Identification of Novel Biomarkers of Homologous Recombination Defect in DNA Repair to Predict Sensitivity of Prostate Cancer Cells to PARP-Inhibitors

Abstract
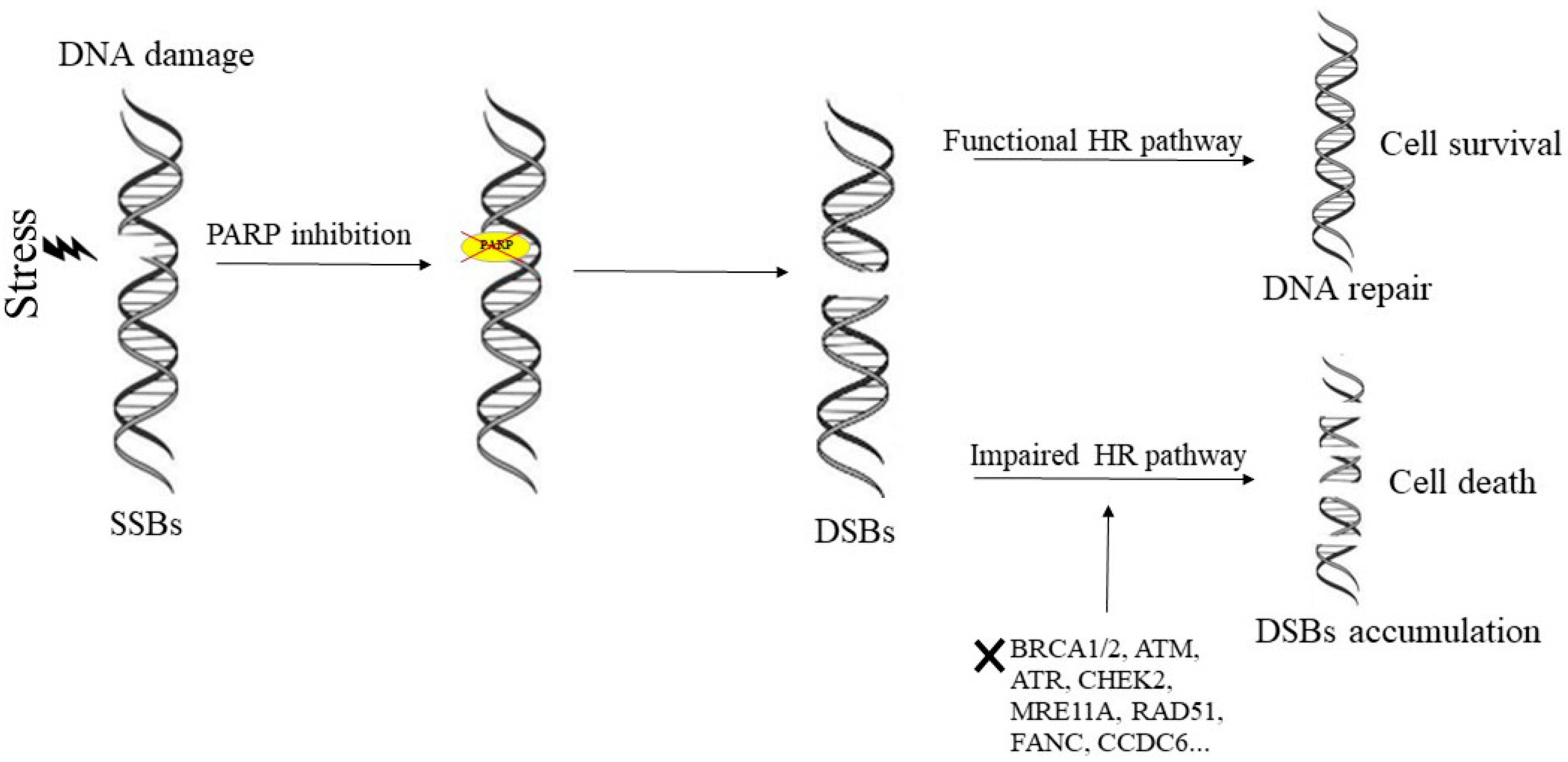
1. Mechanism of Action of PARP-Inhibitors and Rationale for Their Inclusion in Clinical Settings
2. Rationale for Use of Poly(ADP-Ribose) Polymerase Inhibitors in Treatment of Prostate Cancer
3. DNA Repair Deficiency and PARP-Inhibitors Response in Prostate Cancer
4. Ongoing Prostate Cancer Clinical Trials Involving PARP Inhibitors
5. Sensitivity to PARP-Inhibitors Induced in Prostate Cancer with Apparent Integrity of Homologous Recombination Machinery
6. Combinations of PARP-Inhibitors with Immune-Checkpoint Inhibitors
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| mCRPC | metastatic castration resistance prostate cancer |
| PARP | Poly (ADP-ribose) polymerase |
| DDR | DNA damage response and repair |
| FDA | food and drug administration |
| BRCA | Breast cancer |
| ATM | ataxia telengiectasia mutated |
| HR | homologous recombination |
| BER | base excision repair |
| NER | nucleotide excision repair |
| MMR | mismatch repair |
| NAD | nicotinamide adenine dinucleotide |
| SSBs | single strand breaks |
| DSBs | double strand breaks |
| NHEJ | non-homologous end joining |
| PCa | prostate cancer |
| ADT | androgen deprivation therapy |
| AR | androgen receptor |
| CRPC | castration resistant prostate cancer |
| PFS | progression free survival |
| OS | overall survival |
| FANCA | FA Complementation Group A |
| CHEK2 | checkpoint kinase 2 |
| MRE11 | meiotic recombination 11 homolog 1 |
| RAD51 | recombinase 51 |
| CDK12 | cyclin dependent Kinase 12 |
| PALB2 | Partner and localizer of BRCA2 |
| HDAC2 | Histone deacetylase 2 |
| MLH3 | MutL Homolog 3 |
| PTEN | Phosphatase and Tensin Homolog |
| ERG | ETS-Related Gene |
| CCDC6 | coiled coil domain containing 6 |
| FBXW7 | F-box/WD repeat-containing protein 7 |
| USP7 | Ubiquitin-specific-processing protease 7 (USP7) |
| LAPC | Locally, Advanced Prostate Cancer |
| rPC | Recurrent Prostate Cancer |
| mHSPC | Metastatic Hormone-Densitive Prostate Cancer |
| nmCRPC | Non Metastatic Castration-Resistant Prostate Cancer |
| DDRi | DNA Damage Response inhibitors |
| TMB | tumor mutational burden |
| MHC | major histocompatibility complex |
| STING | stimulator of interferon genes |
| PD-1 | Prorammed cell death protein 1 |
| PD-L1 | Ligand of PD-1 |
| NSCLC | Non-Small Cell Lung Cancer |
| HNSCC | Head and Neck Squamous Cell Carcinoma |
| NAMPT | Nicotinamide phosphorybosyl transferase |
| NMN | Nicotinamide mononucleotide |
References
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Barkauskaite, E.; Jankevicius, G.; Ahel, I. Structures and mechanisms of enzymes employed in the synthesis and degradation of PARP-dependent protein ADP-ribosylation. Mol. Cell 2015, 58, 935–946. [Google Scholar] [CrossRef]
