Transcription Profiles Reveal the Regulatory Synthesis of Phenols during the Development of Lotus Rhizome (Nelumbo nucifera Gaertn)

Abstract
1. Introduction
2. Results
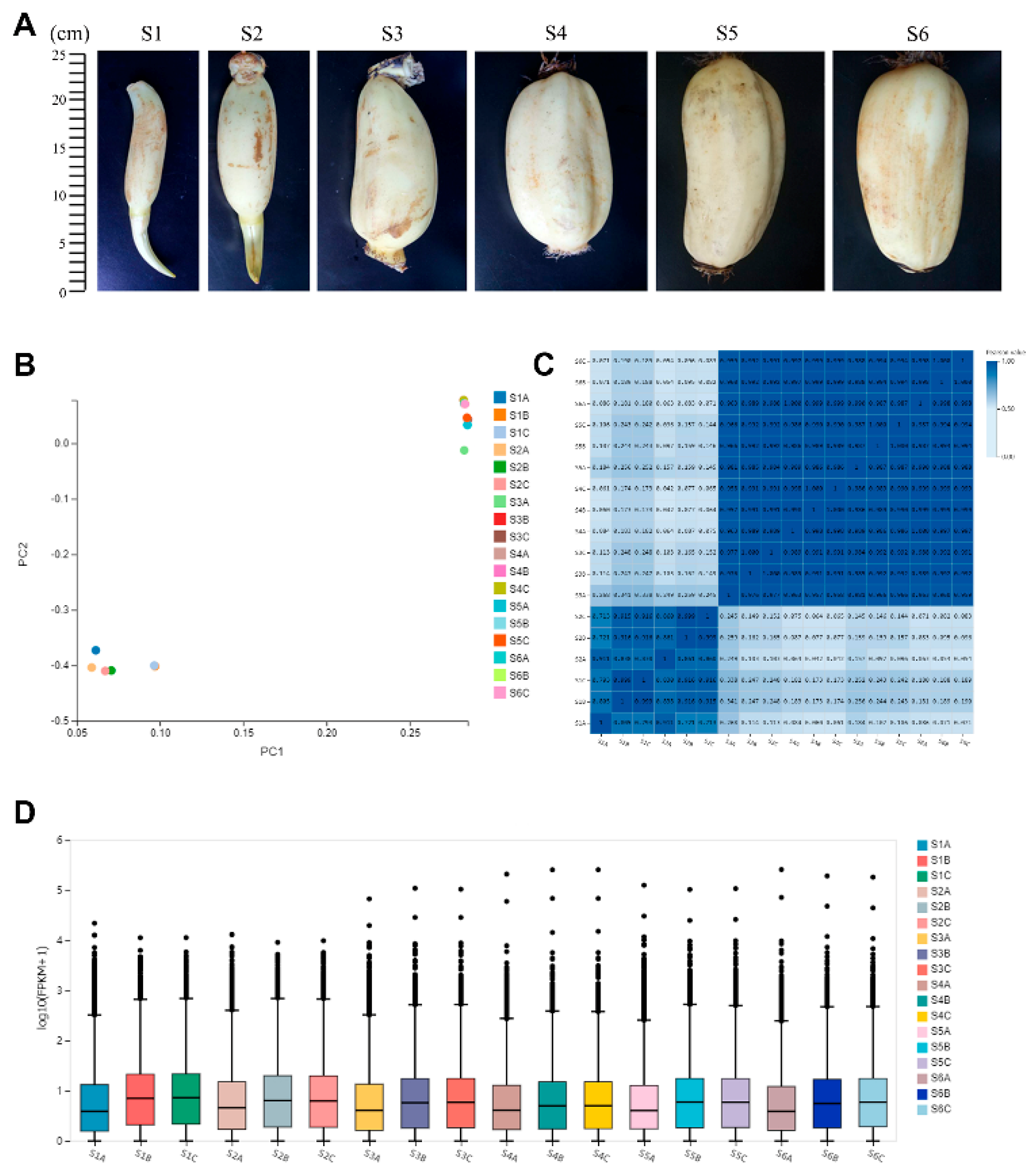
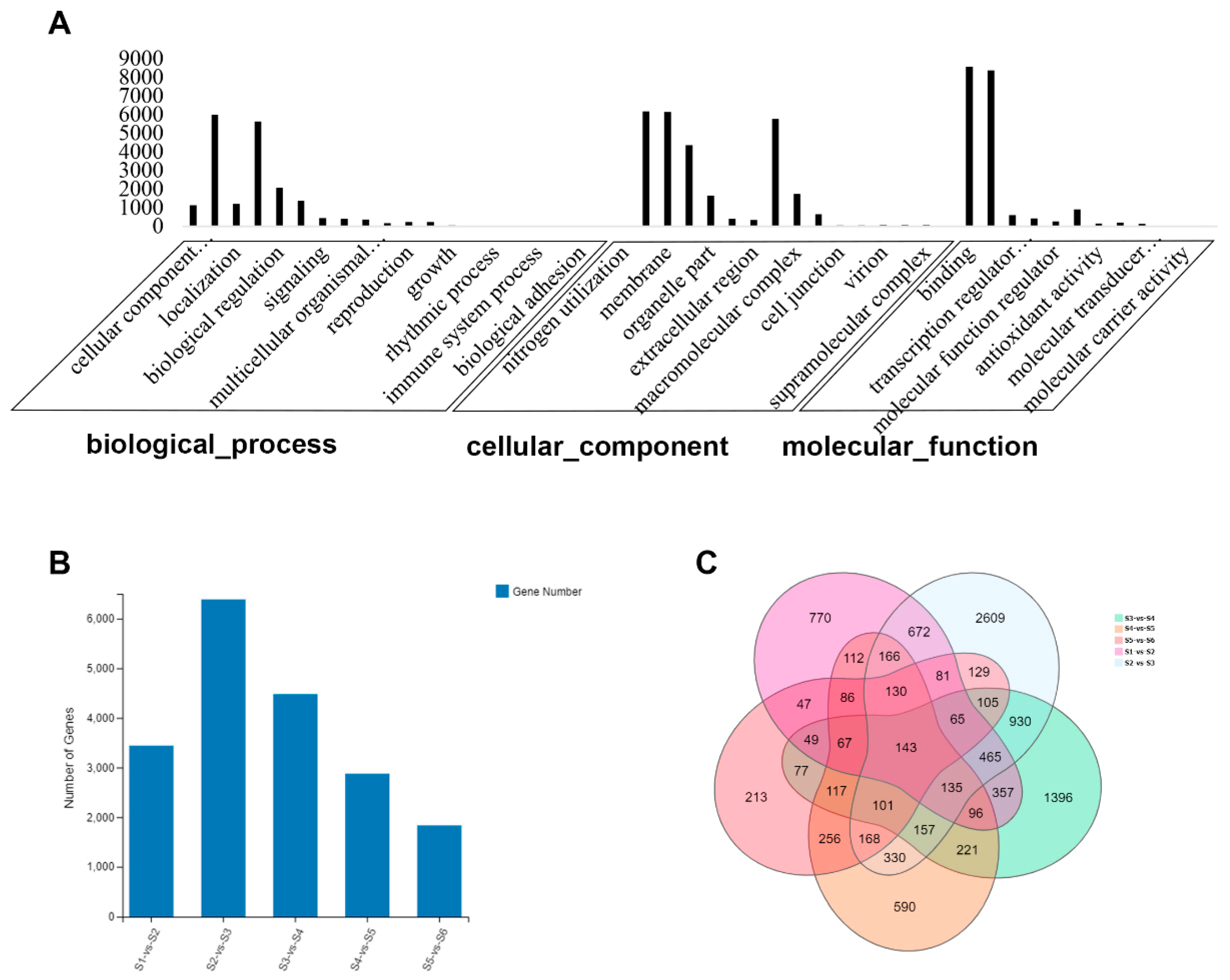
2.1. Transcriptome of Lotus Rhizome
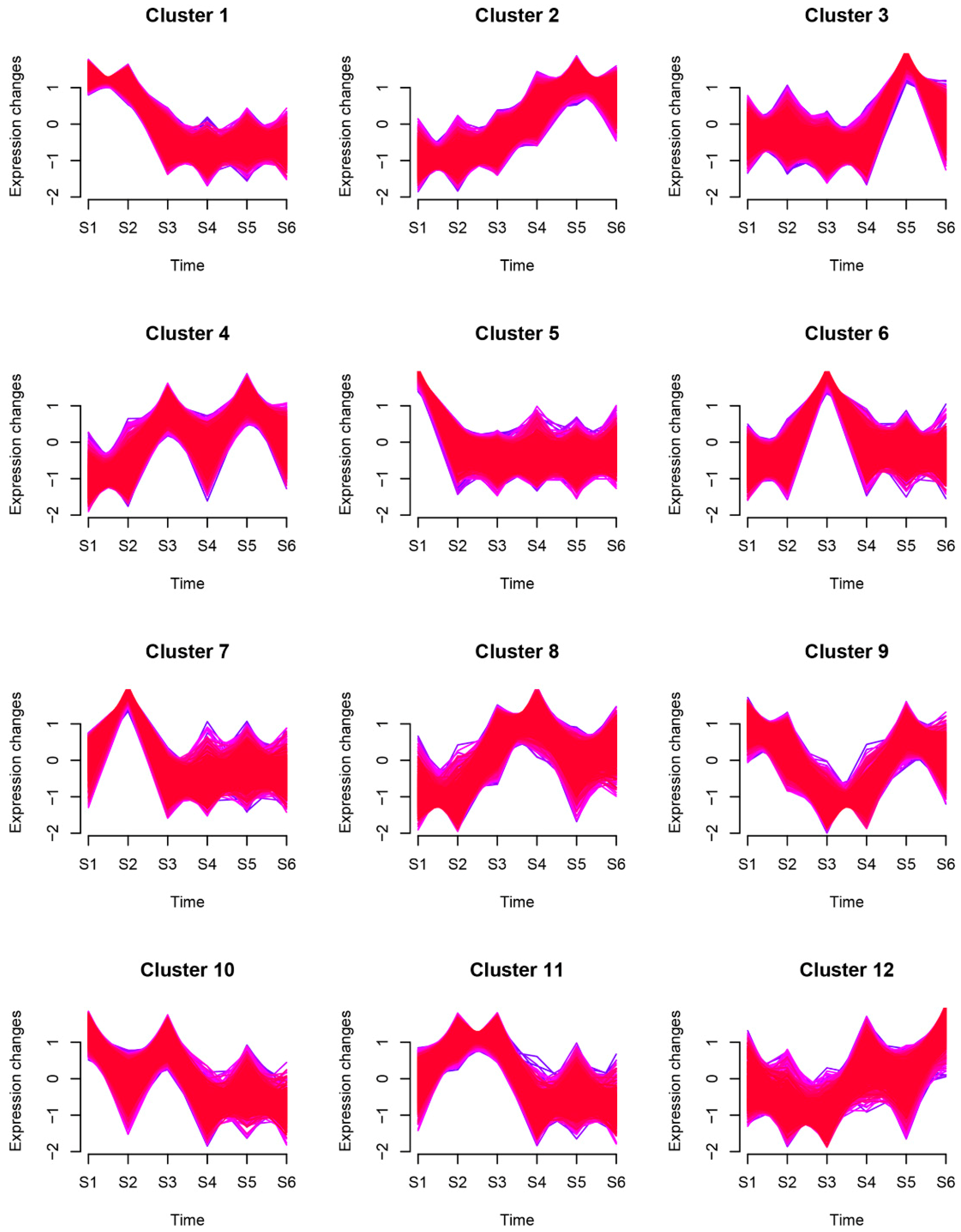
2.2. Genes Expression Pattern during Rhizome Formation
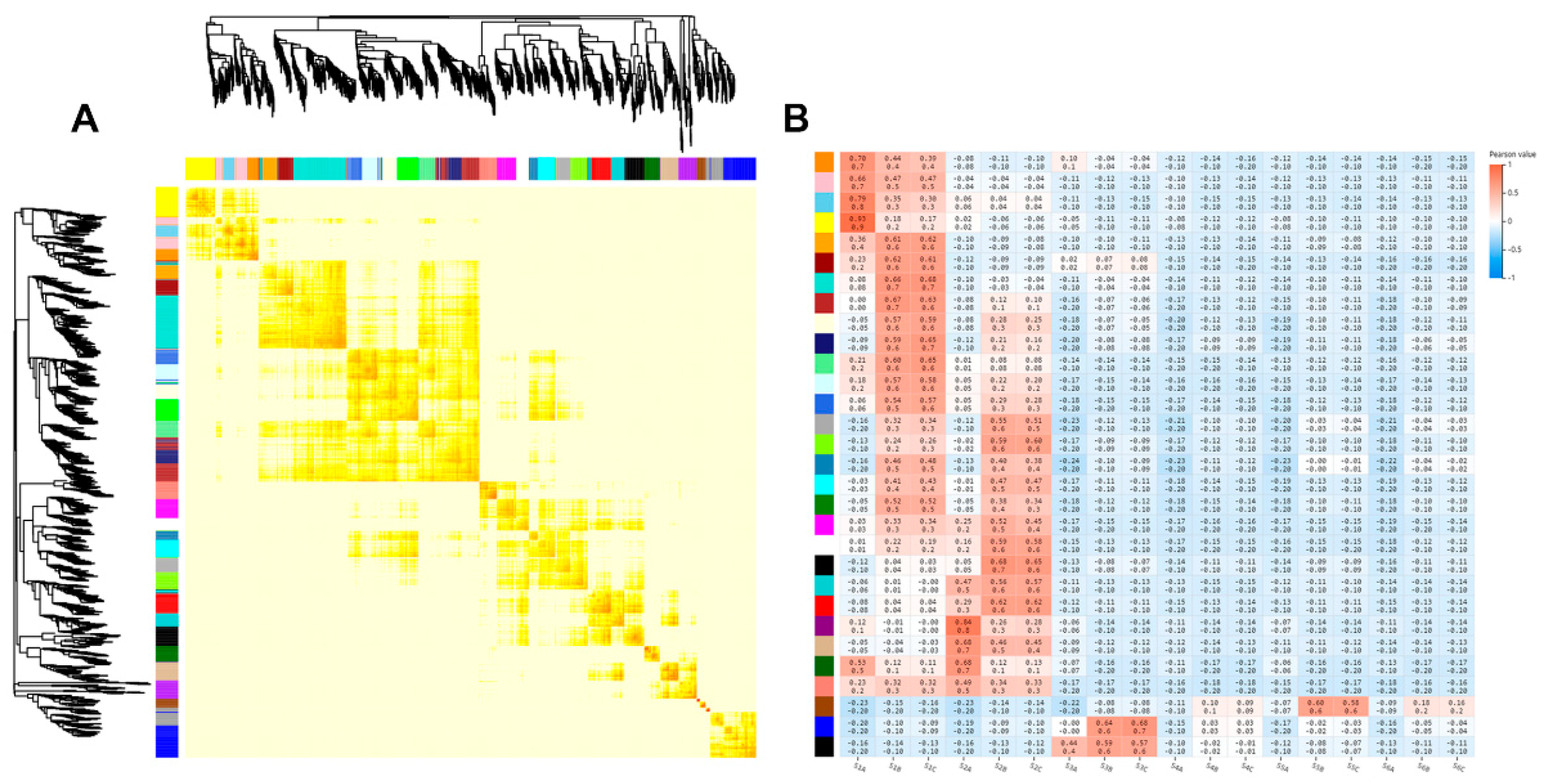
2.3. Correlation between Transcriptomes of Different Stages of Rhizome Development
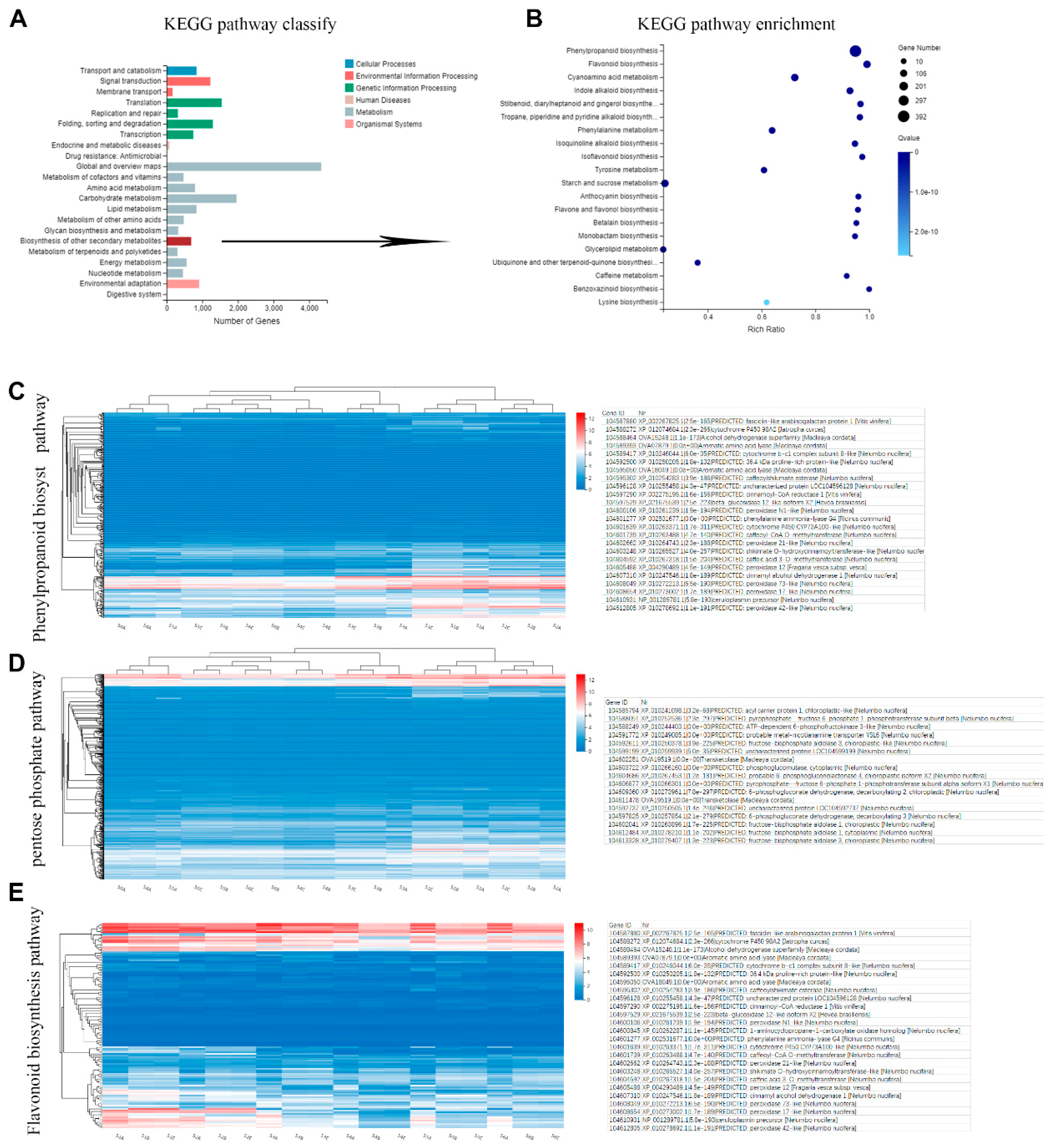
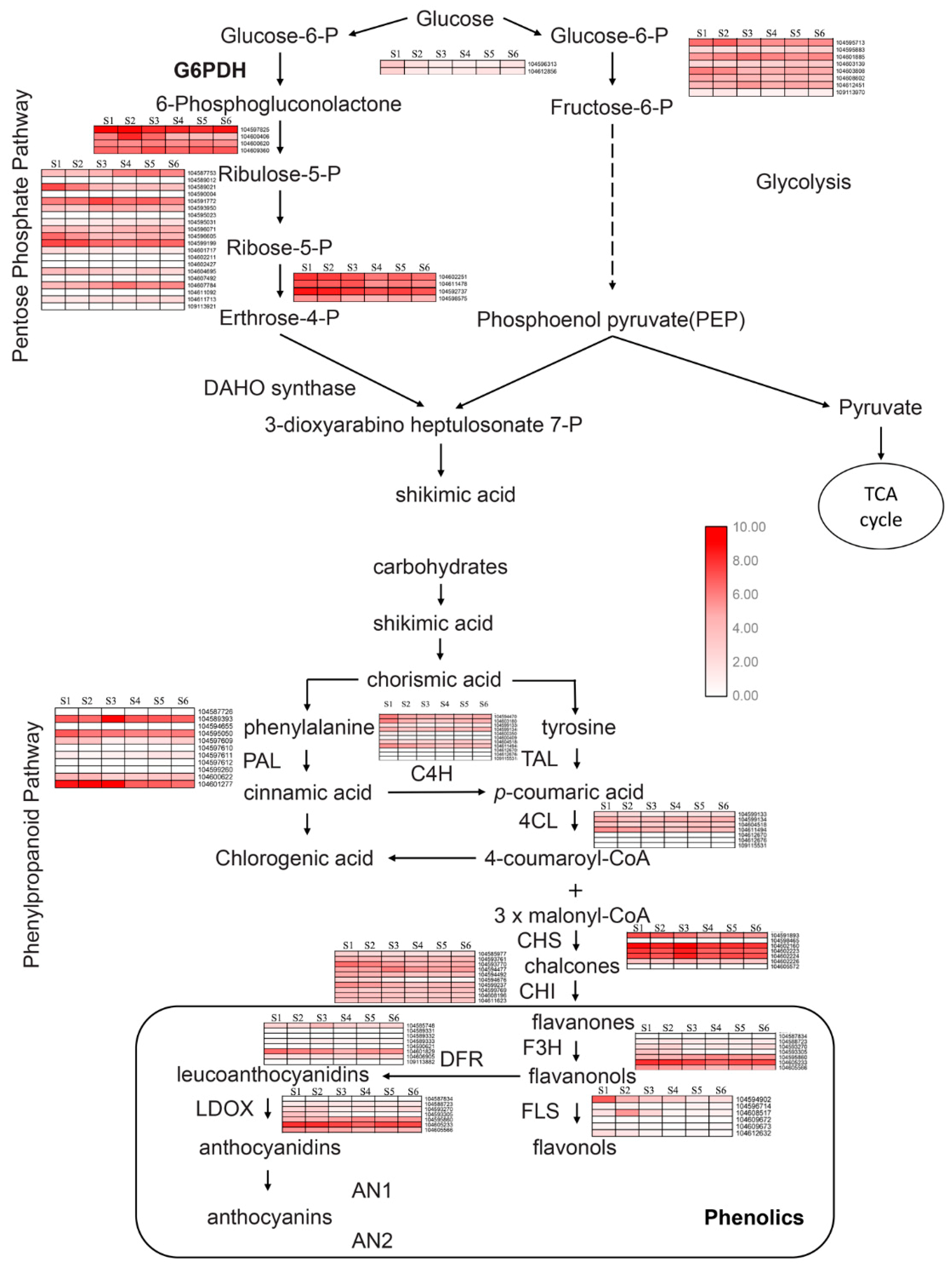
2.4. Metabolic Pathway Analysis of Candidate Genes Involved in Phenols Synthesis
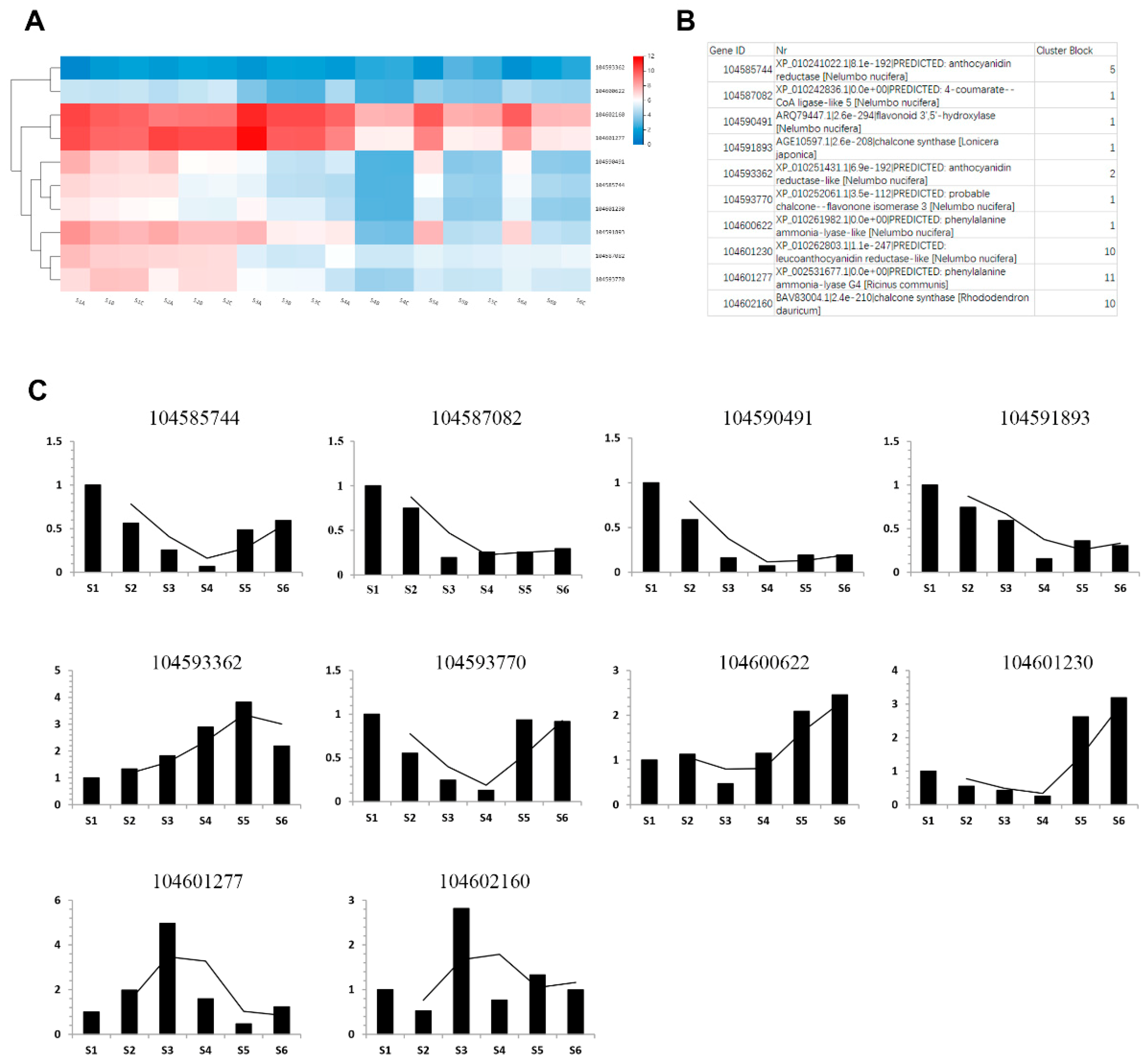
2.5. Key Enzymes Related to Phenols Synthesis during Rhizome Development
2.6. Differentially Expressed Genes (DEGs) during Rhizome Development
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Determination of Total Phenolic Content
4.2. RNA Isolation and Sequencing
4.3. Analysis and Mapping of RNA-Seq Reads
4.4. Differential Expression Analysis
4.5. Gene Ontology and KEGG Ortholog Enrichment Analysis
4.6. Quantitative Real-Time PCR Validation of RNA-Seq Data
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Guo, H.B. Cultivation of lotus (Nelumbo nucifera Gaertn. ssp. nucifera) and its utilization in China. Genet. Resour. Crop. Ev. 2009, 56, 323–330. [Google Scholar] [CrossRef]
