Targeting Angiogenesis in Prostate Cancer



Abstract
1. Introduction
2. Background
2.1. Prostate Cancer
2.2. Treatment Options in Prostate Cancer
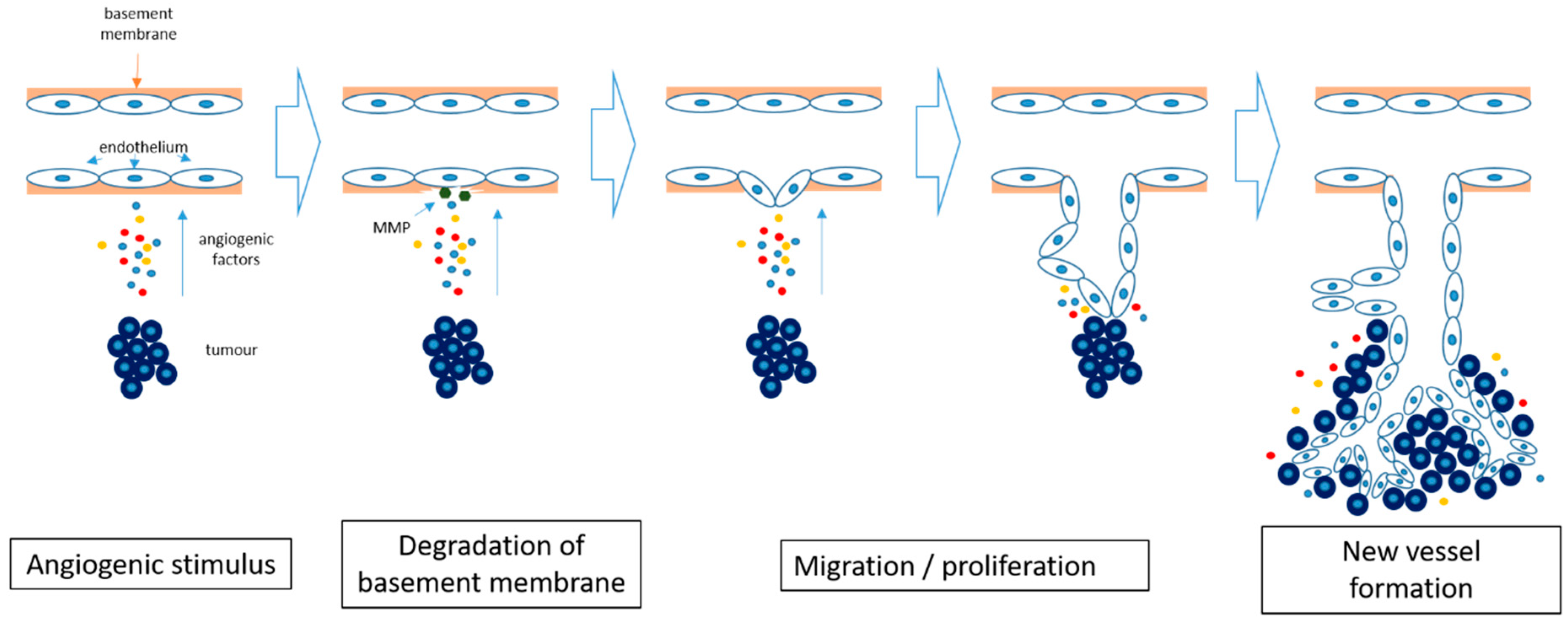
2.3. Angiogenesis in Cancer
2.4. Angiogenesis Inhibition in Cancer
3. Results
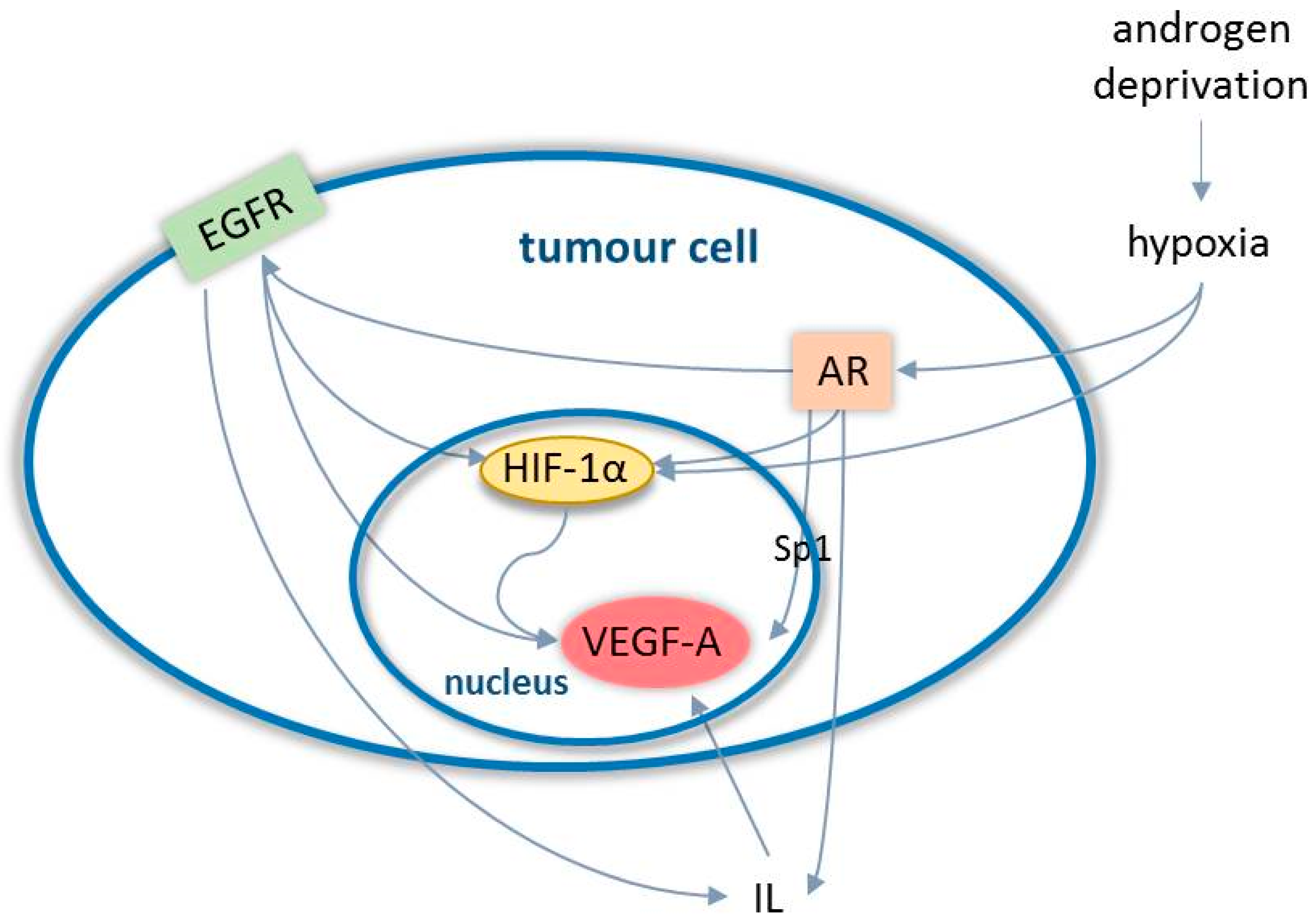
3.1. Angiogenesis in Prostate Cancer
3.2. Anti-Angiogenesis Clinical Studies in Prostate Cancer
4. Discussion
5. Materials and Methods
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- National Cancer Institute. SEER Stat Fact Sheets: Prostate; National Cancer Institute: Bethesda, MD, USA. Available online: https://seer.cancer.gov/statfacts/html/prost.html#prevalence (accessed on 10 March 2018).
- American Cancer Society. Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2018; Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2018/cancer-facts-and-figures-2018.pdf (accessed on 10 March 2018).
- Cancer Research UK. Prostate Cancer Incidence Statistics [Internet]. 2014. Available online: http://www.cancerresearchuk.org/cancer-info/cancerstats/types/prostate/incidence/#age (accessed on 14 August 2018).
- Zlotta, A.R.; Egawa, S.; Pushkar, D.; Govorov, A.; Kimura, T.; Kido, M.; Takahashi, H.; Kuk, C.; Kovylina, M.; Aldaoud, N.; et al. Prevalence of prostate cancer on autopsy: Cross-sectional study on unscreened Caucasian and Asian men. J. Natl. Cancer Inst. 2013, 105, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2012; Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2012/estimated-number-of-new-cancer-cases-and-deaths-by-sex-2012.pdf (accessed on 19 August 2018).
- The National Cancer Registration Service, Eastern Office [Internet]. Available online: http://www.ncras.nhs.uk/ncrs-east/ (accessed on 14 August 2018).
- Zelefsky, M.J.; Eastham, J.A.; Sartor, A.O. Cancer of the prostate. In Cancer: Principles and Practice of Oncology, 9th ed.; De Vita, V.T., Jr., Lawrence, T.S., Rosenberg, S.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011; pp. 1220–7121. [Google Scholar]
- PDQ Adult Treatment Editorial Board. Prostate Cancer Treatment (PDQ®): Patient Version. 30 April 2018. In PDQ Cancer Information Summaries [Internet]; National Cancer Institute (US): Bethesda, MD, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK65915/ (accessed on 19 August 2018).