- Lupo, B.; Trusolino, L. Inhibition of poly(ADP-ribosyl)ation in cancer: Old and new paradigms revisited. Biochim. Biophys. Acta 2014, 1846, 201–215. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Differential trapping of PARP1 and PARP2 by clinical PARP-inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Helleday, T. PARP-inhibitor receives FDA breakthrough therapy designation in castration resistant prostate cancer: Beyond germline BRCA mutations. Ann. Oncol. 2016, 27, 755–757. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef]
- Attard, G.; Parker, C.; Eeles, R.A.; Schröder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; de Bono, J.S. Prostate cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef]
- Gonthier, K.; Poluri, R.T.K.; Audet-Walsh, É. Functional genomic studies reveal the androgen receptor as a master regulator of cellular energy metabolism in prostate cancer. J. Steroid Biochem. Mol. Biol. 2019, 191, 105367. [Google Scholar] [CrossRef]
- Sridhar, S.S.; Freedland, S.J.; Gleave, M.E.; Higano, C.; Mulders, P.; Parker, C.; Sartor, O.; Saad, F. Castration-resistant prostate cancer: From new pathophysiology to new treatment. Eur. Urol. 2014, 65, 289–299. [Google Scholar] [CrossRef]
- Mostaghel, E.A. Abiraterone in the treatment of metastatic castration-resistant prostate cancer. Cancer Manag. Res. 2014, 6, 39–51. [Google Scholar] [CrossRef][Green Version]
- Lavaud, P.; Gravis, G.; Foulon, S.; Joly, F.; Oudard, S.; Priou, F.; Latorzeff, I.; Mourey, L.; Soulié, M.; Delva, R.; et al. Anticancer Activity and Tolerance of Treatments Received Beyond Progression in Men Treated Upfront with Androgen Deprivation Therapy with or Without Docetaxel for Metastatic Castration-naïve Prostate Cancer in the GETUG-AFU 15 Phase 3 Trial. Eur. Urol. 2018, 73, 696–703. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Gleave, M. Targeting the adaptive molecular landscape of castration-resistant prostate cancer. EMBO Mol. Med. 2015, 7, 878–894. [Google Scholar] [CrossRef]
- Lorente, D.; Mateo, J.; Perez-Lopez, R.; de Bono, J.S.; Attard, G. Sequencing of agents in castration-resistant prostate cancer. Lancet Oncol. 2015, 16, e279–e292. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Cato, L.; de Tribolet-Hardy, J.; Lee, I.; Rottenberg, J.T.; Coleman, I.; Melchers, D.; Houtman, R.; Xiao, T.; Li, W.; Uo, T.; et al. ARv7 represses tumor-suppressor genes in castration-resistant prostate cancer. Cancer Cell 2019, 35, 401–413. [Google Scholar] [CrossRef]
- Warner, E.W.; Yip, S.M.; Chi, K.N.; Wyatt, A.W. DNA repair defects in prostate cancer: Impact for screening, prognostication and treatment. BJU Int. 2018, 123, 769–776. [Google Scholar] [CrossRef]
- Akbari, M.R.; Wallis, C.J.; Toi, A.; Trachtenberg, J.; Sun, P.; Narod, S.A.; Nam, R.K. The impact of a BRCA2 mutation on mortality from screen-detected prostate cancer. Br. J. Cancer 2014, 111, 1238–1240. [Google Scholar] [CrossRef]
- Mateo, J.; de Bono, J.S. Targeting DNA damage response systems to impact cancer care. Curr. Probl. Cancer 2017, 41, 247–250. [Google Scholar] [CrossRef]
- Rescigno, P.; Chandler, R.; de Bono, J. Relevance of poly (ADP-ribose) polymerase inhibitors in prostate cancer. Curr. Opin Support Palliat Care 2018, 12, 339–343. [Google Scholar] [CrossRef]
