinase inhibition in leukemia induces an altered metabolic state sensitive to mitochondrial perturbations. Clin. Cancer Res. 2015, 21, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Iommelli, F.; De Rosa, V.; Gargiulo, S.; Panico, M.; Monti, M.; Greco, A.; Gramanzini, M.; Ortosecco, G.; Fonti, R.; Brunetti, A.; et al. Monitoring reversal of met-mediated resistance to egfr tyrosine kinase inhibitors in non-small cell lung cancer using 3’-deoxy-3’-[18f]-fluorothymidine positron emission tomography. Clin. Cancer Res. 2014, 20, 4806–4815. [Google Scholar] [CrossRef] [PubMed]
- Monti, M.; Iommelli, F.; De Rosa, V.; Carriero, M.V.; Miceli, R.; Camerlingo, R.; Di Minno, G.; Del Vecchio, S. Integrin-dependent cell adhesion to neutrophil extracellular traps through engagement of fibronectin in neutrophil-like cells. PLoS ONE 2017, 12, e0171362. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Rosa, V.; Monti, M.; Terlizzi, C.; Fonti, R.; Del Vecchio, S.; Iommelli, F. Coordinate Modulation of Glycolytic Enzymes and OXPHOS by Imatinib in BCR-ABL Driven Chronic Myelogenous Leukemia Cells. Int. J. Mol. Sci. 2019, 20, 3134. https://doi.org/10.3390/ijms20133134
De Rosa V, Monti M, Terlizzi C, Fonti R, Del Vecchio S, Iommelli F. Coordinate Modulation of Glycolytic Enzymes and OXPHOS by Imatinib in BCR-ABL Driven Chronic Myelogenous Leukemia Cells. International Journal of Molecular Sciences. 2019; 20(13):3134. https://doi.org/10.3390/ijms20133134
Chicago/Turabian StyleDe Rosa, Viviana, Marcello Monti, Cristina Terlizzi, Rosa Fonti, Silvana Del Vecchio, and Francesca Iommelli. 2019. "Coordinate Modulation of Glycolytic Enzymes and OXPHOS by Imatinib in BCR-ABL Driven Chronic Myelogenous Leukemia Cells" International Journal of Molecular Sciences 20, no. 13: 3134. https://doi.org/10.3390/ijms20133134
APA StyleDe Rosa, V., Monti, M., Terlizzi, C., Fonti, R., Del Vecchio, S., & Iommelli, F. (2019). Coordinate Modulation of Glycolytic Enzymes and OXPHOS by Imatinib in BCR-ABL Driven Chronic Myelogenous Leukemia Cells. International Journal of Molecular Sciences, 20(13), 3134. https://doi.org/10.3390/ijms20133134