Mast Cells in Viral, Bacterial, and Fungal Infection Immunity


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mast Cells and Flavivirus Infections
2.1. Beneficial Effects of Mast Cells in DENV Infections
2.2. Detrimental Effects of Mast Cells in DENV Infections
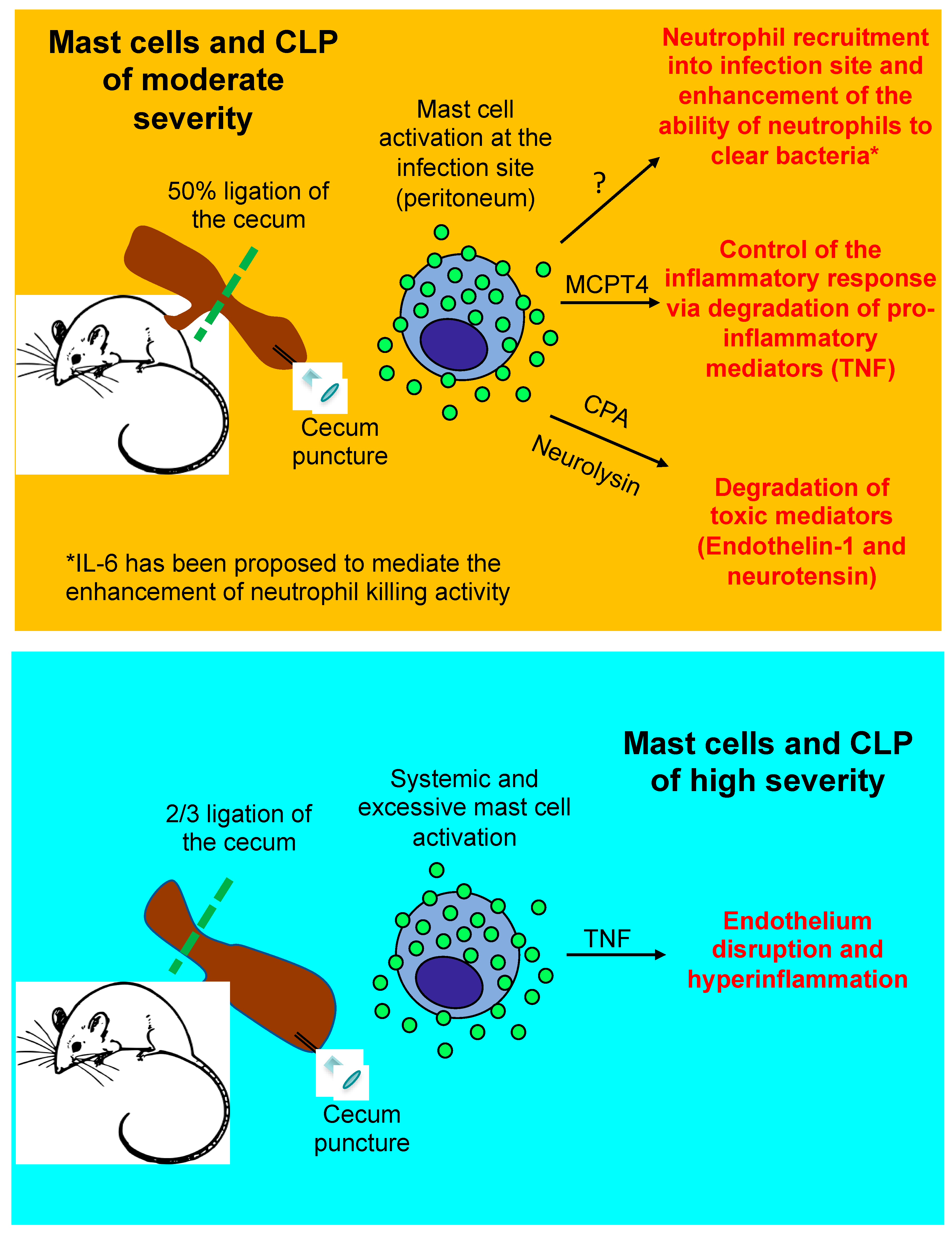
3. Mast Cells in the Cecal Ligation and Puncture (CLP) Model of Sepsis
3.1. Beneficial Role of Mast Cells in Moderately Severe CLP
3.2. Detrimental Role of Mast Cells in Severe CLP
4. Mast Cells and Candida albicans Infection
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Orinska, Z.; Bulanova, E.; Budagian, V.; Metz, M.; Maurer, M.; Bulfone-Paus, S. TLR3-induced activation of mast cells modulates CD8+ T-cell recruitment. Blood 2005, 106, 978–987. [Google Scholar] [CrossRef]
- Dietrich, N.; Rohde, M.; Geffers, R.; Kroger, A.; Hauser, H.; Weiss, S.; Gekara, N.O. Mast cells elicit proinflammatory but not type I interferon responses upon activation of TLRs by bacteria. Proc. Natl. Acad. Sci. USA 2010, 107, 8748–8753. [Google Scholar] [CrossRef] [PubMed]
- Sandig, H.; Bulfone-Paus, S. TLR signaling in mast cells: Common and unique features. Front. Immunol. 2012, 3, 185. [Google Scholar] [CrossRef] [PubMed]
- Arifuzzaman, M.; Mobley, Y.R.; Choi, H.W.; Bist, P.; Salinas, C.A.; Brown, Z.D.; Chen, S.L.; Staats, H.F.; Abraham, S.N. MRGPR-mediated activation of local mast cells clears cutaneous bacterial infection and protects against reinfection. Sci. Adv. 2019, 5, eaav0216. [Google Scholar] [CrossRef] [PubMed]
- McCurdy, J.D.; Olynych, T.J.; Maher, L.H.; Marshall, J.S. Cutting edge: Distinct Toll-like receptor 2 activators selectively induce different classes of mediator production from human mast cells. J. Immunol. 2003, 170, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, H.; Yamada, N.; Matsue, H.; Shimada, S. TLR3-, TLR7-, and TLR9-mediated production of proinflammatory cytokines and chemokines from murine connective tissue type skin-derived mast cells but not from bone marrow-derived mast cells. J. Immunol. 2004, 173, 531–541. [Google Scholar] [CrossRef]
- McNeil, B.D.; Pundir, P.; Meeker, S.; Han, L.; Undem, B.J.; Kulka, M.; Dong, X. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 2015, 519, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Crowle, P.K. Mucosal mast cell reconstitution and Nippostrongylus brasiliensis rejection by W/Wv mice. J. Parasitol. 1983, 69, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Carlos, D.; Frantz, F.G.; Souza-Junior, D.A.; Jamur, M.C.; Oliver, C.; Ramos, S.G.; Quesniaux, V.F.; Ryffel, B.; Silva, C.L.; Bozza, M.T.; et al. TLR2-dependent mast cell activation contributes to the control of Mycobacterium tuberculosis infection. Microbes Infect. 2009, 11, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rodriguez, K.M.; Goenka, A.; Alonso-Rasgado, M.T.; Hernandez-Pando, R.; Bulfone-Paus, S. The Role of Mast Cells in Tuberculosis: Orchestrating Innate Immune Crosstalk? Front Immunol 2017, 8, 1290. [Google Scholar] [CrossRef] [PubMed]