- Marshall, C.H.; Fu, W.; Wang, H.; Baras, A.S.; Lotan, T.L.; Antonarakis, E.S. Prevalence of DNA repair gene mutations in localized prostate cancer according to clinical and pathologic features: Association of Gleason score and tumor stage. Prostate Cancer Prostatic Dis. 2019, 22, 59–65. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Kote-Jarai, Z.; Leongamornlert, D.; Saunders, E.; Tymrakiewicz, M.; Castro, E.; Mahmud, N.; Guy, M.; Edwards, S.; O’Brien, L.; Sawyer, E.; et al. BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: Implications for genetic testing in prostate cancer patients. Br. J. Cancer 2011, 105, 1230–1234. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Cerrato, A.; Morra, F.; Celetti, A. Use of poly ADP-ribose polymerase [PARP] inhibitors in cancer cells bearing DDR defects: The rationale for their inclusion in the clinic. J. Exp. Clin. Cancer Res. 2016, 35, 179. [Google Scholar] [CrossRef]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic Hallmarks of Localized, Non-Indolent Prostate Cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The Mutational Landscape of Lethal Castration-Resistant Prostate Cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Kumar, A.; White, T.A.; MacKenzie, A.P.; Clegg, N.; Lee, C.; Dumpit, R.F.; Coleman, I.; Ng, S.B.; Salipante, S.J.; Rieder, M.J.; et al. Exome Sequencing Identifies a Spectrum of Mutation Frequencies in Advanced and Lethal Prostate Cancers. Proc. Natl. Acad. Sci. USA 2011, 108, 17087–17092. [Google Scholar] [CrossRef]
- Castro, E.; Goh, C.; Olmos, D.; Saunders, E.; Leongamornlert, D.; Tymrakiewicz, M.; Mahmud, N.; Dadaev, T.; Govindasami, K.; Guy, M.; et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J. Clin. Oncol. 2013, 31, 1748–1757. [Google Scholar] [CrossRef]
- Mersch, J.; Jackson, M.A.; Park, M.; Nebgen, D.; Peterson, S.K.; Singletary, C.; Arun, B.K.; Litton, J.K. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 2015, 121, 269–275. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-repair defects and Olaparib in metastatic prostate cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- De Felice, F.; Tombolini, V.; Marampon, F.; Musella, A.; Marchetti, C. Defective DNA repair mechanisms in prostate cancer: Impact of Olaparib. Drug. Des. Devel Ther. 2017, 11, 547–552. [Google Scholar] [CrossRef]
- Cerrato, A.; Merolla, F.; Morra, F.; Celetti, A. CCDC6: The identity of a protein known to be partner in fusion. Int. J. Cancer 2018, 142, 1300–1308. [Google Scholar] [CrossRef]
- Morra, F.; Luise, C.; Visconti, R.; Staibano, S.; Merolla, F.; Ilardi, G.; Guggino, G.; Paladino, S.; Sarnataro, D.; Franco, R.; et al. New therapeutic perspectives in CCDC6 deficient lung cancer cells. Int. J. Cancer 2015, 136, 2146–2157. [Google Scholar] [CrossRef]
- Morra, F.; Luise, C.; Merolla, F.; Poser, I.; Visconti, R.; Ilardi, G.; Paladino, S.; Inuzuka, H.; Guggino, G.; Monaco, R.; et al. FBXW7 and USP7 regulate CCDC6 turnover during the cell cycle and affect cancer drugs susceptibility in NSCLC. Oncotarget 2017, 6, 12697–12709. [Google Scholar] [CrossRef]
- Malapelle, U.; Morra, F.; Ilardi, G.; Visconti, R.; Merolla, F.; Cerrato, A.; Napolitano, V.; Monaco, R.; Guggino, G.; Monaco, G.; et al. USP7 inhibitors, downregulating CCDC6, sensitize lung neuroendocrine cancer cells to PARP-inhibitor drugs. Lung Cancer 2017, 107, 41–49. [Google Scholar] [CrossRef]
