Hiseq Base Molecular Characterization of Soil Microbial Community, Diversity Structure, and Predictive Functional Profiling in Continuous Cucumber Planted Soil Affected by Diverse Cropping Systems in an Intensive Greenhouse Region of Northern China


Abstract
1. Introduction
2. Results
2.1. Soil Properties and Cucumber Yield
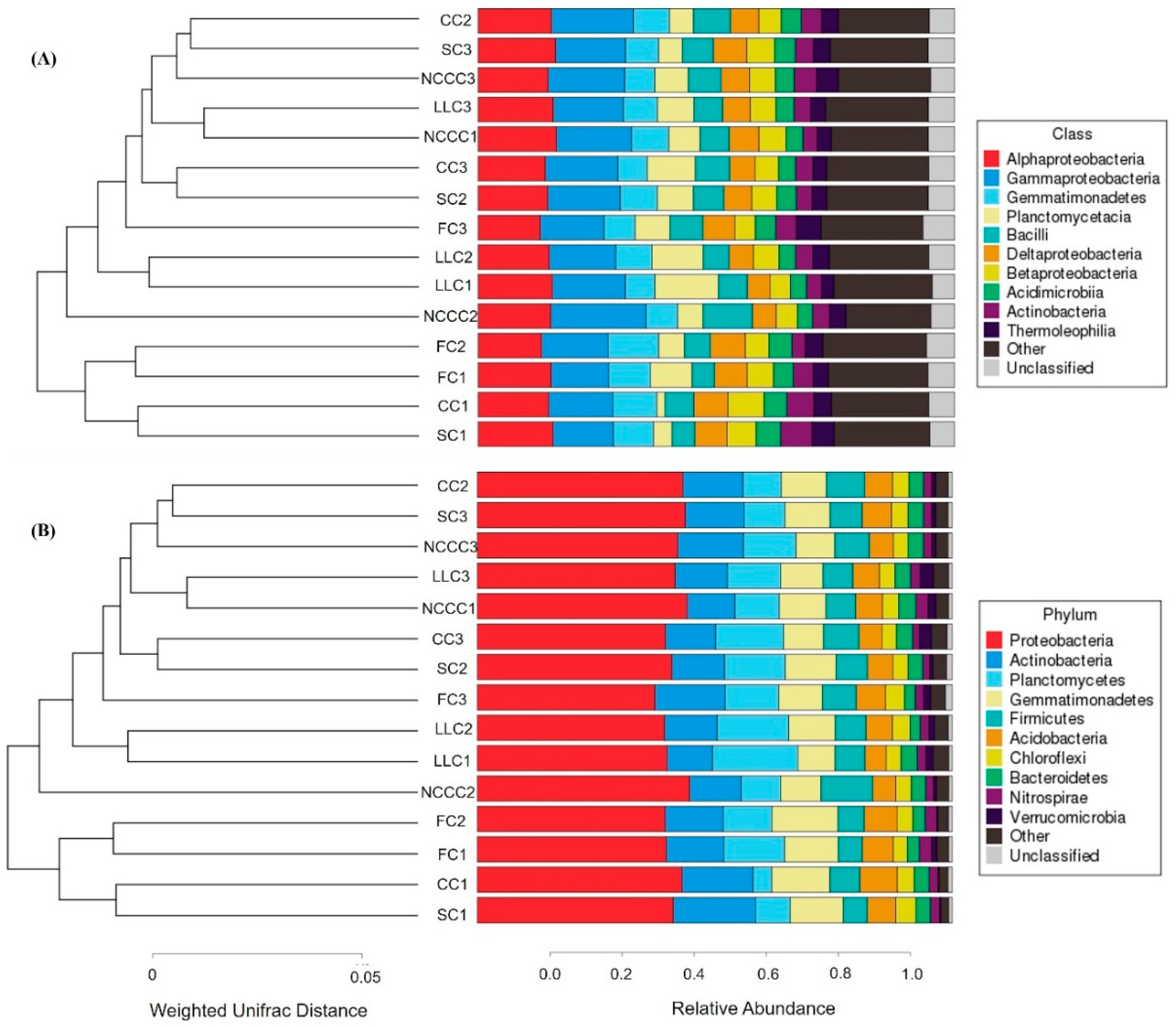
2.2. Taxonomic Characterization of Rhizosphere Microbiota
2.3. Changes in Bacterial Community Composition and Diversity
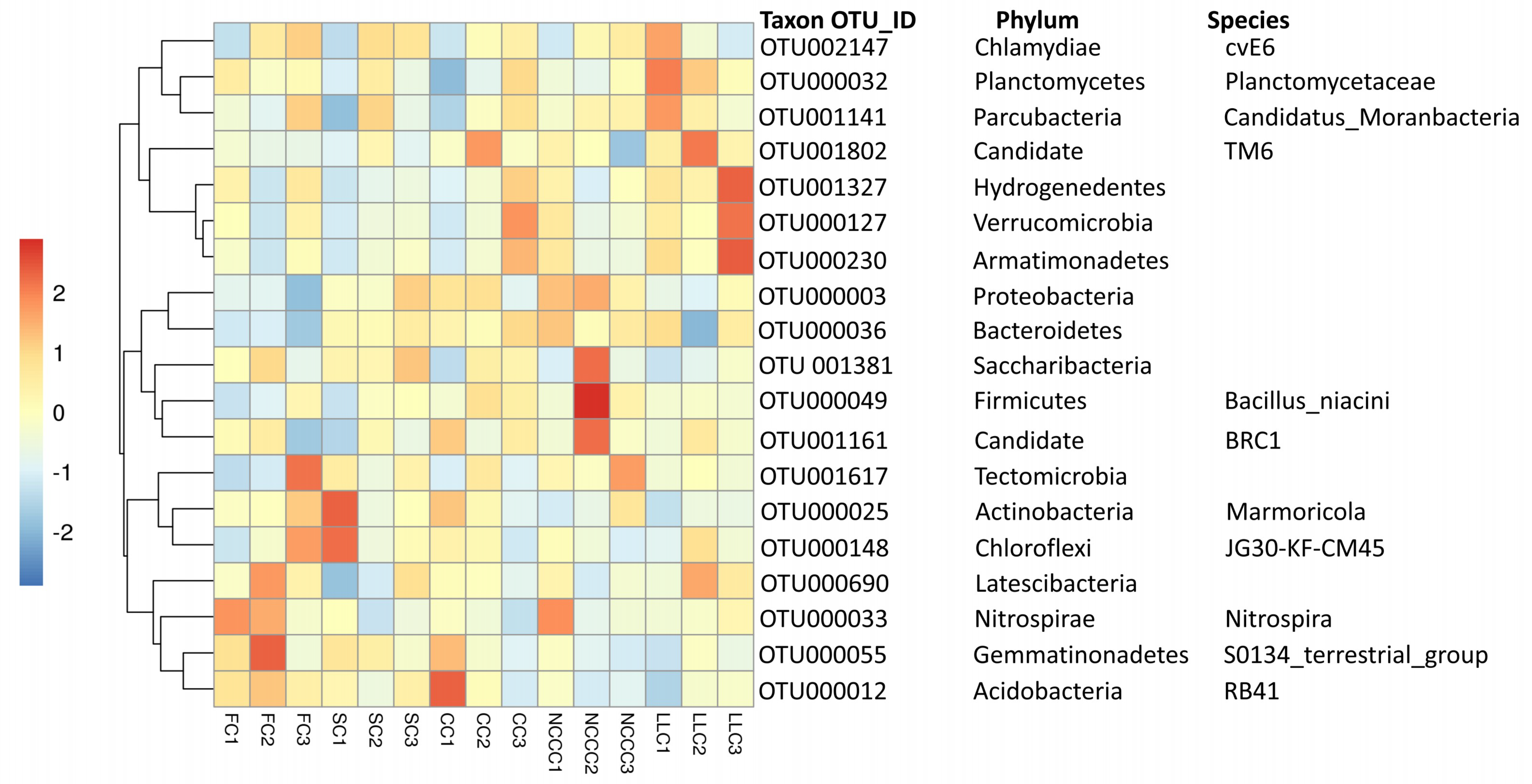
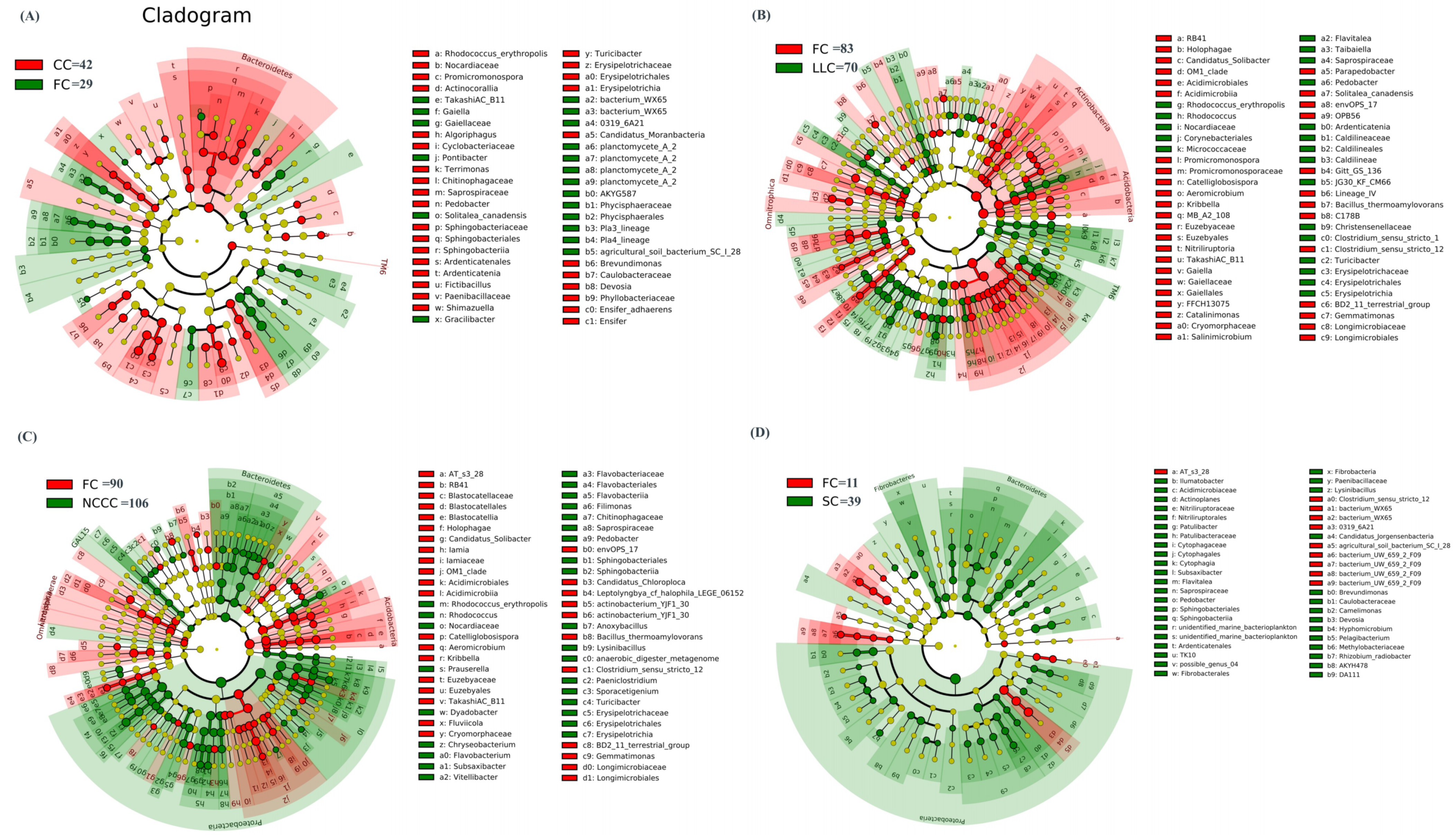
2.4. Comparative Assessment of Microbial Biomarkers
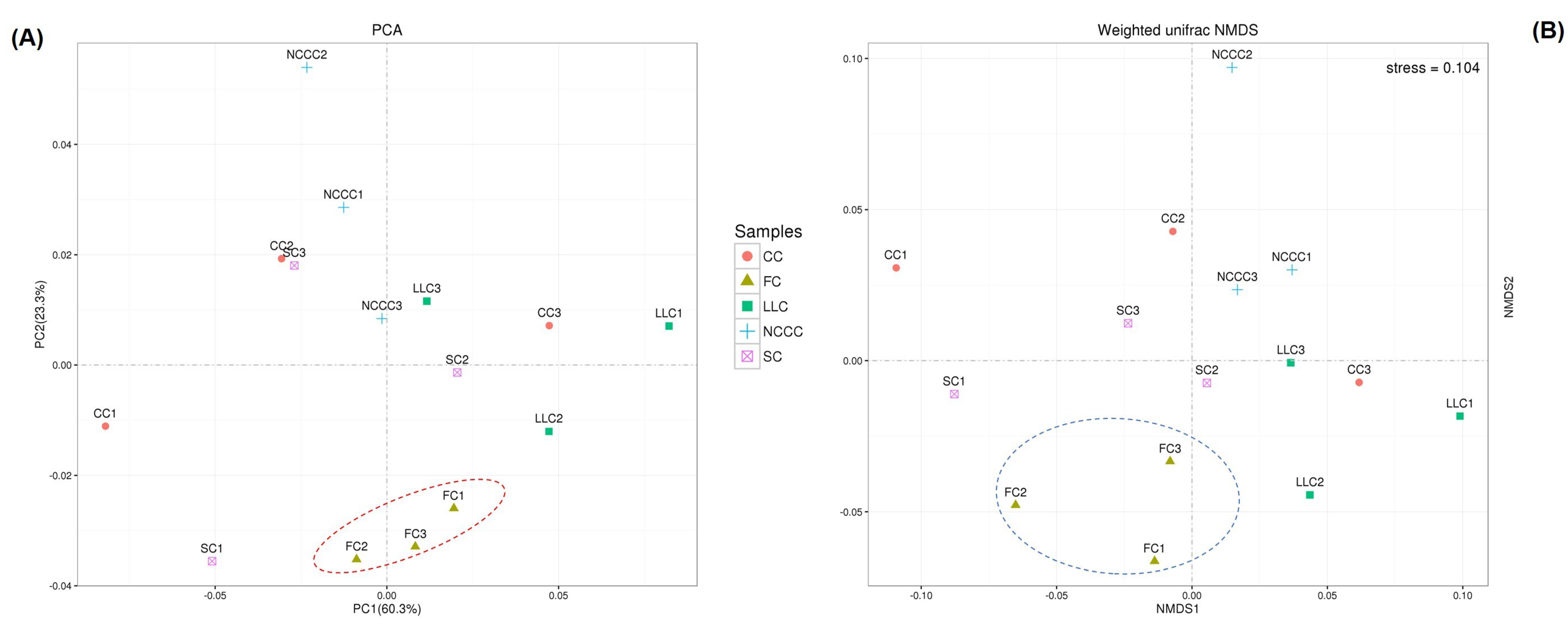
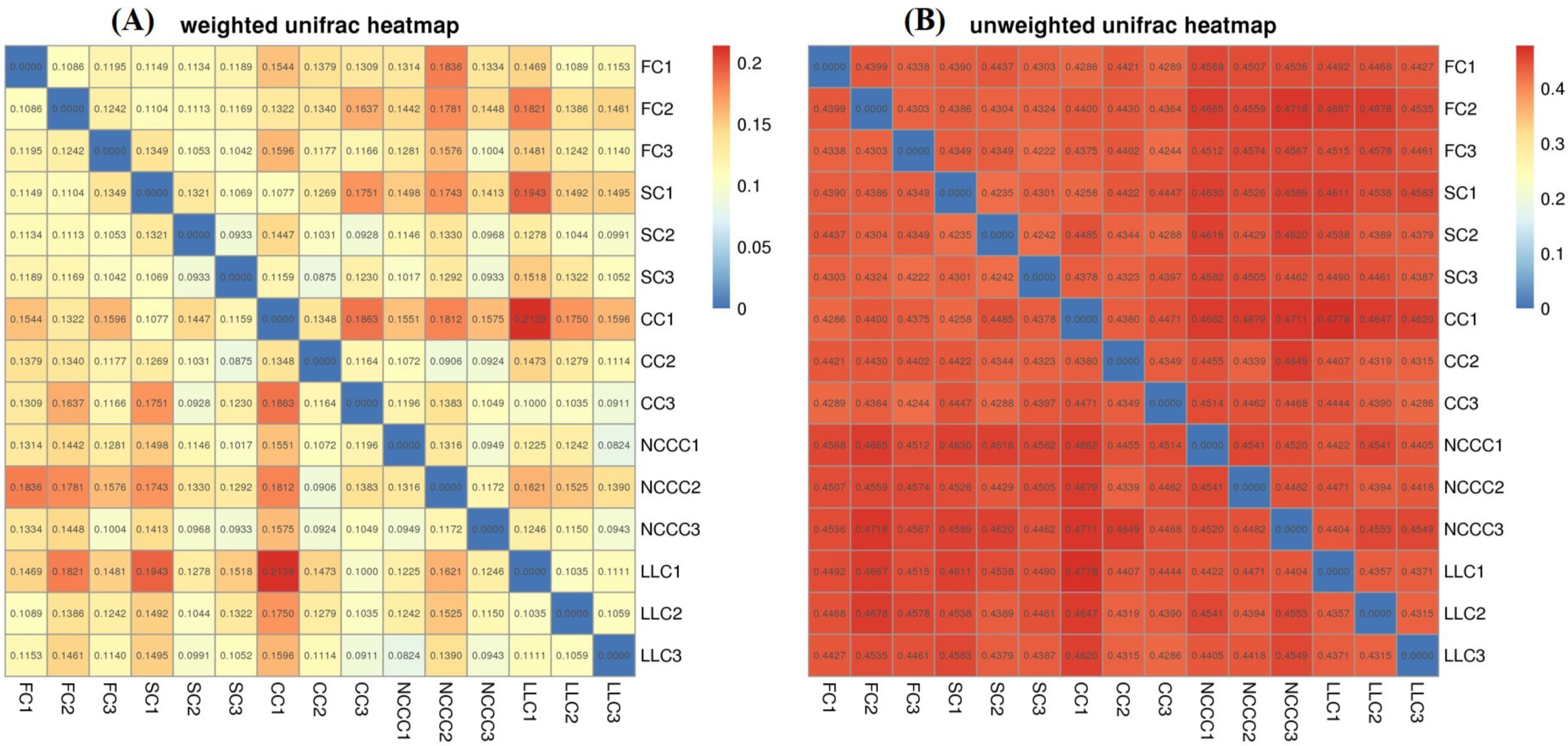
2.5. Soil Bacterial Diversity Responses to Different Planting Types
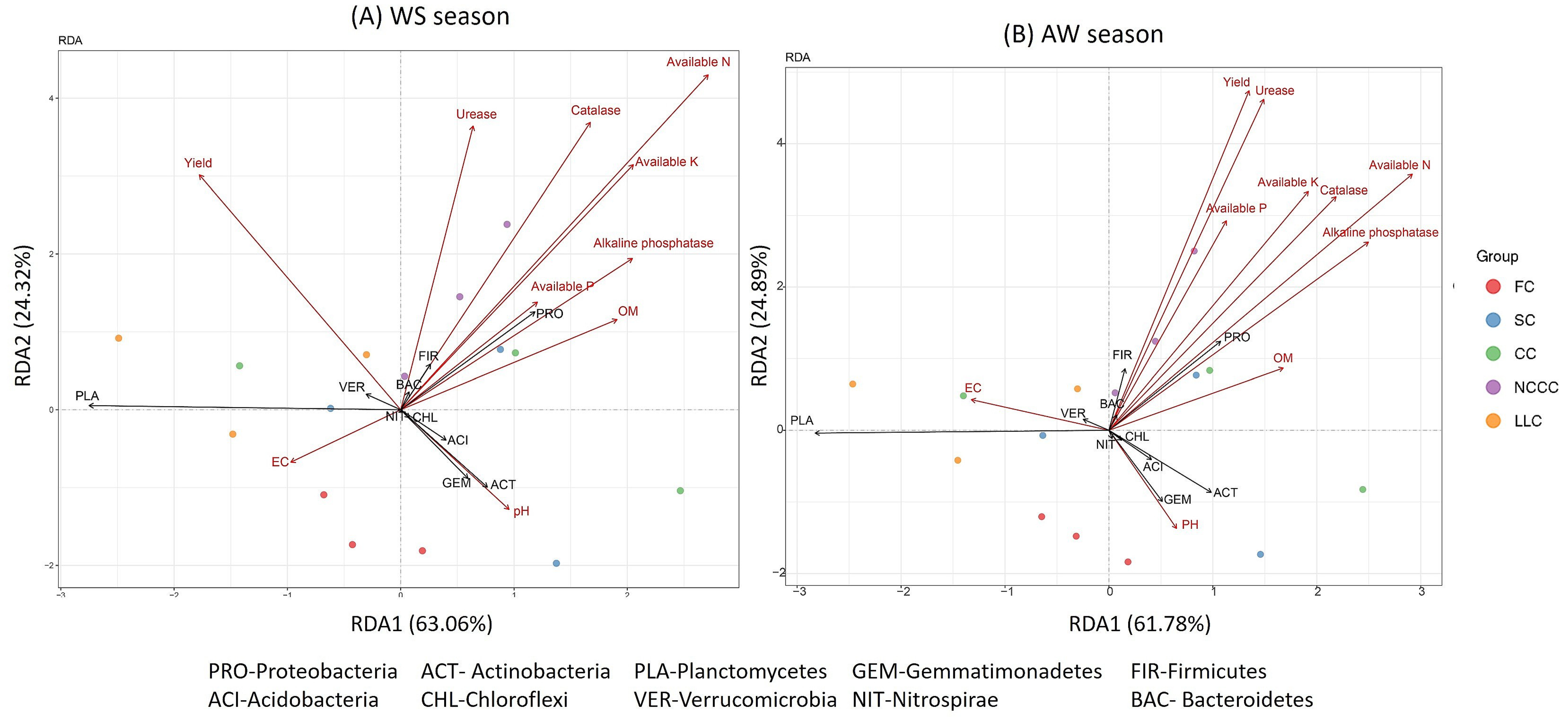
2.6. Linking Bacterial Community to Soil Properties
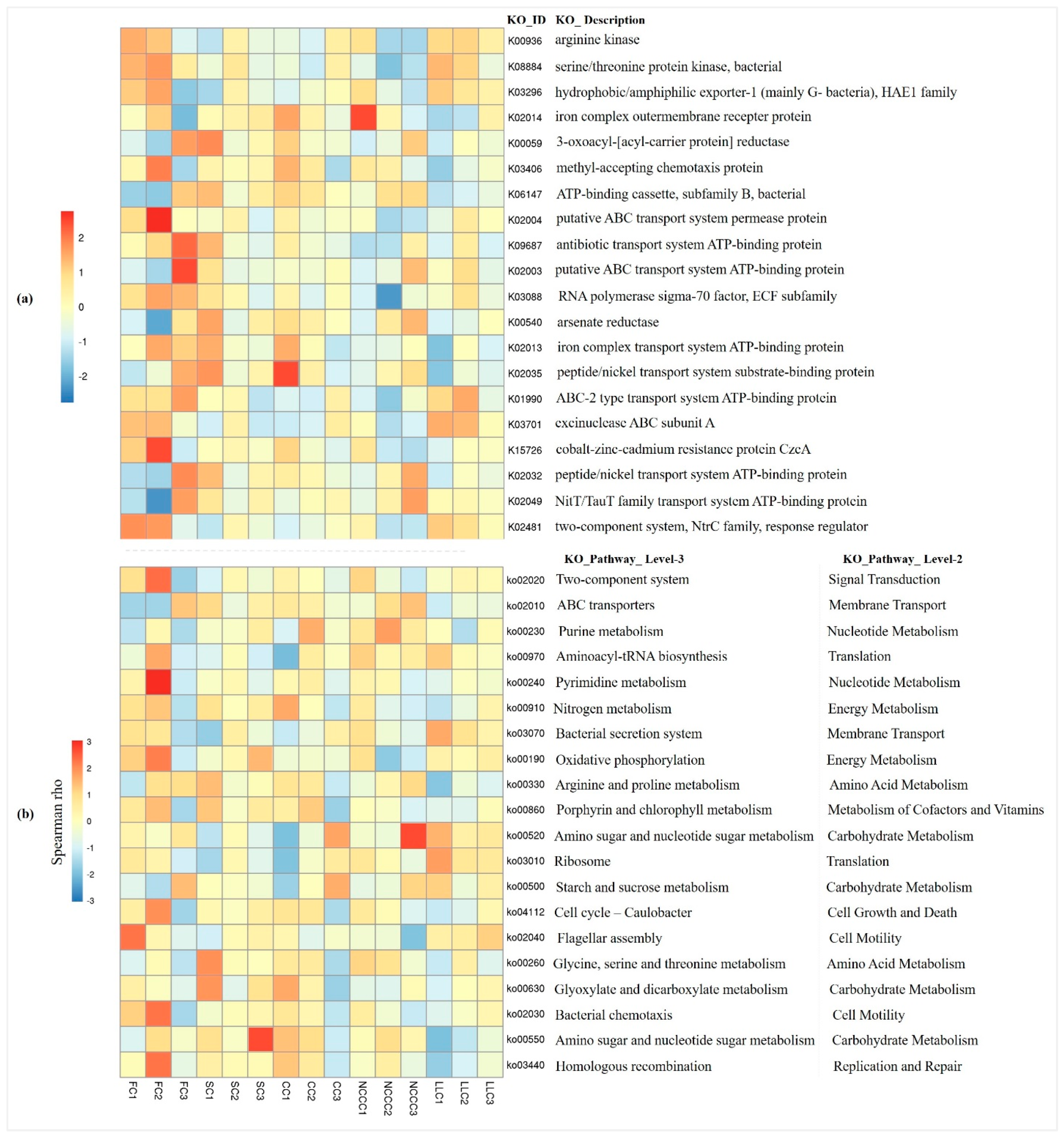
2.7. Predictive Metagenomics Profiling
3. Discussion
3.1. Effects of Different Cropping Systems on Soil Quality and Cucumber Yield
3.2. Effects of Different Cropping Systems on Soil Bacterial Diversity
3.3. Changes in Community Composition and Functional Profiling of Active Microbiome
3.4. Effects of Compositional Shift on Predictive Metabolic Functions
4. Materials and Methods
4.1. Field Description and Experimental Site
4.2. Experimental Design, Crop Establishment and Management
4.3. Soil Sampling and Analysis
4.4. DNA Extraction, Polymerase Chain Reaction (PCR) Amplification, and Metagenomic Sequencing
4.5. Sequence Data Analysis, Bioinformatics
4.6. Predictive Functional Profiling of Microbial Communities Using 16S rRNA Gene
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Raphael, J.P.A.; Calonego, J.C.; Milori, D.M.B.P.; Rosolem, C.A. Soil organic matter in crop rotations under no-till. Soil Tillage Res. 2016, 155, 45–53. [Google Scholar]
- Pare, T.; Chalifour, F.P.; Bourassa, J.; Antoun, H. Residual effects of faba bean and soybean for a 2nd or 3rd succeeding forage-corn crop. Can. J. Plant Sci. 1993, 73, 495–507. [Google Scholar] [CrossRef]
- Chang, C.L.; Fu, X.P.; Zhou, X.G.; Guo, M.Y.; Wu, F.Z. Effects of seven different companion plants on cucumber productivity, soil chemical characteristics and Pseudominas community. J. Integr. Agric. 2017, 16, 2206–2214. [Google Scholar] [CrossRef]
- Li, Y.; Wang, B.; Chang, Y.; Yang, Y.; Yao, C.; Huang, X.; Zhang, J.; Cai, Z.; Zhao, J. Reductive soil disinfestation effectively alleviates the replant failure of Sanqi ginseng through allelochemical degradation and pathogen suppression. Appl. Microbiol. Biotechnol. 2019, 103, 3581–3595. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Wang, W.; Zhang, G. Reducing environmental risk of excessively fertilized soils and improving cucumber growth by Caragana microphylla-straw compost application in long-term continuous cropping systems. Sci. Total Environ. 2016, 544, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Bu, R.; Xie, J.; Yu, J.; Liao, W.; Xiao, X.; Lv, J.; Clderón-Urrea, A. Autotoxicity in cucumber (Cucumis sativus L.) seedlings is alleviated by silicon through an increase in the activity of antioxidant enzymes and by mitigating lipid peroxidation. J. Plant Biol. 2016, 59, 247–259. [Google Scholar] [CrossRef]