- Hamdy, F.C.; Donovan, J.L.; Lane, J.A.; Mason, M.; Metcalfe, C.; Holding, P.; Davis, M.; Peters, T.J.; Turner, E.L.; Martin, R.M.; et al. 10-Year Outcomes after Monitoring, Surgery, or Radiotherapy for Localized Prostate Cancer. N. Engl. J. Med. 2016, 375, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.; Kirkbride, P.; Cann, K.; Hasler, E.; Prettyjohns, M. Prostate cancer:summary of updated NICE guidance. BMJ 2014, 8, 348. [Google Scholar] [CrossRef]
- Ragde, H.; Blasko, J.C.; Grimm, P.D.; Kenny, G.M.; Sylvester, J.E.; Hoak, D.C.; Landin, K.; Cavanagh, W. Interstitial iodine-125 radiation without adjuvant therapy in the treatment of clinically localized prostate carcinoma. Cancer 1997, 80, 442–453. [Google Scholar] [CrossRef]
- The Medical Research Council Prostate Cancer Working Party Investigators Group. Immediate versus deferred treatment for advanced prostatic cancer: Initial results of the Medical Research Council Trial. Br. J. Urol. 1997, 79, 235–426. [Google Scholar]
- Dearnaley, D.P.; Mason, M.D.; Parmar, M.K.; Sanders, K.; Sydes, M.R. Adjuvant therapy with oral sodium clodronate in locally advanced and metastatic prostate cancer: Long-term overall survival results from the MRC PR04 and PR05 randomizedcontrolled trials. Lancet Oncol. 2009, 10, 872–876. [Google Scholar] [CrossRef]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.; Amos, C.L.; Gilson, C.; Jones, R.J. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef]
- De Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomizedopen-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Fizazi, K.; Carducci, M.; Smith, M.; Damião, R.; Brown, J.; Karsh, L.; Milecki, P.; Shore, N.; Rader, M.; Wang, H.; et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomized, double-blind study. Lancet 2011, 377, 813–822. [Google Scholar] [CrossRef]
- Oosterhof, G.O.N.; Roberts, J.T.; de Reijke, T.M.; Engelholm, S.A.; Horenblas, S.; von der Maase, H.; Neymark, N.; Debois, M. ColletteL. Strontium (89) chloride versus palliative local field radiotherapy in patients with hormonal escaped prostate cancer: A phase III study of the European Organisation for Research and Treatment of Cancer, Genitourinary Group. Eur. Urol. 2003, 44, 519–526. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef]
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 21, 34. [Google Scholar] [CrossRef]
- Winkler, F. Hostile takeover: How tumors hijack pre-existing vascular environments to thrive. J. Pathol. 2017, 242, 267–272. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Pavlakovic, H.; Havers, W.; Schweigerer, L. Multiple angiogenesis stimulators in a single malignancy: Implications for anti-angiogenic tumor therapy. Angiogenesis 2001, 4, 259–262. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef]
- Gressett, M.; Shah, S.R. Intricacies of bevacizumab-induced toxicities and their management. Ann. Pharmacother. 2009, 43, 490–501. [Google Scholar] [CrossRef]
- Kamba, T.; McDonald, D.M. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br. J. Cancer 2007, 96, 1788–1795. [Google Scholar] [CrossRef]
- Ferrara, N. VEGF as a therapeutic target in cancer. Oncology 2005, 69 (Suppl. 3), 11–16. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- El-Kenawi, A.E.; El-Remessy, A.B. Angiogenesis inhibitors in cancer therapy: Mechanistic perspective on classification and treatment rationales. Br. J. Pharmacol. 2013, 170, 712–729. [Google Scholar] [CrossRef]
- Mundel, T.M.; Kalluri, R. Type IV collagen-derived angiogenesis inhibitors. Microvasc. Res. 2007, 74, 85–89. [Google Scholar] [CrossRef]
- Kurozumi, K.; Ichikawa, T.; Onishi, M.; Fujii, K.; Date, I. Cilengitide treatment for malignant glioma: Current status and future direction. Neurol. Med. Chir. 2012, 52, 539–547. [Google Scholar] [CrossRef]