- Morra, F.; Merolla, F.; Criscuolo, D.; Insabato, L.; Giannella, R.; Ilardi, G.; Cerrato, A.; Visconti, R.; Staibano, S.; Celetti, A. CCDC6 and USP7 expression levels suggest novel treatment options in high-grade urothelial bladder cancer. J. Exp. Clin. Cancer Res. 2019, 38, 90. [Google Scholar] [CrossRef]
- Morra, F.; Merolla, F.; Napolitano, V.; Ilardi, G.; Miro, C.; Paladino, S.; Staibano, S.; Cerrato, A.; Celetti, A. The combined effect of USP7 inhibitors and PARP-inhibitors in hormone-sensitive and castration-resistant prostate cancer cells. Oncotarget 2017, 8, 31815–31829. [Google Scholar] [CrossRef]
- Christenson, E.S.; Antonarakis, E.S. PARP-inhibitors for homologous recombination-deficient prostate cancer. Expert. Opin. Emerg. Drugs 2018, 23, 123–133. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN. 673 and comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic dissection of PARP1 trapping and the impact on in vivo tolerability and efficacy of PARP inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef]
- Shen, Y.; Aoyagi-scharber, M.; Wang, B. Trapping Poly (ADP-Ribose) Polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 446–457. [Google Scholar] [CrossRef]
- Sun, K.; Mikule, K.; Wang, Z.; Poon, G.; Vaidyanathan, A.; Smith, G.; Zhang, Z.Y.; Hanke, J.; Ramaswamy, S.; Wang, J. A comparative pharmacokinetic study of PARP inhibitors demonstrates favorable properties for Niraparib efficacy in preclinical tumor models. Oncotarget 2018, 9, 37080–37096. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP- ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchel, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malle, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Makvandi, M.; Pantel, A.; Schwartz, L.; Schubert, E.; Xu, K.; Hsieh, C.J.; Hou, C.; Kim, H.; Weng, C.C.; Winters, H.; et al. A PET imaging agent for evaluating PARP-1 expression in ovarian cancer. J. Clin Investig. 2018, 128, 2116–2126. [Google Scholar] [CrossRef]
- Antonarakis, E.S. Abiraterone plus Olaparib in prostate cancer: A new form of synthetic lethality? Lancet Oncol. 2018, 19, 860–861. [Google Scholar] [CrossRef]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signa 2017, 10, eaam7479. [Google Scholar] [CrossRef]
- Dhawan, M.; Ryan, C.J. BRCAness and prostate cancer: Diagnostic and therapeutic considerations. Prostate Cancer Prostatic Dis. 2018, 21, 488–498. [Google Scholar] [CrossRef]
- Hussain, M.; Daignault-Newton, S.; Twardowski, P.W.; Albany, C.; Stein, M.N.; Kunju, L.P. Targeting Androgen Receptor and DNA Repair in Metastatic Castration-Resistant Prostate Cancer: Results from NCI 9012. J. Clin. Oncol. 2018, 36, 991–999. [Google Scholar] [CrossRef]
- Knudsen, K.E.; Feng, F.Y. Expanding Role of Germline DNA Repair Alterations in Prostate Cancer Risk and Early Onset. Eur. Urol. 2019. [Google Scholar] [CrossRef]
- Giri, V.N.; Knudsen, K.E.; Kelly, W.K.; Abida, W.; Andriole, G.L.; Bangma, C.H.; Bekelman, J.E.; Benson, M.C.; Blanco, A.; Burnett, A.; et al. Role of Genetic Testing for Inherited Prostate Cancer Risk: Philadelphia Prostate Cancer Consensus Conference 2017. J. Clin. Oncol. 2018, 36, 414–424. [Google Scholar] [CrossRef]
