The Absence of Endothelial Sodium Channel α (αENaC) Reduces Renal Ischemia/Reperfusion Injury

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
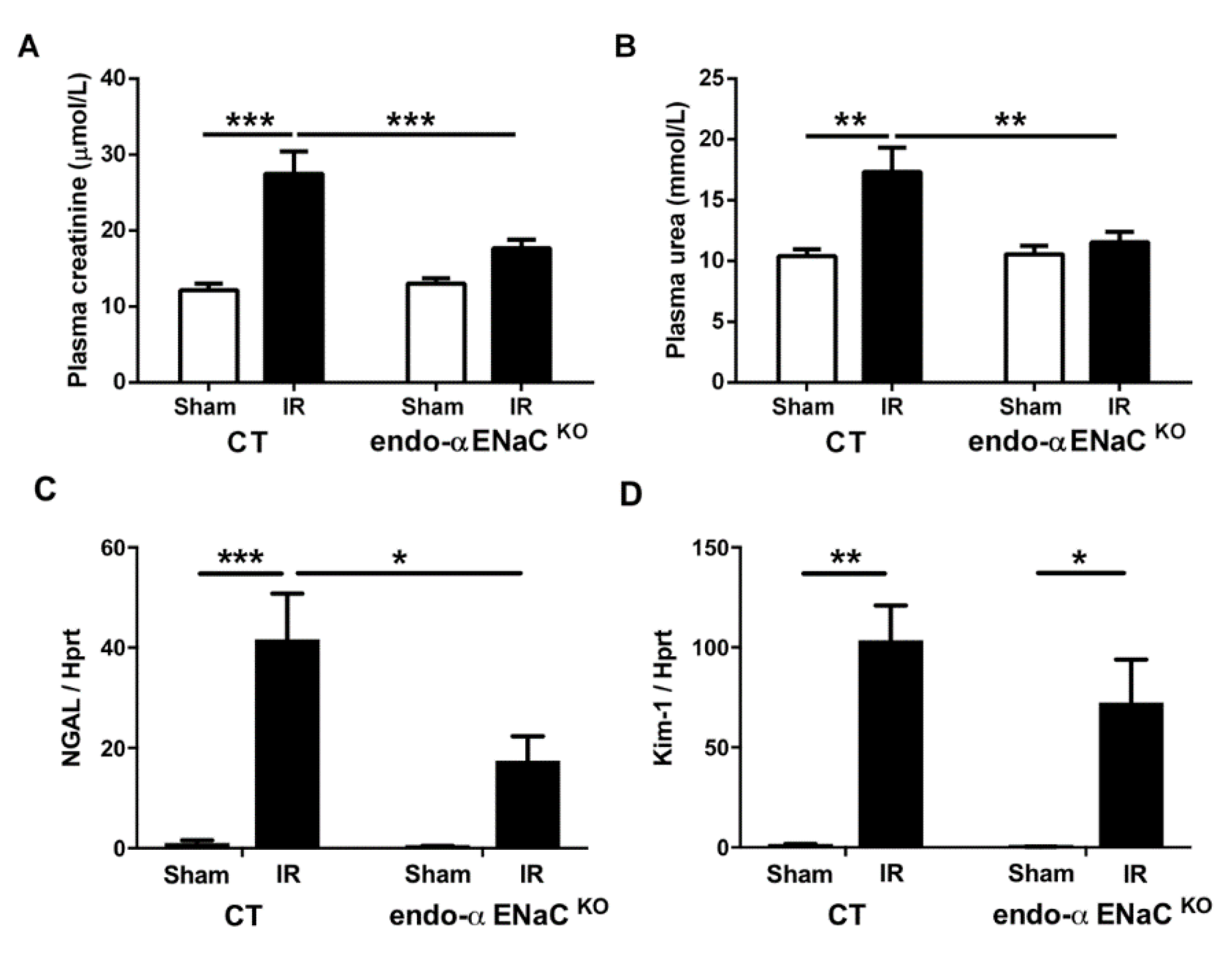
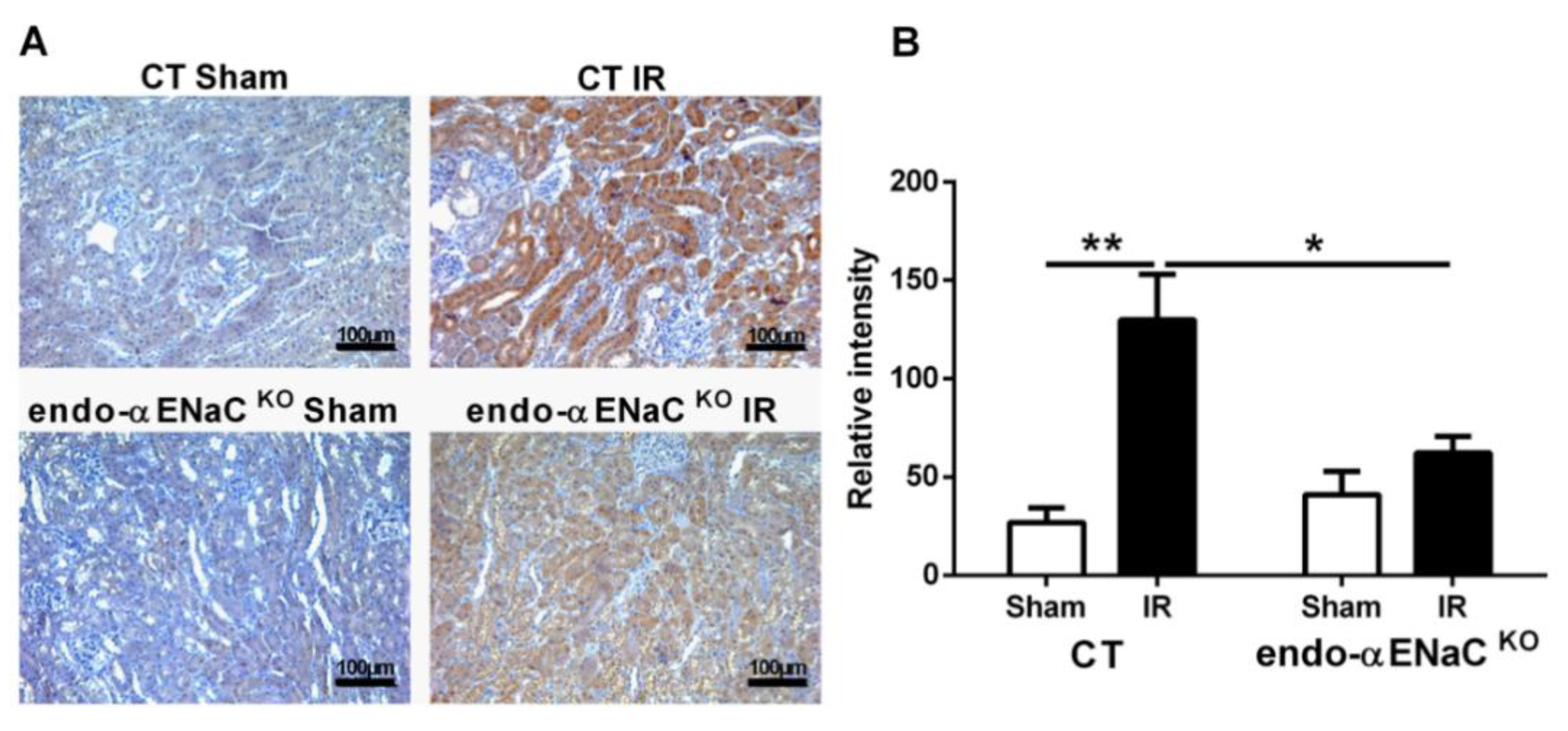
2.1. The Endothelial αENaC Deficiency Reduces Ischemic AKI Severity
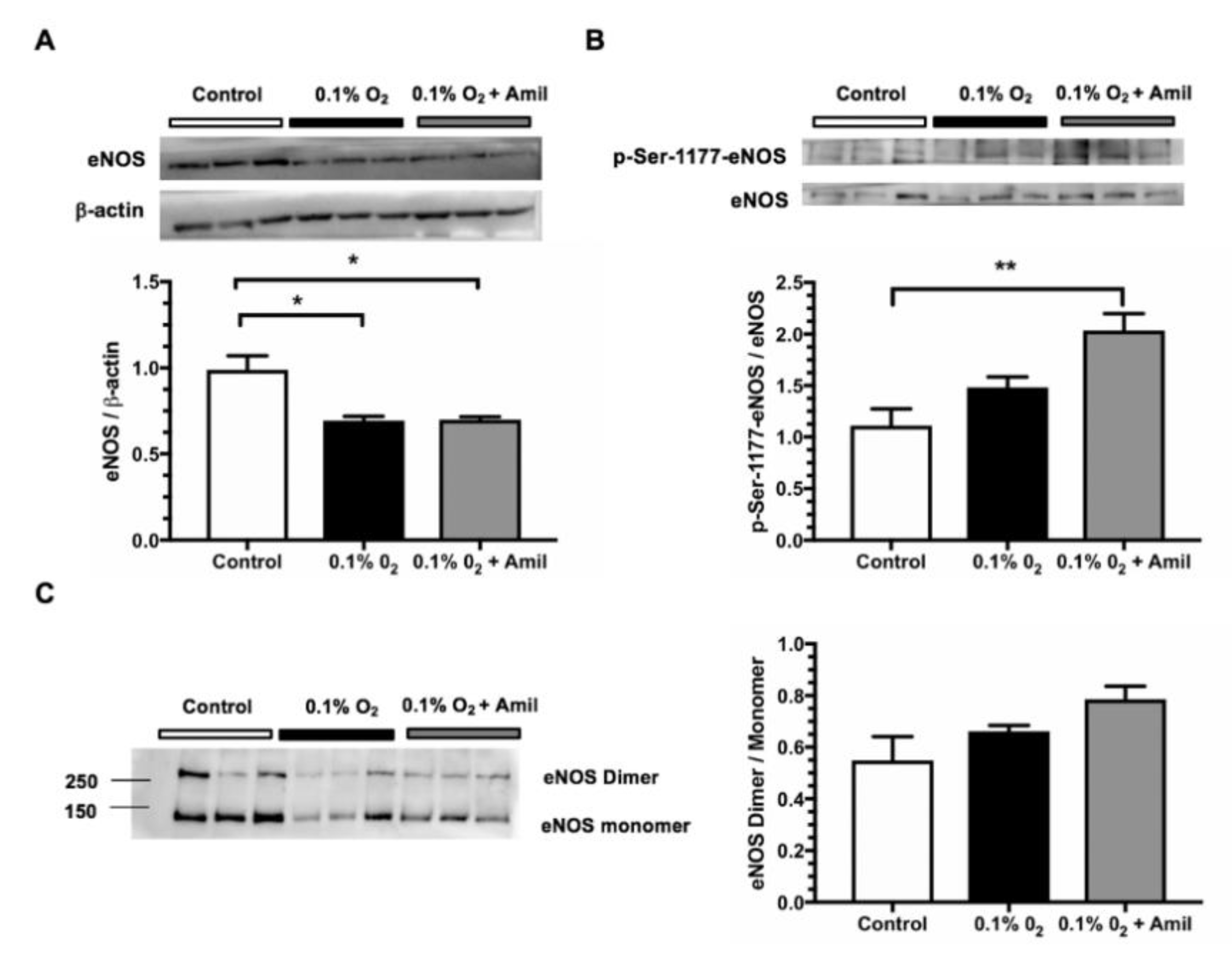
2.2. The Mice with αENaC Deficiency Recover Faster from the Hypoxia Induced by IR
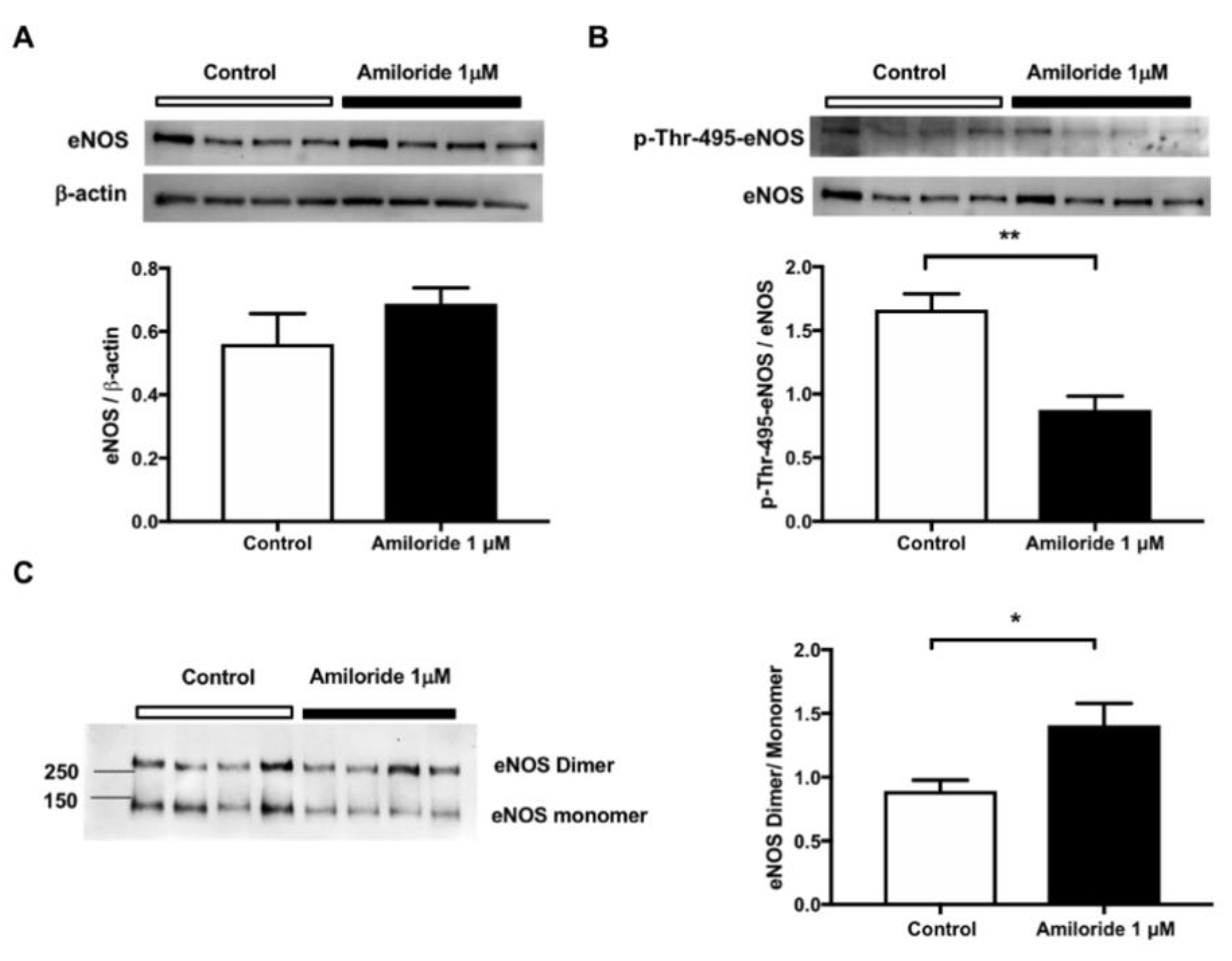
2.3. Pharmacological ENaC Inhibition Promotes eNOS Activation in Human Endothelial Cells
3. Discussion
4. Materials and Methods
4.1. Mouse Models
4.2. Renal Ischemia/Reperfusion Model
4.3. Gene Expression Analysis
4.4. Kidney Histology and Tubular Injury Quantification
4.5. Hypoxia Quantification
4.6. Human Endothelial Cell Culture and ENaC Inhibition
4.7. Western Blot Analysis
4.8. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ENaC | Epithelial sodium channel |
| αENaC | Epithelial sodium channel alpha subunit |
| NO | Nitric oxide |
| eNOS | Endothelial nitric oxide synthase |
| IR | Ischemia / reperfusion |
| Kim-1 | Kidney injury molecule-1 |
| NGAL | Neutrophil gelatinase associated lipocalin |
| Hsp90 | Heat shock protein 90 |
| AKI | Acute kidney injury |
| CKD | Chronic kidney disease |
| CT | Control littermates |
| endoαENaCKO | Epithelial sodium channel alpha subunit knockout in endothelial cells |
References
- Lewington, A.J.; Cerda, J.; Mehta, R.L. Raising awareness of acute kidney injury: A global perspective of a silent killer. Kidney Int. 2013, 84, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.L.; Cerda, J.; Burdmann, E.A.; Tonelli, M.; Garcia-Garcia, G.; Jha, V.; Susantitaphong, P.; Rocco, M.; Vanholder, R.; Sever, M.S.; et al. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): A human rights case for nephrology. Lancet 2015, 385, 2616–2643. [Google Scholar] [CrossRef]