- Vornhagen, J.; Adams Waldorf, K.M.; Rajagopal, L. Perinatal Group B Streptococcal Infections: Virulence Factors, Immunity, and Prevention Strategies. Trends Microbiol. 2017, 25, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Gendrin, C.; Vornhagen, J.; Ngo, L.; Whidbey, C.; Boldenow, E.; Santana-Ufret, V.; Clauson, M.; Burnside, K.; Galloway, D.P.; Adams Waldorf, K.M.; et al. Mast cell degranulation by a hemolytic lipid toxin decreases GBS colonization and infection. Sci. Adv. 2015, 1, e1400225. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lai, Y.; Bernard, J.J.; Macleod, D.T.; Cogen, A.L.; Moss, B.; Di Nardo, A. Skin mast cells protect mice against vaccinia virus by triggering mast cell receptor S1PR2 and releasing antimicrobial peptides. J. Immunol. 2012, 188, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Piliponsky, A.M.; Romani, L. The contribution of mast cells to bacterial and fungal infection immunity. Immunol. Rev. 2018, 282, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, M.K.; Carletti, T.; Marcello, A. Cellular Targets for the Treatment of Flavivirus Infections. Front. Cell Infect Microbiol. 2018, 8, 398. [Google Scholar] [CrossRef]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Wilder-Smith, A.; Ooi, E.E.; Horstick, O.; Wills, B. Dengue. Lancet 2019, 393, 350–363. [Google Scholar] [CrossRef]
- Marchette, N.J.; Halstead, S.B.; Falkler, W.A., Jr.; Stenhouse, A.; Nash, D. Studies on the pathogenesis of dengue infection in monkeys. 3. Sequential distribution of virus in primary and heterologous infections. J. Infect Dis. 1973, 128, 23–30. [Google Scholar] [CrossRef]
- Yauch, L.E.; Zellweger, R.M.; Kotturi, M.F.; Qutubuddin, A.; Sidney, J.; Peters, B.; Prestwood, T.R.; Sette, A.; Shresta, S. A protective role for dengue virus-specific CD8+ T cells. J. Immunol. 2009, 182, 4865–4873. [Google Scholar] [CrossRef]
- Shresta, S.; Kyle, J.L.; Robert Beatty, P.; Harris, E. Early activation of natural killer and B cells in response to primary dengue virus infection in A/J mice. Virology 2004, 319, 262–273. [Google Scholar] [CrossRef]
- Azeredo, E.L.; De Oliveira-Pinto, L.M.; Zagne, S.M.; Cerqueira, D.I.; Nogueira, R.M.; Kubelka, C.F. NK cells, displaying early activation, cytotoxicity and adhesion molecules, are associated with mild dengue disease. Clin. Exp. Immunol. 2006, 143, 345–356. [Google Scholar] [CrossRef] [PubMed]
- St John, A.L.; Rathore, A.P.; Yap, H.; Ng, M.L.; Metcalfe, D.D.; Vasudevan, S.G.; Abraham, S.N. Immune surveillance by mast cells during dengue infection promotes natural killer (NK) and NKT-cell recruitment and viral clearance. Proc. Natl. Acad. Sci. USA 2011, 108, 9190–9195. [Google Scholar] [CrossRef] [PubMed]
- Kurane, I.; Hebblewaite, D.; Brandt, W.E.; Ennis, F.A. Lysis of dengue virus-infected cells by natural cell-mediated cytotoxicity and antibody-dependent cell-mediated cytotoxicity. J. Virol. 1984, 52, 223–230. [Google Scholar] [PubMed]
- Mantri, C.K.; St John, A.L. Immune synapses between mast cells and gammadelta T cells limit viral infection. J. Clin. Investig. 2019, 129, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Murao, L.A.; Lan, N.T.; Huy, N.T.; Huong, V.T.; Thuy, T.T.; Tham, V.D.; Nga, C.T.; Ha, T.T.; Ohmoto, Y.; et al. Association of mast cell-derived VEGF and proteases in Dengue shock syndrome. PLoS Negl. Trop. Dis. 2012, 6, e1505. [Google Scholar] [CrossRef]