- Ramakrishnan Geethakumari, P.; Schiewer, M.J.; Knudsen, K.E.; Kelly, W.K. PARP Inhibitors in Prostate Cancer. Curr. Treat. Options Oncol. 2017, 18, 37. [Google Scholar] [CrossRef]
- Clarke, N.; Wiechno, P.; Alekseev, B.; Sala, N.; Jones, R.; Kocak, I.; Chiuri, V.E.; Jassem, J.; Fléchon, A.; Redfern, C.; et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018, 19, 975–986. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Liang, C.; Wang, H.; Chen, Y.; Silberstein, J.L.; Piana, D.; Lai, Z.; Chen, Y.; et al. Germline DNA-repair Gene Mutations and Outcomes in Men with Metastatic Castration-resistant Prostate Cancer Receiving First-line Abiraterone and Enzalutamide. Eur. Urol. 2018, 74, 218–225. [Google Scholar] [CrossRef]
- Chen, S.T.; Okada, M.; Nakato, R.; Izumi, K.; Bando, M.; Shirahige, K. The Deubiquitinating Enzyme USP7 Regulates Androgen Receptor Activity by Modulating Its Binding to Chromatin. J. Biol. Chem. 2015, 290, 21713–21723. [Google Scholar] [CrossRef]
- Leone, V.; Mansueto, G.; Pierantoni, G.M.; Tornincasa, M.; Merolla, F.; Cerrato, A.; Santoro, M.; Grieco, M.; Scaloni, A.; Celetti, A. CCDC6 represses CREB1 activity by recruiting histone deacetylase 1 and protein phosphatase 1. Oncogene 2010, 29, 4341–4351. [Google Scholar] [CrossRef]
- Leone, V.; Langella, C.; Esposito, F.; Arra, C.; Palma, G.; Rea, D.; Paciello, O.; Merolla, F.; De Biase, D.; Papparella, S. Ccdc6 knock-in mice develop thyroid hyperplasia associated to an enhanced CREB1 activity. Oncotarget 2015, 6, 15628–15638. [Google Scholar] [CrossRef]
- Rawat, R.; Starczynowski, D.T.; Ntziachristos, P. Nuclear deubiquitination in the spotlight: The multifaceted nature of USP7 biology in disease. Curr. Opin. Cell Biol. 2019, 58, 85–94. [Google Scholar] [CrossRef]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, K.G.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef]
- Abramson, H.N. The Multiple Myeloma Drug Pipeline-2018: A Review of Small Molecules and Their Therapeutic Targets. Clin. Lymphoma Myeloma Leuk. 2018, 18, 611–627. [Google Scholar] [CrossRef]
- Feiersinger, G.E.; Trattnig, K.; Leitner, P.D.; Guggenberger, F.; Oberhuber, A.; Peer, S.; Hermann, M.; Skvortsova, I.; Vrbkova, J.; Bouchal, J.; et al. Olaparib is effective in combination with, and as maintenance therapy after, first-line endocrine therapy in prostate cancer cells. Mol. Oncol. 2018, 12, 561–576. [Google Scholar] [CrossRef]
- Criscuolo, D.; Morra, F.; Giannella, R.; Visconti, R.; Cerrato, A.; Celetti, A. New combinatorial strategies to improve the PARP-inhibitors efficacy in the urothelial bladder Cancer treatment. J. Exp. Clin Cancer Res. 2019, 38, 91. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef]
- McGrail, D.J.; Federico, L.; Li, Y.; Dai, H.; Lu, Y.; Mills, G.B.; Yi, S.; Lin, S.Y.; Sahni, N. Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers. Nat. Commun. 2018, 9, 1317. [Google Scholar] [CrossRef]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling by S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2016, 109. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 2012, 24, 207–212. [Google Scholar] [CrossRef]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L.; et al. Activity of durvalumab plus Olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. Immunother. Cancer 2018, 6, 141. [Google Scholar] [CrossRef] [PubMed]