- Zhou, X.; Gao, D.; Liu, J.; Qiao, P.; Zhou, X.; Lu, H.; Wu, X.; Liu, D.; Jin, X.; Wu, F. Changes in rhizosphere soil microbial communities in a continuously monocropped cucumber (Cucumis sativus L.) system. Eur. J. Soil Biol. 2014, 60, 1–8. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, G.; Wu, F. Soil phenolics in a continuously mono-cropped cucumber (Cucumis sativus L.) system and their effects on cucumber seedling growth and soil microbial communities. Eur. J. Soil Sci. 2012, 63, 332–340. [Google Scholar]
- Zhou, X.; Guan, S.; Wu, F. Composition of soil microbial communities in the rhizosphere of cucumber cultivars with differing nitrogen acquisition efficiency. Appl. Soil Ecol. 2015, 95, 90–98. [Google Scholar] [CrossRef]
- Fontaine, S.; Mariotti, A.; Abbadie, L. The priming effect of organic matter: A question of microbial competition? Soil Biol. Biochem. 2003, 35, 837–843. [Google Scholar] [CrossRef]
- Hernanz, J.L.; Sanchez-Giron, V.; Navarrete, L. Soil carbon sequestration and stratification in a cereal/leguminous crop rotation with three tillage systems in semiarid conditions. Agric. Ecosyst. Environ. 2009, 133, 114–122. [Google Scholar] [CrossRef]
- Strock, J.S.; Porter, P.M.; Russelle, M.P. Cover cropping to reduce nitrate loss through subsurface drainage in the northern US Corn Belt. J. Environ. Qual. 2004, 33, 1010–1016. [Google Scholar] [CrossRef]
- Gil, S.V.; Meriles, J.; Conforto, C.; Basanta, M.; Rad, V.; Hagn, A.; Schloter, M.; March, G.J. Response of soil microbial communities to different management practices in surface soils of a soybean agroecosystem in Argentina. Eur. J. Soil Biol. 2011, 47, 55–60. [Google Scholar]
- Yang, L.; Huang, B.; Mao, M.; Yao, L.; Niedermann, S.; Hu, W.; Chen, Y. Sustainability assessment of greenhouse vegetable farming practices from environmental, economic, and socio-institutional perspectives in China. Environ. Sci. Pollut. Res. 2016, 23, 17287–17297. [Google Scholar] [CrossRef]
- Song, X.Z.; Zhao, C.X.; Wang, X.L.; Li, J. Study of nitrate leaching and nitrogen fate under intensive vegetable production pattern in northern China. C. R. Biol. 2009, 332, 385–392. [Google Scholar] [CrossRef]
- Zhou, J.B.; Chen, Z.J.; Liu, X.J.; Zhai, B.N.; Powlson, D.S. Nitrate accumulation in soil profiles under seasonally open ‘sunlight greenhouses’ in northwest China and potential for leaching loss during summer fallow. Soil Use Manag. 2010, 26, 332–339. [Google Scholar] [CrossRef]
- Ju, X.T.; Kou, C.L.; Zhang, F.S.; Christie, P. Nitrogen balance and groundwater nitrate contamination: Comparison among three intensive cropping systems on the North China Plain. Environ. Pollut. 2006, 143, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Huang, B.; Du, N.; Guo, S.; Shu, S.; Sun, J. Effects of Garlic/Cucumber Relay Intercropping on Soil Enzyme Activities and the Microbial Environment in Continuous Cropping. Hortic. Sci. 2017, 52, 78–84. [Google Scholar] [CrossRef]