- Su, J.; Cai, M.; Li, W.; Hou, B.; He, H.; Ling, C.; Huang, T.; Liu, H.; Guo, Y. Molecularly Targeted Drugs Plus Radiotherapy and Temozolomide Treatment for Newly Diagnosed Glioblastoma: A Meta-Analysis and Systematic Review. Oncol. Res. 2016, 24, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Herbert, S.P.; Stainier, D.Y. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell. Biol. 2011, 12, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Margolin, K.; Gordon, M.S.; Holmgren, E.; Gaudreault, J.; Novotny, W.; Fyfe, G.; Adelman, D.; Stalter, S.; Breed, J. Phase Ib trial of intravenous recombinant humanized monoclonal antibody to vascular endothelial growth factor in combination with chemotherapy in patients with advanced cancer: Pharmacologic and long-term safety data. J. Clin. Oncol. 2011, 19, 851–856. [Google Scholar] [CrossRef]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef]
- Li, M.; Kroetz, D.L. Bevacizumab-induced hypertension: Clinical presentation and molecular understanding. Pharmacol. Ther. 2018, 182, 152–160. [Google Scholar] [CrossRef]
- Minder, P.; Zajac, E.; Quigley, J.P.; Deryugina, E.I. EGFR Regulates the Development and Microarchitecture of Intratumoral Angiogenic Vasculature Capable of Sustaining Cancer Cell Intravasation. Neoplasia 2015, 17, 634–649. [Google Scholar] [CrossRef]
- Sharma, S.; Sharma, M.C.; Sarkar, C. Morphology of angiogenesis in human cancer: A conceptual overview, histoprognostic perspective and significance of neoangiogenesis. Histopathology 2005, 46, 481–489. [Google Scholar] [CrossRef]
- Bono, A.V.; Celato, N.; Cova, V.; Salvadore, M.; Chinetti, S.; Novario, R. Microvessel density in prostate carcinoma. Prostate Cancer Prostatic Dis. 2002, 5, 123–127. [Google Scholar] [CrossRef]
- Borre, M.; Offersen, B.V.; Nerstrom, B.; Overgaard, J. Microvessel density predicts survival in prostate cancer patients subjected to watchful waiting. Br. J. Cancer 1998, 78, 940–944. [Google Scholar] [CrossRef]
- Jiang, J.; Chen, Y.; Zhu, Y.; Yao, X.; Qi, J. Contrast-enhanced ultrasonography for the detection and characterization of prostate cancer: Correlation with microvessel density and Gleason score. Clin. Radiol. 2011, 66, 732–737. [Google Scholar] [CrossRef]
- Tretiakova, M.; Antic, T.; Binder, D.; Kocherginsky, M.; Liao, C.; Taxy, J.B.; Oto, A. Microvessel density is not increased in prostate cancer: Digital imaging of routine sections and tissue microarrays. Hum. Pathol. 2013, 44, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Saka, H. Reconsideration of the clinical and histopathological significance of angiogenesis in prostate cancer: Usefulness and limitations of microvessel density measurement. Int. J. Urol. 2015, 22, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Taverna, G.; Grizzi, F.; Colombo, P.; Seveso, M.; Giusti, G.; Proietti, S.; Fiorini, G.; Lughezzani, G.; Casale, P.; Buffi, N.; et al. Two-dimensional neovascular complexity is significantly higher in nontumor prostate tissue than in low-risk prostate cancer. Korean J. Urol. 2015, 56, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Taverna, G.; Grizzi, F.; Colombo, P.; Graziotti, P. Is angiogenesis a hallmark of prostate cancer? Front. Oncol. 2013, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- De Brot, S.; Ntekim, A.; Cardenas, R.; James, V.; Allegrucci, C.; Heery, D.M.; Bates, D.O.; Ødum, N.; Persson, J.L.; Mongan, N.P. Regulation of vascular endothelial growth factor in prostate cancer. Endocr. Relat. Cancer 2015, 22, 107–123. [Google Scholar] [CrossRef]
- Wong, S.Y.; Haack, H.; Crowley, D.; Barry, M.; Bronson, R.T.; Hynes, R.O. Tumor-secreted vascular endothelial growth factor-C is necessary for prostate cancer lymphangiogenesis, but lymphangiogenesis is unnecessary for lymph node metastasis. Cancer Res. 2005, 65, 9789–9798. [Google Scholar] [CrossRef]
- Wegiel, B.; Bjartell, A.; Ekberg, J.; Gadaleanu, V.; Brunhoff, C.; Persson, J.L. A role for cyclin A1 in mediating the autocrine expression of vascular endothelial growth factor in prostate cancer. Oncogene 2005, 24, 6385–6393. [Google Scholar] [CrossRef]