- Zuk, A.; Palevsky, P.M.; Fried, L.; Harrell, F.E., Jr.; Khan, S.; McKay, D.B.; Devey, L.; Chawla, L.; de Caestecker, M.; Kaufman, J.S.; et al. Overcoming Translational Barriers in Acute Kidney Injury: A Report from an NIDDK Workshop. Clin. J. Am. Soc. Nephrol. 2018, 13, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.A.; Fisher, C.J.; Molitoris, B.A. Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int. 2002, 62, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Maringer, K.; Sims-Lucas, S. The multifaceted role of the renal microvasculature during acute kidney injury. Pediatr. Nephrol. 2016, 31, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Molitoris, B.A. Renal endothelial injury and microvascular dysfunction in acute kidney injury. Semin. Nephrol. 2015, 35, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Goligorsky, M.S.; Brodsky, S.V.; Noiri, E. NO bioavailability, endothelial dysfunction, and acute renal failure: New insights into pathophysiology. Semin. Nephrol. 2004, 24, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Salom, M.G.; Ceron, S.N.; Rodriguez, F.; Lopez, B.; Hernandez, I.; Martinez, J.G.; Losa, A.M.; Fenoy, F.J. Heme oxygenase-1 induction improves ischemic renal failure: Role of nitric oxide and peroxynitrite. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3542–H3549. [Google Scholar] [CrossRef]
- Kwon, O.; Hong, S.M.; Ramesh, G. Diminished NO generation by injured endothelium and loss of macula densa nNOS may contribute to sustained acute kidney injury after ischemia-reperfusion. Am. J. Physiol. Renal. Physiol. 2009, 296, F25–F33. [Google Scholar] [CrossRef]