- Xiao, X.; Cheng, Z.; Meng, H.; Khan, M.A.; Li, H. Intercropping with garlic alleviated continuous cropping obstacle of cucumber in plastic tunnel. Acta Agric. Scand. Sect. B Soil Plant Sci. 2012, 62, 696–705. [Google Scholar] [CrossRef]
- Li, S.; Gao, D.; Guo, X.; Zhou, X.; Wu, F. Effects of different summer cover crops and residue management on plant growth and soil microbial community. Int. J. Agric. Biol. 2017, 19, 1350–1356. [Google Scholar]
- Hobbs, P.R.; Sayre, K.; Gupta, R. The role of conservation agriculture in sustainable agriculture. Philos. Trans. R. Soc. B 2008, 363, 543–555. [Google Scholar] [CrossRef]
- Guo, R.Y.; Li, X.L.; Christie, P.; Chen, Q.; Jiang, R.F.; Zhang, F.S. Influence of root zone nitrogen management and a summer catch crop on cucumber yield and soil mineral nitrogen dynamics in intensive production systems. Plant Soil 2008, 313, 55–70. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, X.; Liu, J.; Chen, Q.; Gao, L. Microbial properties of rhizosphere soils as affected by rotation, grafting, and soil sterilization in intensive vegetable production systems. Sci. Hortic. 2009, 123, 139–147. [Google Scholar] [CrossRef]
- Logsdon, D.; Kaspar, T.C.; Meek, D.W.; Prueger, J.H. Nitrate leaching as influenced by cover crops in large soil monoliths. Agron. J. 2002, 94, 807–814. [Google Scholar] [CrossRef]
- Thomsen, I.K.; Christensen, B.T. Nitrogen conserving potential of successive ryegrass catch crops in continuous spring barley. Soil Use Manag. 1999, 15, 195–200. [Google Scholar] [CrossRef]
- Aschi, A.; Aubert, M.; Riah-Anglet, W.; Nélieu, S.; Dubois, C.; Akpa-Vinceslas, M.; Trinsoutrot-Gattin, I. Introduction of Faba bean in crop rotation: Impacts on soil chemical and biological characteristics. Appl. Soil Ecol. 2017, 120, 219–228. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.T.; Yang, X.D.; Zhang, J.J.; Lin, Z.A.; Zhao, B.Q. Microbial community structure and functional metabolic diversity are associated with organic carbon availability in an agricultural soil. J. Int. Agric. 2015, 14, 2500–2511. [Google Scholar] [CrossRef]
- Lopez-Bellido, L.; Lopez-Bellido, R.J.; Redondo, R.; Benitez, J. Faba bean nitrogen fixation in a wheat-based rotation under rainfed Mediterranean conditions: Effect of tillage system. Field Crops Res. 2006, 98, 253–260. [Google Scholar] [CrossRef]
- Zak, D.R.; Holmes, W.E.; White, D.C.; Peacock, A.D.; Tilman, D. Plant diversity, soil microbial communities, and ecosystem function: Are there any links? Ecology 2003, 84, 2042–2050. [Google Scholar] [CrossRef]
- Alvey, S.; Bagayoko, M.; Neumann, G.; Buerkert, A. Cereal/legume rotations affect chemical properties and biological activities in two West African soils. Plant Soil 2003, 231, 45–54. [Google Scholar] [CrossRef]