- St John, A.L.; Rathore, A.P.; Raghavan, B.; Ng, M.L.; Abraham, S.N. Contributions of mast cells and vasoactive products, leukotrienes and chymase, to dengue virus-induced vascular leakage. Elife 2013, 2, e00481. [Google Scholar] [CrossRef] [PubMed]
- Tissera, H.; Rathore, A.P.S.; Leong, W.Y.; Pike, B.L.; Warkentien, T.E.; Farouk, F.S.; Syenina, A.; Eong Ooi, E.; Gubler, D.J.; Wilder-Smith, A.; et al. Chymase Level Is a Predictive Biomarker of Dengue Hemorrhagic Fever in Pediatric and Adult Patients. J. Infect Dis. 2017, 216, 1112–1121. [Google Scholar] [CrossRef]
- Morrison, J.; Rathore, A.P.S.; Mantri, C.K.; Aman, S.A.B.; Nishida, A.; St John, A.L. Transcriptional Profiling Confirms the Therapeutic Effects of Mast Cell Stabilization in a Dengue Disease Model. J. Virol. 2017, 91, e00617-17. [Google Scholar] [CrossRef]
- Hsieh, J.T.; Rathore, A.P.S.; Soundarajan, G.; St John, A.L. Japanese encephalitis virus neuropenetrance is driven by mast cell chymase. Nat. Commun. 2019, 10, 706. [Google Scholar] [CrossRef]
- Masri, M.F.B.; Mantri, C.K.; Rathore, A.P.S.; St John, A.L. Peripheral serotonin causes dengue-induced thrombocytopenia through 5HT2 receptors. Blood 2019. [Google Scholar] [CrossRef]
- Sugiyama, K. Histamine release from rat mast cells induced by Sendai virus. Nature 1977, 270, 614–615. [Google Scholar] [CrossRef] [PubMed]
- Erdei, A.; Pecht, I. Complement peptides and mast cell triggering. Immunol. Lett. 1996, 54, 109–112. [Google Scholar] [CrossRef]
- Avirutnan, P.; Hauhart, R.E.; Marovich, M.A.; Garred, P.; Atkinson, J.P.; Diamond, M.S. Complement-mediated neutralization of dengue virus requires mannose-binding lectin. MBio 2011, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Troupin, A.; Shirley, D.; Londono-Renteria, B.; Watson, A.M.; McHale, C.; Hall, A.; Hartstone-Rose, A.; Klimstra, W.B.; Gomez, G.; Colpitts, T.M. A Role for Human Skin Mast Cells in Dengue Virus Infection and Systemic Spread. J. Immunol. 2016, 197, 4382–4391. [Google Scholar] [CrossRef] [PubMed]
- Rothman, A.L.; Ennis, F.A. Immunopathogenesis of Dengue hemorrhagic fever. Virology 1999, 257, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, L.F.; Hotta, H.; Hotta, S.; Homma, M. Degranulation and histamine release from murine mast cells sensitized with dengue virus-immune sera. Microbiol. Immunol. 1986, 30, 753–759. [Google Scholar] [CrossRef] [PubMed]
- King, C.A.; Marshall, J.S.; Alshurafa, H.; Anderson, R. Release of vasoactive cytokines by antibody-enhanced dengue virus infection of a human mast cell/basophil line. J. Virol. 2000, 74, 7146–7150. [Google Scholar] [CrossRef] [PubMed]
- King, C.A.; Anderson, R.; Marshall, J.S. Dengue virus selectively induces human mast cell chemokine production. J. Virol. 2002, 76, 8408–8419. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.G.; Hermann, L.L.; Issekutz, A.C.; Marshall, J.S.; Rowter, D.; Al-Afif, A.; Anderson, R. Dengue virus infection of mast cells triggers endothelial cell activation. J. Virol. 2011, 85, 1145–1150. [Google Scholar] [CrossRef]
