Cross-Talk between Inflammatory Mediators and the Epithelial Mesenchymal Transition Process in the Development of Thyroid Carcinoma

 , ,
, ,  and
and
Abstract

1. Introduction
2. Origin of Epithelial Thyroid Carcinoma (ETC)
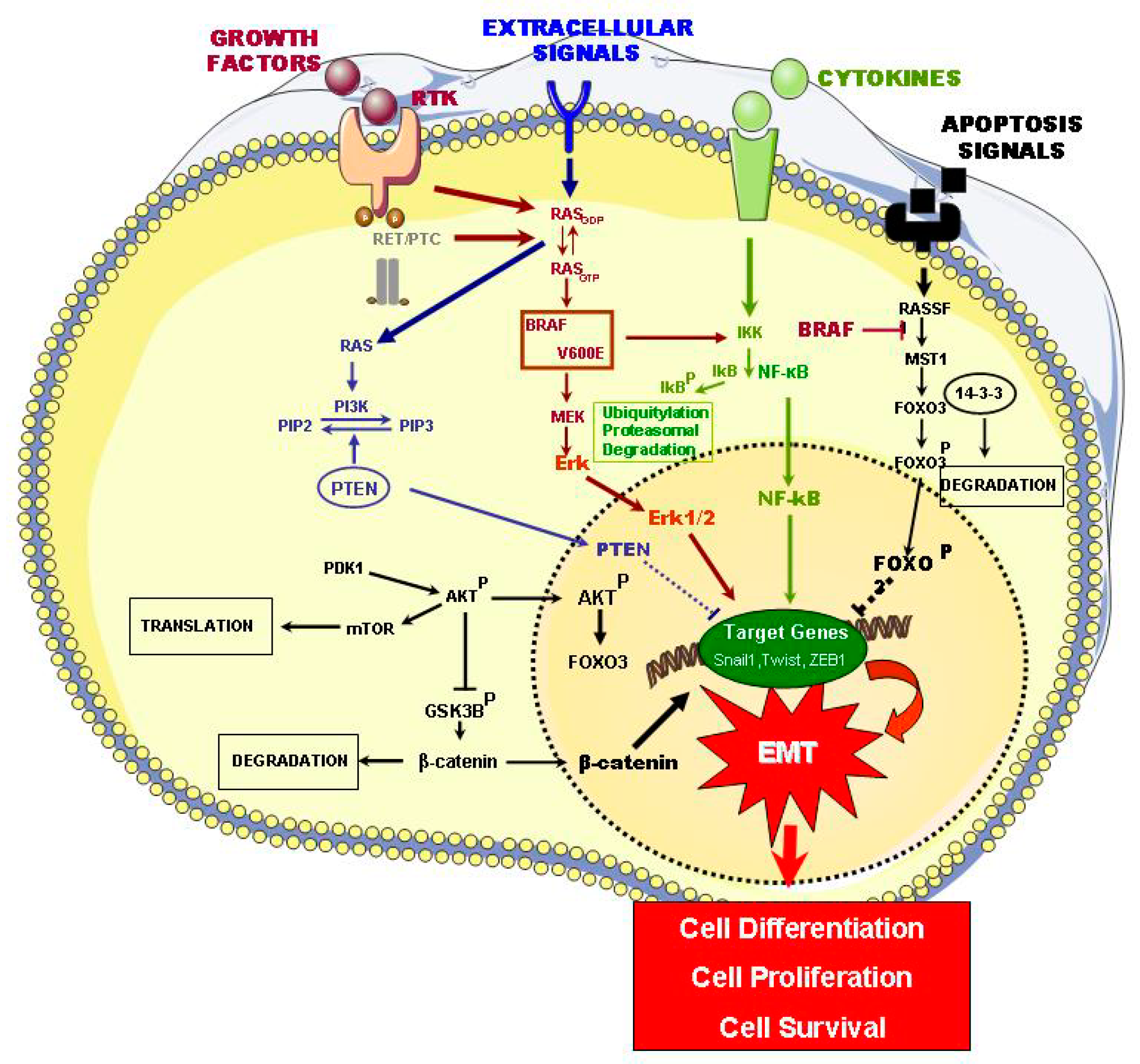
2.1. Signaling Pathways of ETC Oncogenesis
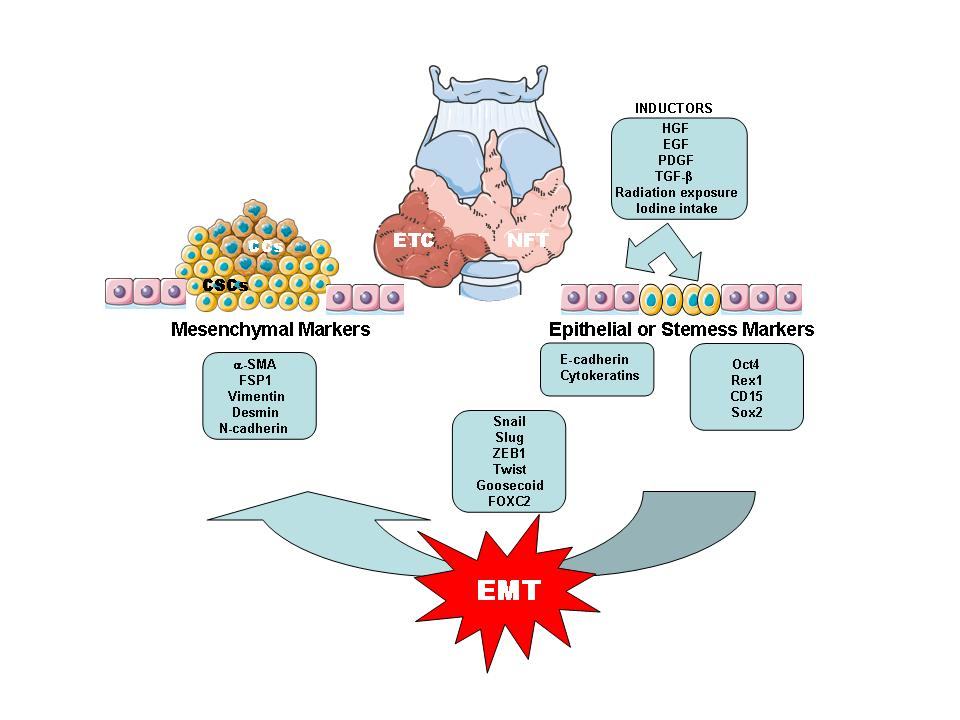
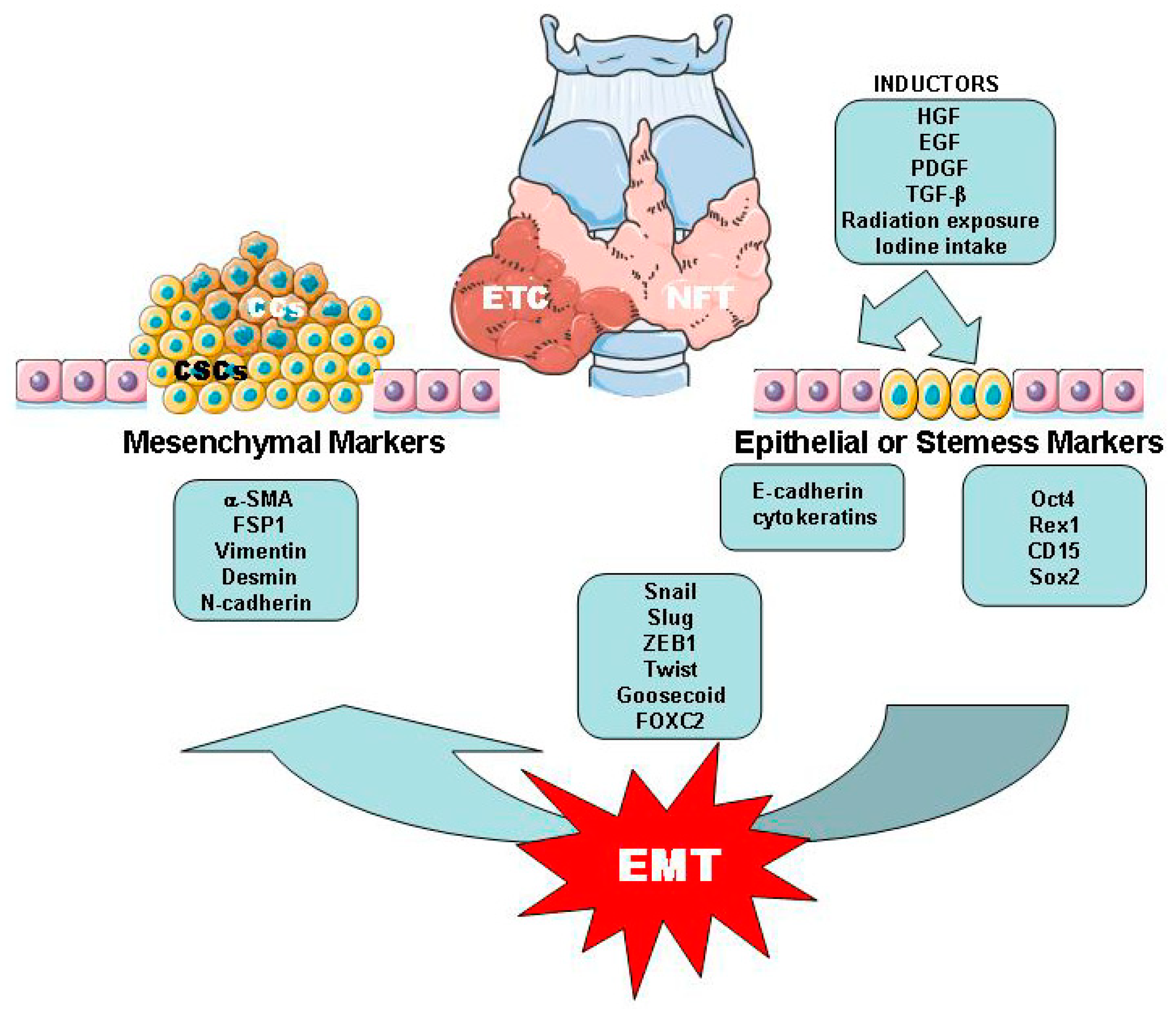
2.2. Thyroid Cancer Cells (CCs) and Cancer Stem Cells (CSCs)
2.3. EMT/MET Processes Related with ETC