- Green, M.M.; Hiley, C.T.; Shanks, J.H.; Bottomley, I.C.; West, C.M.; Cowan, R.A.; Stratford, I.J. Expression of vascular endothelial growth factor (VEGF) in locally invasive prostate cancer is prognostic for radiotherapy outcome. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 84–90. [Google Scholar] [CrossRef]
- Duque, J.L.; Loughlin, K.R.; Adam, R.M.; Kantoff, P.W.; Zurakowski, D.; Freeman, M.R. Plasma levels of vascular endothelial growth factor are increased in patients with metastatic prostate cancer. Urology 1999, 54, 523–527. [Google Scholar] [CrossRef]
- Hrouda, D.; Nicol, D.L.; Gardiner, R.A. The role of angiogenesis in prostate development and the pathogenesis of prostate cancer. Urol. Res. 2003, 30, 347–355. [Google Scholar]
- McKay, R.R.; Zurita, A.J.; Werner, L.; Bruce, J.Y.; Carducci, M.A.; Stein, M.N.; Heath, E.I.; Hussain, A.; Tran, H.T.; Sweeney, C.J.; et al. Randomized Phase II Trial of Short-Course Androgen Deprivation Therapy With or Without Bevacizumab for Patients With Recurrent Prostate Cancer After Definitive Local Therapy. J. Clin. Oncol. 2016, 34, 1913–1920. [Google Scholar] [CrossRef]
- Kelly, W.K.; Halabi, S.; Carducci, M.; George, D.; Mahoney, J.F.; Stadler, W.M.; Morris, M.; Kantoff, P.; Monk, J.P.; Kaplan, E.; et al. Randomized, double-blind, placebo-controlled phase III trial comparing docetaxel and prednisone with or without bevacizumab in men with metastatic castration-resistant prostate cancer: CALGB 90401. J. Clin. Oncol. 2012, 30, 1534–1540. [Google Scholar] [CrossRef]
- Tannock, I.F.; Fizazi, K.; Ivanov, S.; Karlsson, C.T.; Fléchon, A.; Skoneczna, I.; Orlandi, F.; Gravis, G.; Matveev, V.; Bavbek, S.; et al. Aflibercept versus placebo in combination with docetaxel and prednisone for treatment of men with metastatic castration-resistant prostate cancer (VENICE): A phase 3, double-blind randomizedtrial. Lancet Oncol. 2013, 14, 760–768. [Google Scholar] [CrossRef]
- Michaelson, M.D.; Oudard, S.; Ou, Y.C.; Sengeløv, L.; Saad, F.; Houede, N.; Ostler, P.; Stenzl, A.; Daugaard, G.; Jones, R.; et al. Randomized, placebo-controlled, phase III trial of sunitinib plus prednisone versus prednisone alone in progressive, metastatic, castration-resistant prostate cancer. J. Clin. Oncol. 2014, 32, 76–82. [Google Scholar] [CrossRef]
- Keizman, D.; Zahurak, M.; Sinibaldi, V.; Carducci, M.; Denmeade, S.; Drake, C.; Pili, R.; Antonarakis, E.S.; Hudock, S.; Eisenberger, M. Lenalidomide in nonmetastatic biochemically relapsed prostate cancer: Results of a phase I/II double-blinded, randomized study. Clin. Cancer Res. 2010, 16, 5269–5276. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Vogelzang, N.J.; Budnik, N.; Wiechno, P.J.; Sternberg, C.N.; Doner, K.; Bellmunt, J.; Burke, J.M.; de Olza, M.O.; Choudhury, A.; et al. Docetaxel and prednisone with or without lenalidomide in chemotherapy-naivepatients with metastatic castration-resistant prostate cancer (MAINSAIL): Arandomized, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2015, 16, 417–425. [Google Scholar] [CrossRef]
- Mangoni, M.; Vozenin, M.C.; Biti, G.; Deutsch, E. Normal tissues toxicities triggered by combined anti-angiogenic and radiation therapies: Hurdles might be ahead. Br. J. Cancer 2012, 107, 308–314. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ogita, S.; Tejwani, S.; Heilbrun, L.; Fontana, J.; Heath, E.; Freeman, S.; Smith, D.; Baranowski, K.; Vaishampayan, U. Pilot Phase II Trial of Bevacizumab Monotherapy in Nonmetastatic Castrate-Resistant Prostate Cancer. ISRN Oncol. 2012, 2012, 242850. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A. New Insights in Anti-Angiogenesis in Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 2031. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-Q.; Fang, J.-M.; Xiao, Y.-Y.; Zhao, Y.; Cui, R.; Hu, F.; Xu, Q. Prognostic role of vascular endothelial growth factor in prostate cancer: A systematic review and meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 2289–2298. [Google Scholar] [PubMed]