- Cheng, L.; Liu, H.; Jiang, R.; Li, S. A proteomics analysis of adventitious root formation after leaf removal in lotus (Nelumbo nucifera Gaertn.). Z. Für Nat. C 2018, 73, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Abdullakasim, P.; Songchitsomboon, S.; Techagumpuch, M.; Balee, N.; Swatsitang, P.; Sungpuag, P. Antioxidant capacity, total phenolics and sugar content of selected Thai health beverages. Int. J. Food Sci. Nutr. 2007, 58, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yang, M.; Li, L.; Li, H.; Yang, D.; Shi, T.; Yang, P. Whole genome re-sequencing reveals evolutionary patterns of sacred lotus (Nelumbo nucifera). J. Integr. Plant Biol. 2018, 60, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Xingyan, W.Q.Z. Lotus Flower Cultivars in China; China Forestry Publishing House: Beijing, China, 2005. [Google Scholar]
- Cheng, L.; Li, S.; Yin, J.; Li, L.; Chen, X. Genome-Wide Analysis of Differentially Expressed Genes Relevant to Rhizome Formation in Lotus Root (Nelumbo nucifera Gaertn). PLoS ONE 2013, 8, e67116. [Google Scholar] [CrossRef] [PubMed]
- Masuda, J.; Ozaki, Y.; Okubo, H. Rhizome transition to storage organ is under phytochrome control in lotus (Nelumbo nucifera). Planta 2007, 226, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Masuda, J.I.; Ozaki, Y.; Okubo, H. Regulation in rhizome transition to storage organ in Lotus (Nelumbo nucifera Gaertn.) with exogenous gibberellin, gibberellin biosynthesis inhibitors or abscisic Acid. J. Jpn. Soc. Hortic. Sci. 2012, 81, 67–71. [Google Scholar] [CrossRef]
- Yang, M.; Zhu, L.; Pan, C.; Xu, L.; Liu, Y.; Ke, W.; Yang, P. Transcriptomic analysis of the regulation of rhizome formation in temperate and tropical Lotus (Nelumbo nucifera). Sci. Rep. 2015, 5, 13059. [Google Scholar] [CrossRef]