- Yamasowa, H.; Shimizu, S.; Inoue, T.; Takaoka, M.; Matsumura, Y. Endothelial nitric oxide contributes to the renal protective effects of ischemic preconditioning. J. Pharmacol. Exp. Ther. 2005, 312, 153–159. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Perez-Villalva, R.; Ortega, J.A.; Uribe, N.; Gamba, G.; Cortes-Gonzalez, C.; Bobadilla, N.A. Intra-renal transfection of heat shock protein 90 alpha or beta (Hsp90alpha or Hsp90beta) protects against ischemia/reperfusion injury. Nephrol. Dial. Transplant. 2014, 29, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, K.F.; Sandin, J.; Gustafsson, L.E.; Frithiof, R. The novel nitric oxide donor PDNO attenuates ovine ischemia-reperfusion induced renal failure. Intensive Care Med. Exp. 2017, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Chimal, J.; Prince, S.; Fadel, F.; El Moghrabi, S.; Warnock, D.G.; Kolkhof, P.; Jaisser, F. Sulfenic Acid Modification of Endothelin B Receptor is Responsible for the Benefit of a Nonsteroidal Mineralocorticoid Receptor Antagonist in Renal Ischemia. J. Am. Soc. Nephrol. 2016, 27, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Kucuk, A.; Yucel, M.; Erkasap, N.; Tosun, M.; Koken, T.; Ozkurt, M.; Erkasap, S. The effects of PDE5 inhibitory drugs on renal ischemia/reperfusion injury in rats. Mol. Biol. Rep. 2012, 39, 9775–9782. [Google Scholar] [CrossRef] [PubMed]
- Milsom, A.B.; Patel, N.S.; Mazzon, E.; Tripatara, P.; Storey, A.; Mota-Filipe, H.; Sepodes, B.; Webb, A.J.; Cuzzocrea, S.; Hobbs, A.J.; et al. Role for endothelial nitric oxide synthase in nitrite-induced protection against renal ischemia-reperfusion injury in mice. Nitric Oxide 2010, 22, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.E.; Jeong, J.Y.; Lim, B.J.; Chung, S.; Chang, Y.K.; Lee, S.J.; Na, K.R.; Kim, S.Y.; Shin, Y.T.; Lee, K.W. Pretreatment of sildenafil attenuates ischemia-reperfusion renal injury in rats. Am. J. Physiol. Renal. Physiol. 2009, 297, F362–F370. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Berra, L.; Rezoagli, E.; Yu, B.; Dong, H.; Yu, S.; Hou, L.; Chen, M.; Chen, W.; Wang, H.; et al. Nitric Oxide Decreases Acute Kidney Injury and Stage 3 Chronic Kidney Disease after Cardiac Surgery. Am. J. Respir. Crit. Care Med. 2018, 198, 1279–1287. [Google Scholar] [CrossRef]