- Bunemann, E.K.; Bossio, D.A.; Smithson, P.C.; Frossard, E.; Oberson, A. Microbial community composition and substrate use in a highly weathered soil as affected by crop rotation and P fertilization. Soil Biol. Biochem. 2004, 36, 889–901. [Google Scholar] [CrossRef]
- Mbuthia, L.W.; Acosta-Martínez, V.; DeBruyn, J.; Schaeffer, S.; Tyler, D.; Odoi, E.; Mpheshea, M.; Walker, F.; Eash, N. Long term tillage, cover crop, and fertilization effects on microbial community structure, activity: Implications for soil quality. Soil Biol. Biochem. 2015, 89, 24–34. [Google Scholar] [CrossRef]
- Vukicevich, E.; Lowery, T.; Bowen, P.; Úrbez-Torres, J.R.; Hart, M. Cover crops to increase soil microbial diversity and mitigate decline in perennial agriculture. A review. Agron. Sustain. Dev. 2016, 36, 48. [Google Scholar] [CrossRef]
- Ghosh, P.K.; Mohanty, K.; Bandyopadhyay, K.K.; Painuli, D.K.; Misra, A.K. Growth, competition, yields advantage and economics in soybean/pigeonpea intercropping system in semi-arid tropics of India: II. Effect of nutrient management. Field Crops Res. 2006, 96, 90–97. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, X.; Wang, J.; Gao, L. Soil microbial communities associated with the rhizosphere of cucumber under different summer cover crops and residue management: A 4-year field experiment. Sci. Hortic. 2013, 150, 100–109. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, J.; Wu, F. Soil microbial communities in cucumber monoculture and rotation systems and their feedback effects on cucumber seedling growth. Plant Soil 2017, 415, 507–520. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Beiko, R.G. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814. [Google Scholar] [CrossRef]
- Sainju, U.M.; Lenssen, A.; Caesar-Thonthat, T.; Waddell, J. Dryland plant biomass and soil carbon and nitrogen fractions on transient land as influenced by tillage and crop rotation. Soil Tillage Res. 2007, 93, 452–461. [Google Scholar] [CrossRef]
- Nuruzzaman, M.; Lambers, H.; Bolland, M.D.A.; Veneklaas, E.J. Phosphorus uptake by grain legumes and subsequently grown wheat at different levels of residual phosphorus fertilizer. Aust. J. Agric. Res. 2005, 56, 1041–1047. [Google Scholar] [CrossRef]
- Wu, F.; Yu, H.; Yu, G.; Pan, K.; Bao, J. Improved bacterial community diversity and cucumber yields in a rotation with kidney bean–celery–cucumber. Acta Agric. Scand. Sect. B Soil Plant Sci. 2011, 61, 122–128. [Google Scholar] [CrossRef]
- Zhou, X.; Gaobo, Y.; Fengzhi, W. Effects of intercropping cucumber with onion or garlic on soil enzyme activities, microbial communities and cucumber yield. Eur. J. Soil Biol. 2011, 47, 279–287. [Google Scholar] [CrossRef]
- Zhang, X.; Ning, T.; Han, H.; Sun, T.; Li, G.; Li, Z.; Lal, R. Effects of Waxy Maize Relay Intercropping and Residue Retention on Rhizosphere Microbial Communities and Vegetable Yield in a Continuous Cropping System. Pedosphere 2018, 28, 84–93. [Google Scholar] [CrossRef]