- Jiang, Y.; Ng, T.B.; Wang, C.R.; Li, N.; Wen, T.Y.; Qiao, W.T.; Zhang, D.; Cheng, Z.H.; Liu, F. First isolation of tryptophan from edible lotus (Nelumbo nucifera Gaertn) rhizomes and demonstration of its antioxidant effects. Int. J. Food Sci. Nutr. 2010, 61, 346–356. [Google Scholar] [CrossRef]
- Degl Innocenti, E.; Pardossi, A.; Tognoni, F.; Guidi, L. Physiological basis of sensitivity to enzymatic browning in ‘lettuce’, ‘escarole’ and ‘rocket salad’ when stored as fresh-cut products. Food Chem. 2007, 104, 209–215. [Google Scholar] [CrossRef]
- Borgi, W.; Ghedira, K.; Chouchane, N. Antiinflammatory and analgesic activities of Zizyphus lotus root barks. Fitoterapia 2007, 78, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Feng, X.; Zhu, K.; Zhao, X.; Suo, H. Preventive activity of banana peel polyphenols on CCl4-induced experimental hepatic injury in Kunming mice. Exp. Ther. Med. 2016, 11, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Roh, E.; Kim, J.E.; Kwon, J.Y.; Park, J.S.; Bode, A.M.; Dong, Z.; Lee, K.W. Molecular mechanisms of green tea polyphenols with protective effects against skin photoaging. Crit. Rev. Food Sci. Nutr. 2017, 57, 1631–1637. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, Y.; Nagao, K.; Kai, S.; Tsuge, K.; Yoshimura, T.; Koganemaru, K.; Yanagita, T. Polyphenolic extract of lotus root (edible rhizome of Nelumbo nucifera) alleviates hepatic steatosis in obese diabetic db/db mice. Lipids Health Dis. 2011, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Randhir, R.; Lin, Y.; Shetty, K. Stimulation of phenolics, antioxidant and antimicrobial activities in dark germinated mung bean sprouts in response to peptide and phytochemical elicitors. Process. Biochem. 2004, 39, 637–646. [Google Scholar] [CrossRef]