- Siragusa, M.; Fleming, I. The eNOS signalosome and its link to endothelial dysfunction. Pflugers Arch. 2016, 468, 1125–1137. [Google Scholar] [CrossRef]
- Sessa, W.C. eNOS at a glance. J. Cell Sci. 2004, 117, 2427–2429. [Google Scholar] [CrossRef]
- Fels, J.; Callies, C.; Kusche-Vihrog, K.; Oberleithner, H. Nitric oxide release follows endothelial nanomechanics and not vice versa. Pflugers Arch. 2010, 460, 915–923. [Google Scholar] [CrossRef]
- Kusche-Vihrog, K.; Schmitz, B.; Brand, E. Salt controls endothelial and vascular phenotype. Pflugers Arch. 2015, 467, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Warnock, D.G.; Kusche-Vihrog, K.; Tarjus, A.; Sheng, S.; Oberleithner, H.; Kleyman, T.R.; Jaisser, F. Blood pressure and amiloride-sensitive sodium channels in vascular and renal cells. Nat. Rev. Nephrol. 2014, 10, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meng, F.; Mohan, S.; Champaneri, B.; Gu, Y. Functional ENaC channels expressed in endothelial cells: A new candidate for mediating shear force. Microcirculation 2009, 16, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Liang, S.; Wang, S.; Tang, C.; Yao, B.; Wan, W.; Zhang, H.; Jiang, H.; Ahmed, A.; Zhang, Z.; et al. Role of epithelial Na+ channels in endothelial function. J. Cell Sci. 2016, 129, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Kusche-Vihrog, K.; Sobczak, K.; Bangel, N.; Wilhelmi, M.; Nechyporuk-Zloy, V.; Schwab, A.; Schillers, H.; Oberleithner, H. Aldosterone and amiloride alter ENaC abundance in vascular endothelium. Pflugers Arch. 2008, 455, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Drummond, H.A.; Grifoni, S.C.; Jernigan, N.L. A new trick for an old dogma: ENaC proteins as mechanotransducers in vascular smooth muscle. Physiology 2008, 23, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Kusche-Vihrog, K.; Jeggle, P.; Oberleithner, H. The role of ENaC in vascular endothelium. Pflugers Arch. 2014, 466, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Jeggle, P.; Callies, C.; Tarjus, A.; Fassot, C.; Fels, J.; Oberleithner, H.; Jaisser, F.; Kusche-Vihrog, K. Epithelial sodium channel stiffens the vascular endothelium in vitro and in Liddle mice. Hypertension 2013, 61, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.R.; Venegas, F.; Gonzalez, M.; Andres, S.; Vallejos, C.; Riquelme, G.; Sierralta, J.; Michea, L. Endothelial epithelial sodium channel inhibition activates endothelial nitric oxide synthase via phosphoinositide 3-kinase/Akt in small-diameter mesenteric arteries. Hypertension 2009, 53, 1000–1007. [Google Scholar] [CrossRef]
- Tarjus, A.; Maase, M.; Jeggle, P.; Martinez-Martinez, E.; Fassot, C.; Loufrani, L.; Henrion, D.; Hansen, P.B.L.; Kusche-Vihrog, K.; Jaisser, F. The endothelial alphaENaC contributes to vascular endothelial function in vivo. PLoS ONE 2017, 12, e0185319. [Google Scholar] [CrossRef]
- Fels, J.; Oberleithner, H.; Kusche-Vihrog, K. Menage a trois: Aldosterone, sodium and nitric oxide in vascular endothelium. Biochim. Biophys. Acta 2010, 1802, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Kusche-Vihrog, K.; Callies, C.; Fels, J.; Oberleithner, H. The epithelial sodium channel (ENaC): Mediator of the aldosterone response in the vascular endothelium? Steroids 2010, 75, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Habibi, J.; Aroor, A.R.; Hill, M.A.; Yang, Y.; Whaley-Connell, A.; Jaisser, F.; Sowers, J.R. Epithelial Sodium Channel in Aldosterone-Induced Endothelium Stiffness and Aortic Dysfunction. Hypertension 2018, 72, 731–738. [Google Scholar] [CrossRef]