- Slovin, S.F.; Higano, C.S.; Hamid, O.; Tejwani, S.; Harzstark, A.; Alumkal, J.J.; Scher, H.I.; Chin, K.; Gagnier, P.; McHenry, M.B.; et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: Results from an open-label, multicenter phase I/II study. Ann. Oncol. 2013, 24, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; van den Eertwegh, A.J.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef]
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients with Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2017, 35, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.R.; Massard, C.; Ott, P.A.; Haas, N.B.; Lopez, J.S.; Ejadi, S.; Wallmark, J.M.; Keam, B.; Delord, J.P.; Aggarwal, R.; et al. Pembrolizumab for patients with advanced prostate adenocarcinoma: Preliminary results from the KEYNOTE-028 study. Ann. Oncol. 2018, 29, 1807–1813. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.N.; Alumkal, J.J.; Drake, C.G.; Thomas, G.V.; Redmond, W.L.; Farhad, M.; Cetnar, J.P.; Ey, F.S.; Bergan, R.C.; Slottke, R.; et al. Early evidence of anti-PD-1 activity in enzalutamide-resistant prostate cancer. Oncotarget 2016, 7, 52810–52817. [Google Scholar] [CrossRef] [PubMed]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jiménez-García, M.P.; Muñoz-Galvan, S.; Carnero, A. NAMPT Is a Potent Oncogene in Colon Cancer Progression that Modulates Cancer Stem Cell Properties and Resistance to Therapy through Sirt1 and PARP. Clin. Cancer Res. 2018, 24, 1202–1215. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Antonarakis, E.S. PARP inhibition-not all gene mutations are created equal. Nat. Rev. Urol. 2019, 16, 4–6. [Google Scholar] [CrossRef]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene | Functions in DNA Repair | Evidence for PARP Sensitivity in Prostate Cancer Patients | Reference |
|---|---|---|---|
| BRCA1 | Phosphoprotein that assists in 5′ to 3′ resection of DSBs, loading of RAD51 | NCT01682772 | [34] |
| BRCA2 | Phosphoprotein that assists with RAD51 loading on DNA | NCT01682772 | [34] |
| ATM | Serine/threonine protein kinase involved in repair of DSBs | NCT01682772 | [34] |
| FANC A/F | DNA repair protein involved in a post-replication repair | NCT01682772 | [34] |
| CHK2 | Serine/threonine protein kinase involved in repair of DSBs | NCT01682772 | [34] |
| RAD51B/C | Assist the recruitment, stabilization, and loading of RAD51 | NCT01682772 | [34] |
| CDK12 | Cyclin-dependent kinase that regulates the expression of genes involved in DNA repair | NCT01682772 | [34] |
| PARP Inhibitor | ClinicalTrials.gov Identifier | Population | DNA Repair Genes | Treatment |
|---|---|---|---|---|
| Niraparib | NCT02854436 Galahad | mCRPC | BRCA1/2, ATM, FANCA, PALB2, CHEK2, BRIP1, or HDAC2 | Niraparib (single-arm study) |