- Lin, D.; Xiao, M.; Zhao, J.; Li, Z.; Xing, B.; Li, X.; Kong, M.; Li, L.; Zhang, Q.; Liu, Y.; Chen, S. An Overview of plant phenolic compounds and their importance in human nutrition and management of Type 2 Diabetes. Molecules 2016, 21, 1374. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, Z.; Zakrzewska, A.; Agoston, V.C.; Ordas, A.; Racz, P.; Mink, M.; Spaink, H.P.; Meijer, A.H. Deep sequencing of the zebrafish transcriptome response to mycobacterium infection. Mol. Immunol. 2009, 46, 2918–2930. [Google Scholar] [CrossRef] [PubMed]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.; Rothberg, J.M. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef]
- Frishberg, A.; Peshes-Yaloz, N.; Cohn, O.; Rosentul, D.; Steuerman, Y.; Valadarsky, L.; Yankovitz, G.; Mandelboim, M.; Iraqi, F.A.; Amit, I.; Gat-Viks, I. Cell composition analysis of bulk genomics using single-cell data. Nat. Methods 2019. [Google Scholar] [CrossRef]
- Zenoni, S.; Ferrarini, A.; Giacomelli, E.; Xumerle, L.; Fasoli, M.; Malerba, G.; Bellin, D.; Pezzotti, M.; Delledonne, M. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol. 2010, 152, 1787–1795. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jin, Q.; Zhang, X.; Mattson, N.S.; Ren, H.; Cao, J.; Wang, Y.; Yao, D.; Xu, Y. Genome-wide transcriptional analysis of submerged lotus reveals cooperative regulation and gene responses. Sci. Rep. 2018, 8, 9187. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Damaris, R.N.; Shi, T.; Li, J.; Yang, P. Transcriptomic analysis identifies the key genes involved in stamen petaloid in lotus (Nelumbo nucifera). BMC Genom. 2018, 19, 554. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Zhang, D.; Qi, S.; Chen, X.; An, N.; Li, Y.; Zhao, C.; Han, M.; Zhao, J. Transcription profiles reveal the regulatory mechanisms of spur bud changes and flower induction in response to shoot bending in apple (Malus domestica Borkh.). Plant Mol. Biol. 2019, 99, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Singh, V.K.; Rajkumar, M.S.; Kumar, V.; Jain, M. Global transcriptome and coexpression network analyses reveal cultivar-specific molecular signatures associated with seed development and seed size/weight determination in chickpea. Plant J. 2017, 91, 1088–1107. [Google Scholar] [CrossRef]
- Costacou, T.; Mayer-Davis, E.J. Nutrition and prevention of type 2 diabetes. Annu. Rev. Nutr. 2003, 23, 147–170. [Google Scholar] [CrossRef]
- Hung, H.; Joshipura, K.J.; Jiang, R.; Hu, F.B.; Hunter, D.; Smith-Warner, S.A.; Colditz, G.A.; Rosner, B.; Spiegelman, D.; Willett, W.C. Fruit and vegetable intake and risk of major chronic disease. J. Natl. Cancer I. 2004, 96, 1577–1584. [Google Scholar] [CrossRef]
- Nöthlings, U.; Schulze, M.B.; Weikert, C.; Boeing, H.; Van Der Schouw, Y.T.; Bamia, C.; Benetou, V.; Lagiou, P.; Krogh, V.; Beulens, J.W. Intake of vegetables, legumes, and fruit, and risk for all-cause, cardiovascular, and cancer mortality in a European diabetic population. J. Nutr. 2008, 138, 775–781. [Google Scholar] [CrossRef]