- Marschner, P.; Yang, C.H.; Lieberei, R.; Crowley, D.E. Soil and plant specific effects on bacterial community composition in the rhyzosphere. Soil Biol. Biochem. 2001, 33, 1437–1445. [Google Scholar] [CrossRef]
- Wieland, G.; Neumann, R.; Backhaus, H. Variation of microbial communities in soil, rhizosphere, and rhizoplane in response to crop species, soil type, and crop development. Appl. Environ. Microbiol. 2001, 67, 5849–5854. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Y.; Wu, F. Effect of different cultivation modes on cucumber growth and the numbers of culturable rhizosphere soil microorganisms. J. Northeast Agric. Univ. 2012, 7, 95–99. [Google Scholar]
- Chang, C.; Zhou, X.; Fu, X.; Yang, S.; Wu, F. Soil enzymes and bacterial community composition in cucumber (Cucumis sativus L.) monocropping and companion cropping systems. Allelopath. J. 2016, 38, 133–146. [Google Scholar]
- Wagner, M.R.; Lundberg, D.S.; Tijana, G.; Tringe, S.G.; Dangl, J.L.; Mitchell-Olds, T. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 2016, 7, 12151. [Google Scholar] [CrossRef]
- Nevins, C.J.; Cindy, N.; Shalamar, A. Characterization of microbial community response to cover crop residue decomposition. Soil Biol. Biochem. 2018, 127, 39–49. [Google Scholar] [CrossRef]
- Nardi, S.; Concheri, G.; Pizzeghello, D.; Sturaro, A.; Rella, R.; Parvoli, G. Soil organic matter mobilization by root exudates. Chemosphere 2000, 5, 653–658. [Google Scholar] [CrossRef]
- Wang, Y.; Tu, C.; Cheng, L.; Li, C.; Gentry, L.F.; Hoyt, G.D. Long-term impact of farming practices on soil organic carbon and nitrogen pools and microbial biomass and activity. Soil Tillage Res. 2011, 117, 8–16. [Google Scholar] [CrossRef]
- Cai, F.; Pang, G.; Miao, Y.; Li, R.; Li, R.; Shen, Q. The nutrient preference of plants influences their rhizosphere microbiome. Appl. Soil Ecol. 2017, 110, 146–150. [Google Scholar] [CrossRef]
- Li, S.; Wu, F. Diversity and cooccurrence patterns of soil bacterial and fungal communities in seven intercropping systems. Front. Microbiol. 2018, 9, 1521. [Google Scholar] [CrossRef]
- Song, X.; Tao, B.; Guo, J.; Li, J.; Chen, G. Changes in the Microbial Community Structure and Soil Chemical Properties of Vertisols under Different Cropping Systems in Northern China. Front. Environ. Sci. 2018, 6, 132. [Google Scholar] [CrossRef]
- Hu, H.W.; Zhang, L.M.; Dai, Y.; Di, H.J.; He, J.Z. pH-dependent distribution of soil ammonia oxidizers across a large geographical scale as revealed by high-throughput pyrosequencing. J. Soils Sediments 2013, 13, 1439–1449. [Google Scholar] [CrossRef]
- Sharma, S.B.; Sayyed, R.Z.; Trivedi, M.H.; Gobi, T.A. Phosphate solubilizing microbes: Sustainable approach for managing phosphorus deficiency in agricultural soils. SpringerPlus 2013, 2, 587. [Google Scholar] [CrossRef]
- Vikram, S.; Guerrero, L.D.; Makhalanyane, T.P.; Le, P.T.; Seely, M.; Cowan, D.A. Metagenomic analysis provides insights into functional capacity in a hyperarid desert soil niche community. Environ. Microbiol. 2016, 18, 1875–1888. [Google Scholar] [CrossRef]