- Wang, Z.R.; Liu, H.B.; Sun, Y.Y.; Hu, Q.Q.; Li, Y.X.; Zheng, W.W.; Yu, C.J.; Li, X.Y.; Wu, M.M.; Song, B.L.; et al. Dietary salt blunts vasodilation by stimulating epithelial sodium channels in endothelial cells from salt-sensitive Dahl rats. Br. J. Pharmacol. 2018, 175, 1305–1317. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.B.; Zhang, J.; Sun, Y.Y.; Li, X.Y.; Jiang, S.; Liu, M.Y.; Shi, J.; Song, B.L.; Zhao, D.; Ma, H.P.; et al. Dietary salt regulates epithelial sodium channels in rat endothelial cells: Adaptation of vasculature to salt. Br. J. Pharmacol. 2015, 172, 5634–5646. [Google Scholar] [CrossRef] [PubMed]
- Sternak, M.; Bar, A.; Adamski, M.G.; Mohaissen, T.; Marczyk, B.; Kieronska, A.; Stojak, M.; Kus, K.; Tarjus, A.; Jaisser, F.; et al. The Deletion of Endothelial Sodium Channel alpha (alphaENaC) Impairs Endothelium-Dependent Vasodilation and Endothelial Barrier Integrity in Endotoxemia in Vivo. Front. Pharmacol. 2018, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Ashley, Z.; Mugloo, S.; McDonald, F.J.; Fronius, M. Epithelial Na(+) channel differentially contributes to shear stress-mediated vascular responsiveness in carotid and mesenteric arteries from mice. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H1022–H1032. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lemus, L.A.; Aroor, A.R.; Ramirez-Perez, F.I.; Jia, G.; Habibi, J.; DeMarco, V.G.; Barron, B.; Whaley-Connell, A.; Nistala, R.; Sowers, J.R. Amiloride Improves Endothelial Function and Reduces Vascular Stiffness in Female Mice Fed a Western Diet. Front. Physiol. 2017, 8, 456. [Google Scholar] [CrossRef]