- Liu, J.; Yang, C.Q.; Zhang, Q.; Lou, Y.; Wu, H.J.; Deng, J.C.; Yang, F.; Yang, W.Y. Partial improvements in the flavor quality of soybean seeds using intercropping systems with appropriate shading. Food Chem. 2016, 207, 107–114. [Google Scholar] [CrossRef]
- Hoff, J.E.; Singleton, K.I. A method for determination of tannins in foods by means of immobilized protein. J. Food Sci. 1977, 42, 1566–1569. [Google Scholar] [CrossRef]
- Alu Datt, M.H.; Rababah, T.; Alhamad, M.N.; Al-Mahasneh, M.A.; Almajwal, A.; Gammoh, S.; Ereifej, K.; Johargy, A.; Alli, I. A review of phenolic compounds in oil-bearing plants: Distribution, identification and occurrence of phenolic compounds. Food Chem. 2017, 218, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Oomah, B.D.; Kenaschuk, E.O.; Mazza, G. Phenolic acids in Flaxseed. J. Agric. Food Chem. 1995, 43, 2016–2019. [Google Scholar] [CrossRef]
- Lorenc-Kukula, K.; Amarowicz, R.; Oszmianski, J.; Doermann, P.; Starzycki, M.; Skala, J.; Zuk, M.; Kulma, A.; Szopa, J. Pleiotropic effect of phenolic compounds content increases in transgenic flax plant. J. Agric. Food Chem. 2005, 53, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Maga, J.A.; Lorenz, K. Gas-liquid chromatography separation of the free phenolic acid fractions in various oilseed protein sources. J. Sci. Food Agric. 1974, 25, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, K.J.; Sosulski, F.W. Composition of free and hydrolyzable phenolic acids in defatted flours of ten oilseeds. J. Agric. Food Chem. 1984, 32, 128–130. [Google Scholar] [CrossRef]
- Chen, J.Y.; He, L.H.; Jiang, J.F. Expression of PAL and HSPs in fresh-cut banana fruit. Environ. Exp. Bot. 2009, 66, 31–37. [Google Scholar] [CrossRef]