| Olaparib | NCT03432897 BrUOG 337 | LAPC | BRCA1, BRCA 2, ATM, CHEK1, CHEK2, FANCONIS ANEMIA (FANCL), HDAC2, PALB2, BARD1, BRIP1, CDK12, PPP2R2A, RAD51B, RAD51C, RAD51D, RAD54L | Olaparib prior to prostatectomy (single-arm study) |
| NCT03012321 | mCRPC | ATM, BRCA1, BRCA2, FANCA, PALB2, RAD51, ERCC3, MRE11, NBN, MLH3, CDK12, CHEK2, HDAC2, ATR, PMS2, GEN1, MSH2, MSH6, BRIP1, or FAM175A | Abiraterone/Prednisone, Olaparib or Abiraterone/Prednisone + Olaparib | |
| NCT03047135 | rPC | ATM, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D | Olaparib following prostatectomy (single-arm study) | |
| Rucaparib | NCT03413995 TRIUMPH | mHSPC | BRCA1, BRCA2, ATM, CHEK2, NBN, RAD50, RAD51C, RAD51D, PALB2, MRE11, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM | Rucaparib (single-arm study) |
| NCT02952534 TRITON2 | mCRPC | BRCA1, BRCA2, ATM, BARD1, BRIP1, CDK12 CHEK2, FANCA, NBN PALB2, RAD51, RAD51B RAD51C, RAD51D, RAD54L | Rucaparib (single-arm study) | |
| NCT02975934 TRITON3 | mCRPC | BRCA1, BRCA2, ATM | Rucaparib vs abiraterone, enzalutamide or docetaxel | |
| NCT03533946 ROAR | nmCRPC | ATM, ATR, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, ERCC3, FAM175A, FANCA, FANCL, GEN1, HDAC2, MLH1, MRE11, NBN, PALB2, PPP2R2A, RAD51, RAD54L | Rucaparib (single-arm study) | |
| Talazoparib | NCT03148795 | mCRPC | BRCA1, BRCA2 | Talazoparib (single-arm study) |
| ClinicalTrials.gov Identifier | Patients | Immune Checkpoint Inhibitors | Reference |
|---|---|---|---|
| NCT00323882 | Metastatic hormone refractory prostate cancer | Ipilimumab | [82] |
| NCT00861614 | Castration Resistant Prostate Cancer | Ipilimumab | [83] |
| NCT01057810 | Metastatic Chemotherapy-Naïve Castration Resistant Prostate Cancer | Ipilimumab | [84] |
| NCT02054806 | Advanced Adenocarcinoma | Pembrolizumab | [85] |
| NCT02312557 | Metastatic Castration Resistant Prostate Cancer | Pembrolizumab | [86] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Criscuolo, D.; Morra, F.; Giannella, R.; Cerrato, A.; Celetti, A. Identification of Novel Biomarkers of Homologous Recombination Defect in DNA Repair to Predict Sensitivity of Prostate Cancer Cells to PARP-Inhibitors. Int. J. Mol. Sci. 2019, 20, 3100. https://doi.org/10.3390/ijms20123100
Criscuolo D, Morra F, Giannella R, Cerrato A, Celetti A. Identification of Novel Biomarkers of Homologous Recombination Defect in DNA Repair to Predict Sensitivity of Prostate Cancer Cells to PARP-Inhibitors. International Journal of Molecular Sciences. 2019; 20(12):3100. https://doi.org/10.3390/ijms20123100
Chicago/Turabian StyleCriscuolo, Daniela, Francesco Morra, Riccardo Giannella, Aniello Cerrato, and Angela Celetti. 2019. "Identification of Novel Biomarkers of Homologous Recombination Defect in DNA Repair to Predict Sensitivity of Prostate Cancer Cells to PARP-Inhibitors" International Journal of Molecular Sciences 20, no. 12: 3100. https://doi.org/10.3390/ijms20123100
APA StyleCriscuolo, D., Morra, F., Giannella, R., Cerrato, A., & Celetti, A. (2019). Identification of Novel Biomarkers of Homologous Recombination Defect in DNA Repair to Predict Sensitivity of Prostate Cancer Cells to PARP-Inhibitors. International Journal of Molecular Sciences, 20(12), 3100. https://doi.org/10.3390/ijms20123100