- Brown, M.G.; King, C.A.; Sherren, C.; Marshall, J.S.; Anderson, R. A dominant role for FcgammaRII in antibody-enhanced dengue virus infection of human mast cells and associated CCL5 release. J. Leukoc. Biol. 2006, 80, 1242–1250. [Google Scholar] [CrossRef]
- Fang, Y.T.; Wan, S.W.; Lu, Y.T.; Yao, J.H.; Lin, C.F.; Hsu, L.J.; Brown, M.G.; Marshall, J.S.; Anderson, R.; Lin, Y.S. Autophagy facilitates antibody-enhanced dengue virus infection in human pre-basophil/mast cells. PLoS ONE 2014, 9, e110655. [Google Scholar] [CrossRef]
- Remick, D.G.; Newcomb, D.E.; Bolgos, G.L.; Call, D.R. Comparison of the mortality and inflammatory response of two models of sepsis: Lipopolysaccharide vs. cecal ligation and puncture. Shock 2000, 13, 110–116. [Google Scholar] [CrossRef]
- Hubbard, W.J.; Choudhry, M.; Schwacha, M.G.; Kerby, J.D.; Rue, L.W., 3rd; Bland, K.I.; Chaudry, I.H. Cecal ligation and puncture. Shock 2005, 24, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Echtenacher, B.; Mannel, D.N.; Hultner, L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature 1996, 381, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Supajatura, V.; Ushio, H.; Nakao, A.; Okumura, K.; Ra, C.; Ogawa, H. Protective roles of mast cells against enterobacterial infection are mediated by Toll-like receptor 4. J. Immunol. 2001, 167, 2250–2256. [Google Scholar] [CrossRef] [PubMed]
- Tchougounova, E.; Pejler, G.; Abrink, M. The chymase, mouse mast cell protease 4, constitutes the major chymotrypsin-like activity in peritoneum and ear tissue. A role for mouse mast cell protease 4 in thrombin regulation and fibronectin turnover. J. Exp. Med. 2003, 198, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Seeley, E.J.; Sutherland, R.E.; Kim, S.S.; Wolters, P.J. Systemic mast cell degranulation increases mortality during polymicrobial septic peritonitis in mice. J. Leukoc. Biol. 2011, 90, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Piliponsky, A.M.; Chen, C.C.; Nishimura, T.; Metz, M.; Rios, E.J.; Dobner, P.R.; Wada, E.; Wada, K.; Zacharias, S.; Mohanasundaram, U.M.; et al. Neurotensin increases mortality and mast cells reduce neurotensin levels in a mouse model of sepsis. Nat. Med. 2008, 14, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Piliponsky, A.M.; Chen, C.C.; Rios, E.J.; Treuting, P.M.; Lahiri, A.; Abrink, M.; Pejler, G.; Tsai, M.; Galli, S.J. The chymase mouse mast cell protease 4 degrades TNF, limits inflammation, and promotes survival in a model of sepsis. Am. J. Pathol. 2012, 181, 875–886. [Google Scholar] [CrossRef]
- De Filippo, K.; Dudeck, A.; Hasenberg, M.; Nye, E.; van Rooijen, N.; Hartmann, K.; Gunzer, M.; Roers, A.; Hogg, N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 2013, 121, 4930–4937. [Google Scholar] [CrossRef] [PubMed]
- Prodeus, A.P.; Zhou, X.; Maurer, M.; Galli, S.J.; Carroll, M.C. Impaired mast cell-dependent natural immunity in complement C3-deficient mice. Nature 1997, 390, 172–175. [Google Scholar] [CrossRef]
- Maurer, M.; Wedemeyer, J.; Metz, M.; Piliponsky, A.M.; Weller, K.; Chatterjea, D.; Clouthier, D.E.; Yanagisawa, M.M.; Tsai, M.; Galli, S.J. Mast cells promote homeostasis by limiting endothelin-1-induced toxicity. Nature 2004, 432, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Echtenacher, B.; Hultner, L.; Kollias, G.; Mannel, D.N.; Langley, K.E.; Galli, S.J. The c-kit ligand, stem cell factor, can enhance innate immunity through effects on mast cells. J. Exp. Med. 1998, 188, 2343–2348. [Google Scholar] [CrossRef]