- Yi, Y.; Sun, J.; Xie, J.; Min, T.; Wang, L.; Wang, H. Phenolic Profiles and Antioxidant Activity of Lotus Root Varieties. Molecules 2016, 21, 863. [Google Scholar] [CrossRef]
- Tohma, Y.; Tokunaga, K.; Nagase, S.; Murata, Y. Structural approach to the estimation of the number of residual software faults based on the hyper-geometric distribution. IEEE Trans. Softw. Eng. 1989, 15, 345–355. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Raw Reads (M) | Total Clean Reads (M) | Total Clean Bases (Gb) | Clean Reads Q20 (%) | Clean Reads Q30 (%) | Clean Reads Ratio (%) | Total Mapping (%) | Uniquely Mapping (%) |
|---|---|---|---|---|---|---|---|---|
| S1A | 25.94 | 25.54 | 1.28 | 98.32 | 91.52 | 98.47 | 88.28 | 76.82 |
| S1B | 21.94 | 21.66 | 1.08 | 98.32 | 90.64 | 98.73 | 88.71 | 77.37 |
| S1C | 21.94 | 21.77 | 1.09 | 98.47 | 91.5 | 99.2 | 88.42 | 77.16 |
| S2A | 25.74 | 25.5 | 1.27 | 98.4 | 91.55 | 99.05 | 86.9 | 75.51 |
| S2B | 21.94 | 21.81 | 1.09 | 98.32 | 90.71 | 99.38 | 88.41 | 76.93 |
| S2C | 21.94 | 21.82 | 1.09 | 98.31 | 90.71 | 99.42 | 88.73 | 77.04 |
| S3A | 25.75 | 25.37 | 1.27 | 98.36 | 91.51 | 98.49 | 75.22 | 64.29 |
| S3B | 21.94 | 21.78 | 1.09 | 98.3 | 90.54 | 99.25 | 86.11 | 73 |
| S3C | 21.94 | 21.83 | 1.09 | 98.39 | 90.97 | 99.47 | 85.73 | 72.65 |
| S4A | 26.18 | 24.85 | 1.24 | 95.33 | 83.71 | 94.93 | 83.75 | 67.79 |
| S4B | 21.94 | 21.77 | 1.09 | 98.4 | 90.93 | 99.22 | 86.99 | 69.13 |
| S4C | 21.94 | 21.84 | 1.09 | 98.34 | 90.6 | 99.53 | 88.11 | 70.06 |
| S5A | 28.4 | 25.62 | 1.28 | 96.62 | 86.76 | 90.2 | 80.08 | 66.9 |