- Ashworth, A.J.; DeBruyn, J.V.M.; Allen, F.L.; Radosevich, M.; Owens, P.R. Microbial community structure is affected by cropping sequences and poultry litter under long-term no-tillage. Soil Biol. Biochem. 2017, 114, 210–219. [Google Scholar] [CrossRef]
- Li, X.; Rui, J.; Xiong, J.; Li, J.; He, Z.; Zhou, J.; Yannarell, A.C.; Mackie, R.I. Functional potential of soil microbial communities in the maize rhizosphere. PLoS ONE 2014, 9, e112609. [Google Scholar] [CrossRef]
- Orlandini, V.; Maida, I.; Fondi, M.; Perrin, E.; Papaleo, M.C.; Bosi, E. Genomic analysis of three sponge-associated Arthrobacter Antarctic strains, inhibiting the growth of Burkholderia cepacia complex bacteria by synthesizing volatile organic compounds. Microbiol. Res. 2014, 169, 593–601. [Google Scholar] [CrossRef]
- McCarty, G.W.; Shogren, D.R.; Bremner, J.M. Regulation of urease production in soil by microbial assimilation of nitrogen. Biol. Fertil. Soils 1992, 12, 261–264. [Google Scholar] [CrossRef]
- Wang, H.B.; Zhang, Z.X.; Li, H.; He, H.B.; Fang, C.X. Characterization of metaproteomics in crop rhizospheric soil. J. Proteome Res. 2010, 10, 932–940. [Google Scholar] [CrossRef]
- Marschner, P.; Crowley, D.; Yang, C.H. Development of specific rhizosphere bacterial communities in relation to plant species, nutrition and soil type. Plant Soil 2004, 261, 199–208. [Google Scholar] [CrossRef]
- Arslan, B.; Djamgoz, M.B.A.; Akun, E. Arsenic: A review on exposure pathways, accumulation, mobility and transmission into human food chain. Rev. Environ. Contam. Toxicol. 2017, 243, 27–51. [Google Scholar]
- Batista, B.L.; Nigar, M.; Mestrot, A.; Rocha, B.A.; Júnior, F.B.; Price, A.H. Identification and quantification of phytochelatins in roots of rice to long-term exposure: Evidence of individual role on arsenic accumulation and translocation. J. Exp. Bot. 2014, 65, 1467–1479. [Google Scholar] [CrossRef]
- Shah, K.; Nongkynrih, J.M. Metal hyperaccumulation and bioremediation. Biol. Plant. 2007, 51, 618–634. [Google Scholar] [CrossRef]
- Abedin, M.J.; Cresser, M.S.; Meharg, A.A.; Feldmann, J.; Cotter-Howells, J. Arsenic accumulation and metabolism in rice (Oryza sativa L.). Environ. Sci. Technol. 2002, 36, 962–968. [Google Scholar] [CrossRef]
- Ding, H.; Ali, A.; Cheng, Z. Dynamics of a Soil Fungal Community in a Three-Year Green Garlic/Cucumber Crop Rotation System in Northwest China. Sustainability 2018, 10, 1391. [Google Scholar] [CrossRef]
- Bao, S.D. Soil and Agricultural Chemistry Analysis; Chinese Agriculture Press: Beijing, China, 2000. [Google Scholar]
- Akhtar, K.; Weiyu, W.; Guangxin, R.; Ahmad, K.; Yongzhong, F.; Gaih, Y. Changes in soil enzymes, soil properties, and maize crop productivity under wheat straw mulching in Guanzhong, China. Soil Tillage Res. 2018, 182, 94–102. [Google Scholar] [CrossRef]