- Malaviya, R.; Ikeda, T.; Ross, E.; Abraham, S.N. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature 1996, 381, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Piliponsky, A.M.; Chen, C.C.; Grimbaldeston, M.A.; Burns-Guydish, S.M.; Hardy, J.; Kalesnikoff, J.; Contag, C.H.; Tsai, M.; Galli, S.J. Mast cell-derived TNF can exacerbate mortality during severe bacterial infections in C57BL/6-KitW-sh/W-sh mice. Am. J. Pathol. 2010, 176, 926–938. [Google Scholar] [CrossRef]
- Thakurdas, S.M.; Melicoff, E.; Sansores-Garcia, L.; Moreira, D.C.; Petrova, Y.; Stevens, R.L.; Adachi, R. The mast cell-restricted tryptase mMCP-6 has a critical immunoprotective role in bacterial infections. J. Biol. Chem. 2007, 282, 20809–20815. [Google Scholar] [CrossRef]
- Sutherland, R.E.; Olsen, J.S.; McKinstry, A.; Villalta, S.A.; Wolters, P.J. Mast cell IL-6 improves survival from Klebsiella pneumonia and sepsis by enhancing neutrophil killing. J. Immunol. 2008, 181, 5598–5605. [Google Scholar] [CrossRef]
- Malaviya, R.; Ikeda, T.; Abraham, S.N. Contribution of mast cells to bacterial clearance and their proliferation during experimental cystitis induced by type 1 fimbriated E. coli. Immunol. Lett. 2004, 91, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Echtenacher, B.; Falk, W.; Mannel, D.N.; Krammer, P.H. Requirement of endogenous tumor necrosis factor/cachectin for recovery from experimental peritonitis. J. Immunol. 1990, 145, 3762–3766. [Google Scholar] [PubMed]
- Piliponsky, A.M.; Shubin, N.J.; Lahiri, A.K.; Truong, P.; Clauson, M.; Niino, K.; Tsuha, A.L.; Nedospasov, S.A.; Karasuyama, H.; Reber, L.L.; et al. Basophil-derived tumor necrosis factor can enhance survival in a sepsis model in mice. Nat. Immunol. 2019, 20, 129–140. [Google Scholar] [CrossRef]
- Dalrymple, S.A.; Slattery, R.; Aud, D.M.; Krishna, M.; Lucian, L.A.; Murray, R. Interleukin-6 is required for a protective immune response to systemic Escherichia coli infection. Infect. Immun. 1996, 64, 3231–3235. [Google Scholar] [PubMed]
- Van der Poll, T.; Keogh, C.V.; Guirao, X.; Buurman, W.A.; Kopf, M.; Lowry, S.F. Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia. J. Infect. Dis. 1997, 176, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Reimer, J.M.; Enoksson, M.; Samollow, P.B.; Hellman, L. Extended substrate specificity of opossum chymase--implications for the origin of mast cell chymases. Mol. Immunol. 2008, 45, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Reimer, J.M.; Samollow, P.B.; Hellman, L. High degree of conservation of the multigene tryptase locus over the past 150-200 million years of mammalian evolution. Immunogenetics 2010, 62, 369–382. [Google Scholar] [CrossRef]
- Reynolds, D.S.; Gurley, D.S.; Austen, K.F.; Serafin, W.E. Cloning of the cDNA and gene of mouse mast cell protease-6. Transcription by progenitor mast cells and mast cells of the connective tissue subclass. J. Biol. Chem. 1991, 266, 3847–3853. [Google Scholar]