| S5B | 21.94 | 21.8 | 1.09 | 98.42 | 91.03 | 99.37 | 85.09 | 71.41 |
| S5C | 21.94 | 21.85 | 1.09 | 98.55 | 91.5 | 99.58 | 85.77 | 71.73 |
| S6A | 25.31 | 24.44 | 1.22 | 98.15 | 90.73 | 96.57 | 83.23 | 65.4 |
| S6B | 21.94 | 21.75 | 1.09 | 98.4 | 91.03 | 99.13 | 87.41 | 71.06 |
| S6C | 21.94 | 21.82 | 1.09 | 98.38 | 90.82 | 99.44 | 87.32 | 71.43 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, T.; Bao, Y.; Zhou, B.; Yi, Y.; Wang, L.; Hou, W.; Ai, Y.; Wang, H. Transcription Profiles Reveal the Regulatory Synthesis of Phenols during the Development of Lotus Rhizome (Nelumbo nucifera Gaertn). Int. J. Mol. Sci. 2019, 20, 2735. https://doi.org/10.3390/ijms20112735
Min T, Bao Y, Zhou B, Yi Y, Wang L, Hou W, Ai Y, Wang H. Transcription Profiles Reveal the Regulatory Synthesis of Phenols during the Development of Lotus Rhizome (Nelumbo nucifera Gaertn). International Journal of Molecular Sciences. 2019; 20(11):2735. https://doi.org/10.3390/ijms20112735
Chicago/Turabian StyleMin, Ting, Yinqiu Bao, Baixue Zhou, Yang Yi, Limei Wang, Wenfu Hou, Youwei Ai, and Hongxun Wang. 2019. "Transcription Profiles Reveal the Regulatory Synthesis of Phenols during the Development of Lotus Rhizome (Nelumbo nucifera Gaertn)" International Journal of Molecular Sciences 20, no. 11: 2735. https://doi.org/10.3390/ijms20112735
APA StyleMin, T., Bao, Y., Zhou, B., Yi, Y., Wang, L., Hou, W., Ai, Y., & Wang, H. (2019). Transcription Profiles Reveal the Regulatory Synthesis of Phenols during the Development of Lotus Rhizome (Nelumbo nucifera Gaertn). International Journal of Molecular Sciences, 20(11), 2735. https://doi.org/10.3390/ijms20112735