- Jia, G.; Habibi, J.; Aroor, A.R.; Hill, M.A.; DeMarco, V.G.; Lee, L.E.; Ma, L.; Barron, B.J.; Whaley-Connell, A.; Sowers, J.R. Enhanced endothelium epithelial sodium channel signaling prompts left ventricular diastolic dysfunction in obese female mice. Metabolism 2018, 78, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Rubera, I.; Loffing, J.; Palmer, L.G.; Frindt, G.; Fowler-Jaeger, N.; Sauter, D.; Carroll, T.; McMahon, A.; Hummler, E.; Rossier, B.C. Collecting duct-specific gene inactivation of alphaENaC in the mouse kidney does not impair sodium and potassium balance. J. Clin. Investig. 2003, 112, 554–565. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarjus, A.; González-Rivas, C.; Amador-Martínez, I.; Bonnard, B.; López-Marure, R.; Jaisser, F.; Barrera-Chimal, J. The Absence of Endothelial Sodium Channel α (αENaC) Reduces Renal Ischemia/Reperfusion Injury. Int. J. Mol. Sci. 2019, 20, 3132. https://doi.org/10.3390/ijms20133132
Tarjus A, González-Rivas C, Amador-Martínez I, Bonnard B, López-Marure R, Jaisser F, Barrera-Chimal J. The Absence of Endothelial Sodium Channel α (αENaC) Reduces Renal Ischemia/Reperfusion Injury. International Journal of Molecular Sciences. 2019; 20(13):3132. https://doi.org/10.3390/ijms20133132
Chicago/Turabian StyleTarjus, Antoine, Cecilia González-Rivas, Isabel Amador-Martínez, Benjamin Bonnard, Rebeca López-Marure, Frédéric Jaisser, and Jonatan Barrera-Chimal. 2019. "The Absence of Endothelial Sodium Channel α (αENaC) Reduces Renal Ischemia/Reperfusion Injury" International Journal of Molecular Sciences 20, no. 13: 3132. https://doi.org/10.3390/ijms20133132
APA StyleTarjus, A., González-Rivas, C., Amador-Martínez, I., Bonnard, B., López-Marure, R., Jaisser, F., & Barrera-Chimal, J. (2019). The Absence of Endothelial Sodium Channel α (αENaC) Reduces Renal Ischemia/Reperfusion Injury. International Journal of Molecular Sciences, 20(13), 3132. https://doi.org/10.3390/ijms20133132