- He, S.; Walls, A.F. Human mast cell chymase induces the accumulation of neutrophils, eosinophils and other inflammatory cells in vivo. Br. J. Pharmacol. 1998, 125, 1491–1500. [Google Scholar] [CrossRef]
- Tani, K.; Ogushi, F.; Kido, H.; Kawano, T.; Kunori, Y.; Kamimura, T.; Cui, P.; Sone, S. Chymase is a potent chemoattractant for human monocytes and neutrophils. J. Leukoc. Biol. 2000, 67, 585–589. [Google Scholar] [CrossRef]
- Orinska, Z.; Maurer, M.; Mirghomizadeh, F.; Bulanova, E.; Metz, M.; Nashkevich, N.; Schiemann, F.; Schulmistrat, J.; Budagian, V.; Giron-Michel, J.; et al. IL-15 constrains mast cell-dependent antibacterial defenses by suppressing chymase activities. Nat. Med. 2007, 13, 927–934. [Google Scholar] [CrossRef]
- Curzen, N.P.; Mitchell, J.A.; Jourdan, K.B.; Griffiths, M.J.; Evans, T.W. Endothelin-1-induced contraction of pulmonary arteries from endotoxemic rats is attenuated by the endothelin-A receptor antagonist, BQ123. Crit. Care Med. 1996, 24, 2007–2013. [Google Scholar] [CrossRef]
- Lilla, J.N.; Chen, C.C.; Mukai, K.; BenBarak, M.J.; Franco, C.B.; Kalesnikoff, J.; Yu, M.; Tsai, M.; Piliponsky, A.M.; Galli, S.J. Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood 2011, 118, 6930–6938. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Tumanov, A.V.; Liepinsh, D.J.; Kruglov, A.A.; Marakusha, B.I.; Shakhov, A.N.; Murakami, T.; Drutskaya, L.N.; Forster, I.; Clausen, B.E.; et al. Distinct and nonredundant in vivo functions of TNF produced by t cells and macrophages/neutrophils: Protective and deleterious effects. Immunity 2005, 22, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Dahdah, A.; Gautier, G.; Attout, T.; Fiore, F.; Lebourdais, E.; Msallam, R.; Daeron, M.; Monteiro, R.C.; Benhamou, M.; Charles, N.; et al. Mast cells aggravate sepsis by inhibiting peritoneal macrophage phagocytosis. J. Clin. Investig. 2014, 124, 4577–4589. [Google Scholar] [CrossRef] [PubMed]
- Saluja, R.; Metz, M.; Maurer, M. Role and relevance of mast cells in fungal infections. Front. Immunol. 2012, 3, 146. [Google Scholar] [CrossRef] [PubMed]
- Frossi, B.; Mion, F.; Tripodo, C.; Colombo, M.P.; Pucillo, C.E. Rheostatic Functions of Mast Cells in the Control of Innate and Adaptive Immune Responses. Trends Immunol. 2017, 38, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, J.; Tsai, M.; Galli, S.J. Roles of mast cells and basophils in innate and acquired immunity. Curr. Opin. Immunol. 2000, 12, 624–631. [Google Scholar] [CrossRef]
- Metz, M.; Maurer, M. Mast cells--key effector cells in immune responses. Trends Immunol. 2007, 28, 234–241. [Google Scholar] [CrossRef]
- Nieto-Patlan, A.; Campillo-Navarro, M.; Rodriguez-Cortes, O.; Munoz-Cruz, S.; Wong-Baeza, I.; Estrada-Parra, S.; Estrada-Garcia, I.; Serafin-Lopez, J.; Chacon-Salinas, R. Recognition of Candida albicans by Dectin-1 induces mast cell activation. Immunobiology 2015, 220, 1093–1100. [Google Scholar] [CrossRef]