- Jing, X.; Sanders, N.J.; Shi, Y.; Chu, H.; Classen, A.T.; Zhao, K. The links between ecosystem multifunctionality and above- and belowground biodiversity are mediated by climate. Nat. Commun. 2015, 6, 8159. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Sathish, S.; Jeremiah, J.; Dirk, G.; Jeffrey, I.; Gordon, R.K.; David, A.M.; Gregory, C. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical and Biological Factors | Treatments | ||||
|---|---|---|---|---|---|
| FC | SC | CC | NCCC. | LLC | |
| Soil pH | 7.74 ± 0.12 a | 7.76 ± 0.05 a | 7.76 ± 0.03 a | 7.71 ± 0.05 a | 7.73 ± 0.03 a |
| Electrical conductivity (EC) (µs·cm−1) | 627.7 ± 5.00 bc | 649.9 ± 5.90 a | 621.6 ± 3.51 c | 632.40 ± 4.79 bc | 641.40 ± 1.79 ab |
| Organic matter (OM) (g·kg−1) | 18.63 ± 1.28 c | 22.94 ± 1.09 a | 22.71 ± 1.23 a | 21.55 ± 1.29 ab | 19.45 ± 1.59 bc |
| Available N (mg·kg−1) | 122.8 ± 3.03 c | 144.4 ± 1.18 a | 137.44 ± 1.59 ab | 146.39 ± 1.77 a | 132.69 ± 3.83 b |
| Available P (mg·kg−1) | 64.39 ± 2.00 c | 78.29 ± 0.75 a | 73.25 ± 2.25 ab | 69.52 ± 0.19 bc | 68.12 ± 1.39 bc |
| Available K (mg·kg−1) | 345.4 ± 1.73 d | 354.78 ± 2.25 bc | 363.06 ± 0.89 a | 359.89 ± 1.85 ab | 351.49 ± 3.82 cd |
| Soil invertase (mg·g−1 soil d−1) | 43.50 ± 1.36 b | 45.38 ± 1.07 b | 63.86 ± 1.39 a | 59.34 ± 3.67 a | 46.64 ± 1.17 b |
| Urease (mg·g−1 soil h−1) | 3.61 ± 0.17 d | 5.54 ± 0.11 a | 4.46 ± 0.26 c | 5.46 ± 0.65 ab | 4.96 ± 0.41 bc |
| Catalase (mg·g−1 20 min−1) | 6.29 ± 0.22 c | 12.04 ± 0.85 a | 9.82 ± 0.27 b | 11.85 ± 0.67 a | 9.15 ± 0.24 b |
| Alkaline phosphatase (mg g−1 soil h−1) | 21.43 ± 0.60 d | 34.66 ± 1.09 a | 29.65 ± 1.39 b | 27.87 ± 0.38 b | 25.21 ± 1.10 c |
| Physicochemical and Biological Factors | Treatments | ||||
|---|---|---|---|---|---|
| FC | SC | CC | NCCC | LLC | |
| Soil pH | 7.73 ± 0.12 a | 7.71 ± 0.04 a | 7.73 ± 0.03 a | 7.70 ± 0.02 a | 7.73 ± 0.04 a |
| EC (µs·cm−1) | 631.04 ± 6.81 bc | 651.04 ± 5.46 a | 627.82 ± 5.38 c | 638.83 ± 3.7abc | 645.51 ± 2.25 ab |
| OM (g·kg−1) | 21.18 ± 1.58 ab | 23.27 ± 0.99 ab | 23.20 ± 0.54 ab | 24.72 ± 1.00 a | 20.09 ± 0.42 b |
| Available N (mg·kg−1) | 117.28 ± 4.74 b | 136.2 ± 5.43 a | 138.9 ± 1.48 a | 142.16 ± 1.67 a | 123.89 ± 0.37 b |
| Available P (mg·kg−1) | 57.72 ± 1.32 b | 65.81 ± 2.60 ab | 69.94 ± 2.00 a | 72.39 ± 0.91 a | 61.53 ± 5.03 b |
| Available K (mg·kg−1) | 350.08 ± 1.76 cd | 358.06 ± 0.91bc | 365.84 ± 1.79 ab | 368.58 ± 2.01 a | 347.69 ± 5.40 d |
| Soil invertase (mg·g−1 soil d−1) | 40.00 ± 0.56 b | 41.86 ± 2.54 b | 57.13 ± 2.51a | 54.86 ± 2.08 a | 38.99 ± 1.44 b |
| Urease (mg·g−1 soil h−1) | 3.74 ± 0.25 c | 5.88 ± 0.42 b | 6.13 ± 0.43 ab | 6.90 ± 0.16 a | 4.760.19 c |
| Catalase (mg·g−1 20 min−1) | 5.72 ± 0.12 c | 7.72 ± 1.23 bc | 8.50 ± 0.84 ab | 10.90 ± 0.86 a | 7.61 ± 0.72 bc |
| Alkaline phosphatase (mg· g−1 soil h−1) | 19.57 ± 0.57 c | 21.87 ± 0.36 bc | 24.28 ± 1.93 ab | 25.68 ± 1.43 a | 22.54 ± 0.70 ab |
| Treatments | WS Season-2017 | AW Season-2017 |
|---|---|---|
| FC | 46.21 ± 2.08 b | 4.24 ± 0.46 bc |
| SC | 55.23 ± 3.18 a | 4.45 ± 0.59 bc |
| CC | 50.82 ± 3.36 ab | 5.36 ± 0.17 ab |