- Wang, K.; Peng, H.L.; Li, L.K. Prognostic value of vascular endothelial growth factorexpression in patients with prostate cancer: A systematic review withmeta-analysis. Asian Pac. J. Cancer Prev. 2012, 13, 5665–5669. [Google Scholar] [CrossRef] [PubMed]
- Scholz, A.; Harter, P.N.; Cremer, S.; Yalcin, B.H.; Gurnik, S.; Yamaji, M.; Di Tacchio, M.; Sommer, K.; Baumgarten, P.; Bähr, O.; et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol. Med. 2016, 8, 39–57. [Google Scholar] [CrossRef]
- Lindholm, E.M.; Krohn, M.; Iadevaia, S.; Kristian, A.; Mills, G.B.; Mælandsmo, G.M.; Engebraaten, O. Proteomic characterization of breast cancer xenografts identifies early and late bevacizumab-induced responses and predicts effective drug combinations. Clin. Cancer Res. 2014, 20, 404–412. [Google Scholar] [CrossRef]
- Madan, R.A.; Karzai, F.H.; Ning, Y.-M.; Adesunloye, B.A.; Huang, X.; Harold, N.; Couvillon, A.; Chun, G.; Cordes, L.; Sissung, T.; et al. Phase II trial of docetaxel, bevacizumab, lenalidomide and prednisone in patients with metastatic castration-resistant prostate cancer. BJU Int. 2016, 118, 590–597. [Google Scholar] [CrossRef]
- Brauer, M.J.; Zhuang, G.; Schmidt, M.; Yao, J.; Wu, X.; Kaminker, J.S.; Jurinka, S.S.; Kolumam, G.; Chung, A.S.; Jubb, A.; et al. Identification and analysis of in vivo VEGF downstream markers link VEGF pathway activity with efficacy of anti-VEGF therapies. Clin. Cancer Res. 2013, 19, 3681–3692. [Google Scholar] [CrossRef]
- De Haas, S.; Delmar, P.; Bansal, A.T.; Moisse, M.; Miles, D.W.; Leighl, N.; Escudier, B.; Van Cutsem, E.; Carmeliet, P.; Scherer, S.J.; et al. Genetic variability of VEGF pathway genes in six randomized Phase III trials assessing the addition of bevacizumab to standard therapy. Angiogenesis 2014, 17, 909–920. [Google Scholar] [CrossRef]
- Golovine, K.; Kutikov, A.; Teper, E.; Simhan, J.; Makhov, P.B.; Canter, D.J.; Uzzo, R.G.; Kolenko, V.M. Modulation of Akt/mTOR signalling overcomes sunitinib resistance in renal and prostate cancer cells. Mol. Cancer Ther. 2012, 11, 1510–1517. [Google Scholar]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef]
- Wang, Y.; Kreisberg, J.I.; Ghosh, P.M. Cross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr. Cancer Drug Targets 2007, 7, 591–604. [Google Scholar] [CrossRef]
- Yamamoto, Y.; A De Velasco, M.; Kura, Y.; Nozawa, M.; Hatanaka, Y.; Oki, T.; Ozeki, T.; Shimizu, N.; Minami, T.; Yoshimura, K.; et al. Evaluation of in vivo responses of sorafenib therapy in a preclinical mouse model of PTEN-deficient of prostate cancer. J. Transl. Med. 2015, 13, 150. [Google Scholar] [CrossRef]
- De Velasco, M.A.; Kura, Y.; Yoshikawa, K.; Nishio, K.; Davies, B.R.; Uemura, H. Efficacy of targeted AKT inhibition in genetically engineered mouse models of PTEN-deficient prostate cancer. Oncotarget 2016, 7, 15959–15976. [Google Scholar] [CrossRef]
- Sordello, S.; Bertrand, N.; Plouet, J. Vascular endothelial growth factor is up-regulated in vitro and in vivo by androgens. Biochem. Biophys. Res. Commun. 1998, 251, 287–290. [Google Scholar] [CrossRef]
- Eisermann, K.; Fraizer, G. The Androgen Receptor and VEGF: Mechanisms of Androgen-Regulated Angiogenesis in Prostate Cancer. Cancers 2017, 9, 32. [Google Scholar] [CrossRef]
- Kashyap, V.; Ahmad, S.; Nilsson, E.M.; Helczynski, L.; Kenna, S.; Persson, J.L.; Gudas, L.J.; Mongan, N.P. The lysine specific demethylase-1 (LSD1/KDM1A) regulates VEGF-A expression in prostate cancer. Mol. Oncol. 2013, 7, 555–566. [Google Scholar] [CrossRef]
- Deng, X.; Shao, G.; Zhang, H.; Li, C.; Zhang, D.; Cheng, L.; Elzey, B.; Pili, R.; Ratliff, T.; Huang, J. Proteinarginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene 2016, 36, 1223–1231. [Google Scholar] [CrossRef]
- Eisermann, K.; Broderick, C.J.; Bazarov, A.; Moazam, M.M.; Fraizer, G.C. Androgen up-regulates vascular endothelial growth factor expression in prostate cancer cells via an Sp1 binding site. Mol. Cancer 2013, 12, 7. [Google Scholar] [CrossRef]