- Netea, M.G.; Brown, G.D.; Kullberg, B.J.; Gow, N.A. An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 2008, 6, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Shoham, S.; Levitz, S.M. The immune response to fungal infections. Br. J. Haematol. 2005, 129, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.P.; Stylianou, M.; Nilsson, G.; Urban, C.F. Opportunistic pathogen Candida albicans elicits a temporal response in primary human mast cells. Sci. Rep. 2015, 5, 12287. [Google Scholar] [CrossRef] [PubMed]
- Schlapbach, C.; Gehad, A.; Yang, C.; Watanabe, R.; Guenova, E.; Teague, J.E.; Campbell, L.; Yawalkar, N.; Kupper, T.S.; Clark, R.A. Human TH9 cells are skin-tropic and have autocrine and paracrine proinflammatory capacity. Sci. Transl Med. 2014, 6, 219ra8. [Google Scholar] [CrossRef] [PubMed]
- Pinke, K.H.; Lima, H.G.; Cunha, F.Q.; Lara, V.S. Mast cells phagocyte Candida albicans and produce nitric oxide by mechanisms involving TLR2 and Dectin-1. Immunobiology 2016, 221, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, E.; Vita, F.; Medic, N.; Soranzo, M.R.; Zabucchi, G.; Borelli, V. Mast cells kill Candida albicans in the extracellular environment but spare ingested fungi from death. Inflammation 2014, 37, 2174–2189. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Sugita, R.; Miki, A.; Takemura, N.; Kawabata, J.; Watanabe, J.; Sonoyama, K. Gastrointestinal Candida colonisation promotes sensitisation against food antigens by affecting the mucosal barrier in mice. Gut 2006, 55, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Gurish, M.F.; Austen, K.F. Developmental origin and functional specialization of mast cell subsets. Immunity 2012, 37, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Reber, L.L.; Sibilano, R.; Mukai, K.; Galli, S.J. Potential effector and immunoregulatory functions of mast cells in mucosal immunity. Mucosal. Immunol. 2015, 8, 444–463. [Google Scholar] [CrossRef] [PubMed]
- Renga, G.; Moretti, S.; Oikonomou, V.; Borghi, M.; Zelante, T.; Paolicelli, G.; Costantini, C.; De Zuani, M.; Villella, V.R.; Raia, V.; et al. IL-9 and Mast Cells Are Key Players of Candida albicans Commensalism and Pathogenesis in the Gut. Cell Rep. 2018, 23, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Renga, G.; Borghi, M.; Oikonomou, V.; Mosci, P.; Bartoli, A.; Renauld, J.C.; Romani, L.; Costantini, C. IL-9 Integrates the Host-Candida Cross-Talk in Vulvovaginal Candidiasis to Balance Inflammation and Tolerance. Front. Immunol. 2018, 9, 2702. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piliponsky, A.M.; Acharya, M.; Shubin, N.J. Mast Cells in Viral, Bacterial, and Fungal Infection Immunity. Int. J. Mol. Sci. 2019, 20, 2851. https://doi.org/10.3390/ijms20122851
Piliponsky AM, Acharya M, Shubin NJ. Mast Cells in Viral, Bacterial, and Fungal Infection Immunity. International Journal of Molecular Sciences. 2019; 20(12):2851. https://doi.org/10.3390/ijms20122851
Chicago/Turabian StylePiliponsky, Adrian M., Manasa Acharya, and Nicholas J. Shubin. 2019. "Mast Cells in Viral, Bacterial, and Fungal Infection Immunity" International Journal of Molecular Sciences 20, no. 12: 2851. https://doi.org/10.3390/ijms20122851
APA StylePiliponsky, A. M., Acharya, M., & Shubin, N. J. (2019). Mast Cells in Viral, Bacterial, and Fungal Infection Immunity. International Journal of Molecular Sciences, 20(12), 2851. https://doi.org/10.3390/ijms20122851