| NCCC | 51.09 ± 2.54 ab | 6.16 ± 0.06 a |
| LLC | 48.65 ± 2.44 ab | 3.72 ± 0.23 c |
| Sample ID | OTUs | Ace | Chao 1 | Shannon | Simpson | Coverage % |
|---|---|---|---|---|---|---|
| FC1 | 4644 | 6134 | 6229 | 9.74 | 0.99 | 0.97 |
| FC2 | 4726 | 6423 | 6319 | 9.48 | 0.98 | 0.96 |
| FC3 | 4593 | 6027 | 5993 | 9.88 | 0.99 | 0.97 |
| Average | 4654.33 c | 6195 c | 6181 c | 9.70 c | 0.99 a | 0.97 a |
| SC1 | 5407 | 7586 | 7452 | 10.07 | 0.99 | 0.96 |
| SC2 | 4993 | 6785 | 6809 | 9.93 | 0.99 | 0.96 |
| SC3 | 4958 | 6771 | 6788 | 9.92 | 0.99 | 0.96 |
| Average | 5119.33 a | 7048 a | 7016 a | 9.98 a | 0.99 a | 0.96 a |
| CC1 | 4853 | 6860 | 6856 | 9.85 | 0.99 | 0.96 |
| CC2 | 4982 | 6798 | 6768 | 9.68 | 0.99 | 0.97 |
| CC3 | 4574 | 6081 | 6118 | 9.79 | 0.99 | 0.97 |
| Average | 4803 b | 6580 b | 6581 b | 9.77 b | 0.99 a | 0.97 a |
| NCCC1 | 5021 | 6989 | 6860 | 9.99 | 0.99 | 0.96 |
| NCCC2 | 4955 | 6865 | 6918 | 9.80 | 0.99 | 0.96 |
| NCCC3 | 4667 | 6221 | 6146 | 9.95 | 0.99 | 0.96 |
| Average | 4821 b | 6691 b | 6441 b | 9.91 a | 0.99 a | 0.96 a |
| LLC1 | 4270 | 5517 | 5597 | 9.76 | 0.99 | 0.97 |
| LLC2 | 4603 | 6202 | 6188 | 9.89 | 0.99 | 0.97 |
| LLC3 | 4554 | 6028 | 5955 | 9.81 | 0.99 | 0.97 |
| Average | 4475.66 d | 5915 c | 5913 d | 9.82 b | 0.99 | 0.97 a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Imran Ghani, M.; Li, Y.; Ding, H.; Meng, H.; Cheng, Z. Hiseq Base Molecular Characterization of Soil Microbial Community, Diversity Structure, and Predictive Functional Profiling in Continuous Cucumber Planted Soil Affected by Diverse Cropping Systems in an Intensive Greenhouse Region of Northern China. Int. J. Mol. Sci. 2019, 20, 2619. https://doi.org/10.3390/ijms20112619
Ali A, Imran Ghani M, Li Y, Ding H, Meng H, Cheng Z. Hiseq Base Molecular Characterization of Soil Microbial Community, Diversity Structure, and Predictive Functional Profiling in Continuous Cucumber Planted Soil Affected by Diverse Cropping Systems in an Intensive Greenhouse Region of Northern China. International Journal of Molecular Sciences. 2019; 20(11):2619. https://doi.org/10.3390/ijms20112619
Chicago/Turabian StyleAli, Ahmad, Muhammad Imran Ghani, Yuhong Li, Haiyan Ding, Huanwen Meng, and Zhihui Cheng. 2019. "Hiseq Base Molecular Characterization of Soil Microbial Community, Diversity Structure, and Predictive Functional Profiling in Continuous Cucumber Planted Soil Affected by Diverse Cropping Systems in an Intensive Greenhouse Region of Northern China" International Journal of Molecular Sciences 20, no. 11: 2619. https://doi.org/10.3390/ijms20112619
APA StyleAli, A., Imran Ghani, M., Li, Y., Ding, H., Meng, H., & Cheng, Z. (2019). Hiseq Base Molecular Characterization of Soil Microbial Community, Diversity Structure, and Predictive Functional Profiling in Continuous Cucumber Planted Soil Affected by Diverse Cropping Systems in an Intensive Greenhouse Region of Northern China. International Journal of Molecular Sciences, 20(11), 2619. https://doi.org/10.3390/ijms20112619