- Antonarakis, E.; Armstrong, A.; Dehm, S.; Luo, J. Androgen receptor variant-driven prostate cancer: Clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis. 2016, 19, 231–241. [Google Scholar] [CrossRef]
- Fernandez, E.V.; Reece, K.M.; Ley, A.M.; Troutman, S.M.; Sissung, T.M.; Price, D.K.; Chau, C.H.; Figg, W.D. Dual targeting of the androgen receptor and hypoxia-inducible factor 1α pathways synergistically inhibits castration-resistant prostate cancer cells. Mol. Pharmacol. 2015, 87, 1006–1012. [Google Scholar] [CrossRef]
- Pignon, J.C.; Koopmansch, B.; Nolens, G.; Delacroix, L.; Waltregny, D.; Winkler, R. Androgen receptor controls EGFR and ERBB2 gene expression at different levels in prostate cancer cell lines. Cancer Res. 2009, 69, 2941–2949. [Google Scholar] [CrossRef]
- Zheng, Y.; Izumi, K.; Yao, J.L.; Miyamoto, H. Dihydrotestosterone upregulates the expression of epidermal growth factor receptor and ERBB2 in androgen receptor-positive bladder cancer cells. Endocr. Relat. Cancer 2011, 18, 451–464. [Google Scholar] [CrossRef]
- Tabernero, J. The role of VEGF and EGFR inhibition: Implications for combining anti-VEGF and anti-EGFR agents. Mol. Cancer Res. 2007, 5, 203–220. [Google Scholar] [CrossRef]
- Mabjeesh, N.J.; Willard, M.T.; Frederickson, C.E.; Zhong, H.; Simons, J.W. Androgens stimulate hypoxia-inducible factor 1 activation via autocrine loop of tyrosine kinase receptor/phosphatidylinositol 3′-kinase/protein kinase B in prostate cancer cells. Clin. Cancer Res. 2003, 9, 2416–2425. [Google Scholar]
- Cereda, V.; Formica, V.; Roselli, M. Issues and promises of bevacizumab in prostate cancer treatment. Exp. Opin. Biol. Ther. 2018, 18, 707–717. [Google Scholar] [CrossRef]
- Shabsigh, A.; Ghafar, M.A.; De La Taille, A.; Burchardt, M.; Kaplan, S.A.; Anastasiadis, A.G.; Buttyan, R. Biomarker analysis demonstrates a hypoxic environment in the castrated rat ventral prostate gland. J. Cell Biochem. 2001, 81, 437–444. [Google Scholar] [CrossRef]
- Halin, S.; Hammarsten, P.; Wikström, P.; Bergh, A. Androgen-insensitive prostate cancer cells transiently respond to castration treatment when growing in an androgen-dependent prostate environment. Prostate 2007, 67, 370–377. [Google Scholar] [CrossRef]
- Mitani, T.; Harada, N.; Nakano, Y.; Inui, H.; Yamaji, R. Coordinated action of hypoxia-inducible factor-1α and β-catenin in androgen receptor signaling. J. Biol. Chem. 2012, 287, 33594–33606. [Google Scholar] [CrossRef]
- Horii, K.; Suzuki, Y.; Kondo, Y.; Akimoto, M.; Nishimura, T.; Yamabe, Y.; Sakaue, M.; Sano, T.; Kitagawa, T.; Himeno, S.; et al. Androgen-dependent gene expression of prostate-specific antigen is enhanced synergistically by hypoxia in human prostate cancer cells. Mol. Cancer Res. 2007, 5, 383–391. [Google Scholar] [CrossRef]
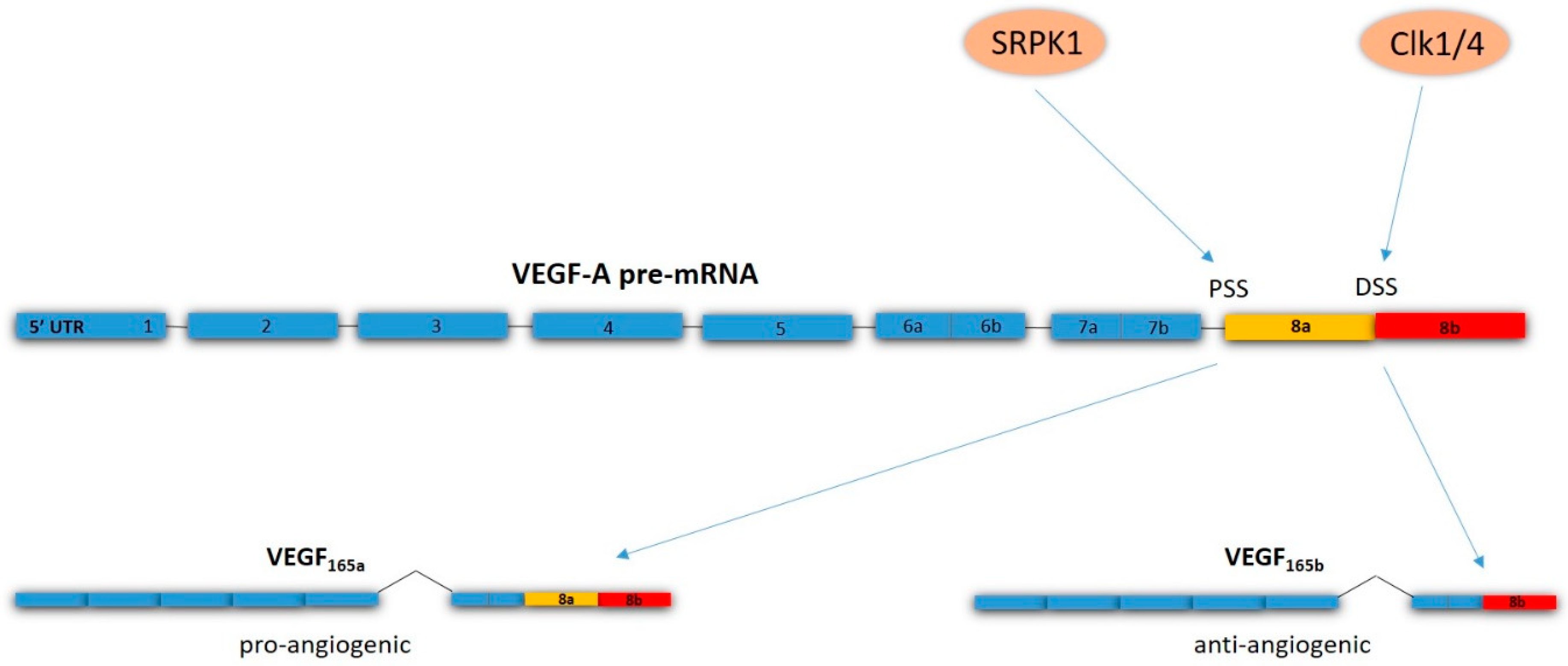
- Bates, D.; Cui, T.-G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131. [Google Scholar]
- Woolard, J.; Wang, W.Y.; Bevan, H.S.; Qiu, Y.; Morbidelli, L.; Pritchard-Jones, R.O.; Cui, T.G.; Sugiono, M.; Waine, E.; Perrin, R.; et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: Mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004, 64, 7822–7835. [Google Scholar] [CrossRef]
- Oltean, S.; Gammons, M.; Hulse, R.; Hamdollah-Zadeh, M.; Mavrou, A.; Donaldson, L.; Salmon, A.H.; Harper, S.J.; Ladomery, M.R.; Bates, D.O. SRPK1 inhibition in vivo: Modulation of VEGF splicing and potential treatment for multiple diseases. Biochem. Soc. Trans. 2012, 40, 831–835. [Google Scholar] [CrossRef]
- Auboeuf, D.; Dowhan, D.H.; Kang, Y.K.; Larkin, K.; Lee, J.W.; Berget, S.M.; O’Malley, B.W. Differential recruitment of nuclear receptor coactivators may determine alternative RNA splice site choice in target genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2270–2274. [Google Scholar] [CrossRef]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.-K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef]
- Nowak, D.G.; Woolard, J.; Amin, E.M.; Konopatskaya, O.; Saleem, M.A.; Churchill, A.J.; Ladomery, M.R.; Harper, S.J.; Bates, D.O. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J. Cell Sci. 2008, 121, 3487–3495. [Google Scholar] [CrossRef]
- Nowak, D.G.; Amin, E.M.; Rennel, E.S.; Hoareau-Aveilla, C.; Gammons, M.; Damodoran, G.; Hagiwara, M.; Harper, S.J.; Woolard, J.; Ladomery, M.R.; et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: A novel therapeutic strategy for angiogenesis. J. Biol. Chem. 2010, 285, 5532–5540. [Google Scholar] [CrossRef]
- Mavrou, A.; Brakspear, K.; Hamdollah-Zadeh, M.; Damodaran, G.; Babaei-Jadidi, R.; Oxley, J.; Gillatt, D.A.; Ladomery, M.R.; Harper, S.J.; Bates, D.O.; Oltean, S. Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 2015, 34, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
- Mavrou, A.; Oltean, S. SRPK1 inhibition in prostate cancer: A novel anti-angiogenic treatment through modulation of VEGF alternative splicing. Pharmacol. Res. 2016, 107, 276–281. [Google Scholar] [CrossRef]
- Van den Brûle, F.A.; Waltregny, D.; Castronovo, V. Increased expression of galectin-1 in carcinoma-associated stroma predicts poor outcome in prostate carcinoma patients. J. Pathol. 2001, 193, 80–87. [Google Scholar] [CrossRef]
- Stanley, P. Galectin-1 Pulls the Strings on VEGFR2. Cell 2014, 156, 625–626. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, F.M.; Gentilini, L.D.; Gueron, G.; Meiss, R.P.; Ortiz, E.G.; Berguer, P.M.; Ahmed, A.; Navone, N.; Rabinovich, G.A.; Compagno, D.; et al. In Vivo Hemin Conditioning Targets the Vascular and Immunologic Compartments and Restrains Prostate Tumor Development. Clin. Cancer Res. 2017, 23, 5135–5148. [Google Scholar] [CrossRef] [PubMed]
- Laderach, D.J.; Gentilini, L.D.; Giribaldi, L.; Delgado, V.C.; Nugnes, L.; Croci, D.O.; Al Nakouzi, N.; Sacca, P.; Casas, G.; Mazza, O.; et al. A Unique Galectin Signature in Human Prostate Cancer Progression Suggests Galectin-1 as a Key Target for Treatment of Advanced Disease. Cancer Res. 2013, 73, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Goud, N.S.; Soukya, P.S.L.; Ghouse, M.; Komal, D.; Alvala, R.; Alvala, M. Human Galectin-1 and its inhibitors: Privileged target for cancer and HIV. Mini Rev. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Level of Risk | PSA Level (ng/mL) | Gleason Score | Clinical Stage | ||
|---|---|---|---|---|---|
| Low risk | <10 | and | ≤6 | and | T1–T2a |
| Intermediate risk | 10–20 | or | 7 | or | T2b |
| High risk | >20 | or | 8–10 | or | ≥T2c |
| Drug | Mechanism of Action | Phase of the Clinical Trial | Number of Patients | Outcome |
|---|---|---|---|---|
| Bevacizumab | Recombinant humanized monoclonal antibody that blocks VEGF-A | II | 99 | Improved relapse-free survival [54] |
| III | 1050 | No improvement in overall survival [55] | ||
| Aflibercept | Binds to circulating VEGF-A | III | 1224 | No improvement in overall survival [56] |
| Sunitinib | Receptor tyrosine kinase inhibitor | III | 873 | No improvement in overall survival [57] |
| Lenalidomide | Multiple mechanisms, including inhibition of VEGF-induced PI3K-Akt pathway signalling | I/II | 60 | Disease stabilisation, decrease in PSA [58] |
| III | 1059 | Worse overall survival [59] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melegh, Z.; Oltean, S. Targeting Angiogenesis in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2676. https://doi.org/10.3390/ijms20112676
Melegh Z, Oltean S. Targeting Angiogenesis in Prostate Cancer. International Journal of Molecular Sciences. 2019; 20(11):2676. https://doi.org/10.3390/ijms20112676
Chicago/Turabian StyleMelegh, Zsombor, and Sebastian Oltean. 2019. "Targeting Angiogenesis in Prostate Cancer" International Journal of Molecular Sciences 20, no. 11: 2676. https://doi.org/10.3390/ijms20112676
APA StyleMelegh, Z., & Oltean, S. (2019). Targeting Angiogenesis in Prostate Cancer. International Journal of Molecular Sciences, 20(11), 2676. https://doi.org/10.3390/ijms20112676