3. Tissue and Inflammatory Mediators in Relation to the EMT Process in ETC
3.1. Adipose Tissue
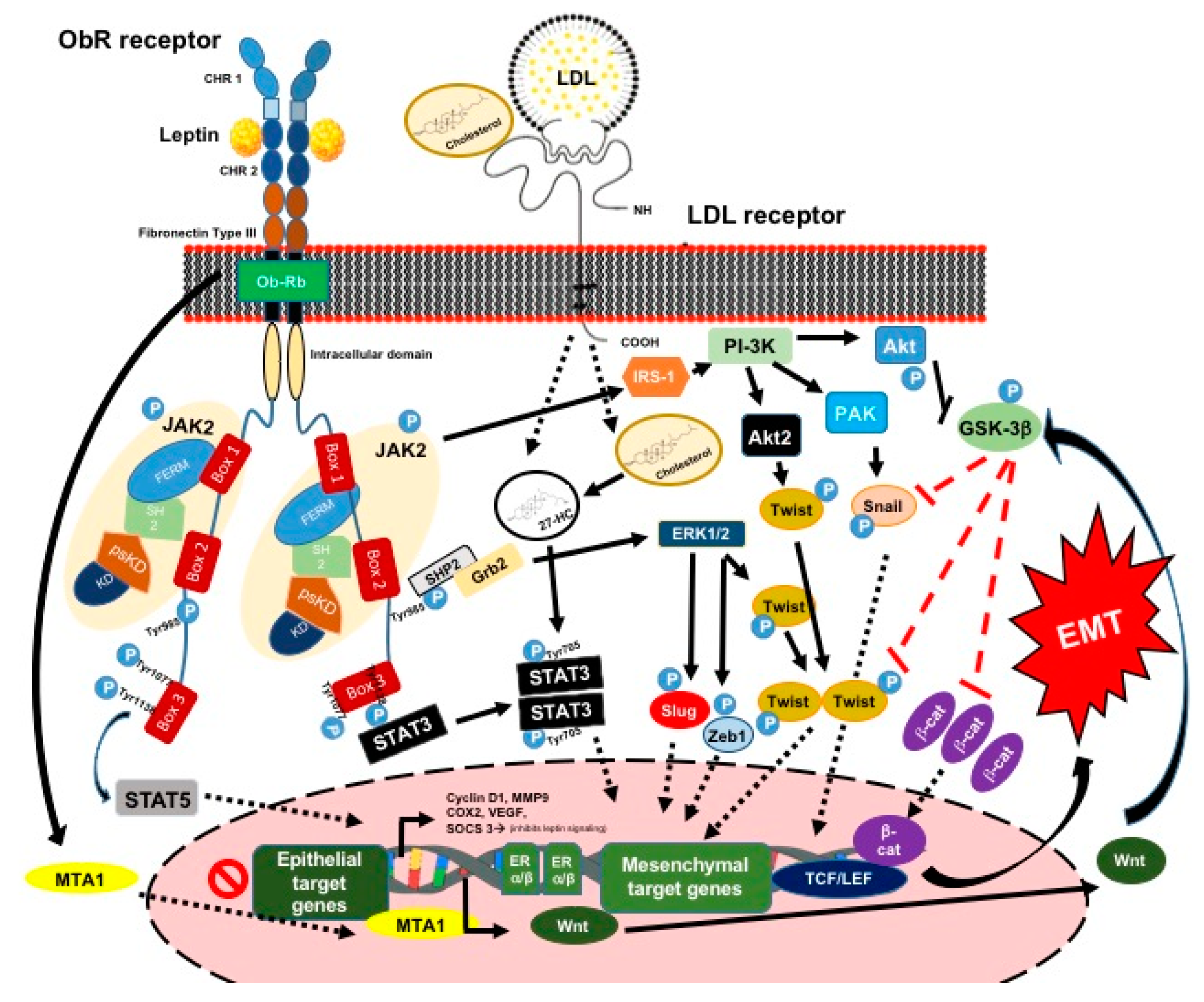
3.1.1. Leptin and Its Signaling Pathway
3.1.2. Leptin and the EMT Process and ETC
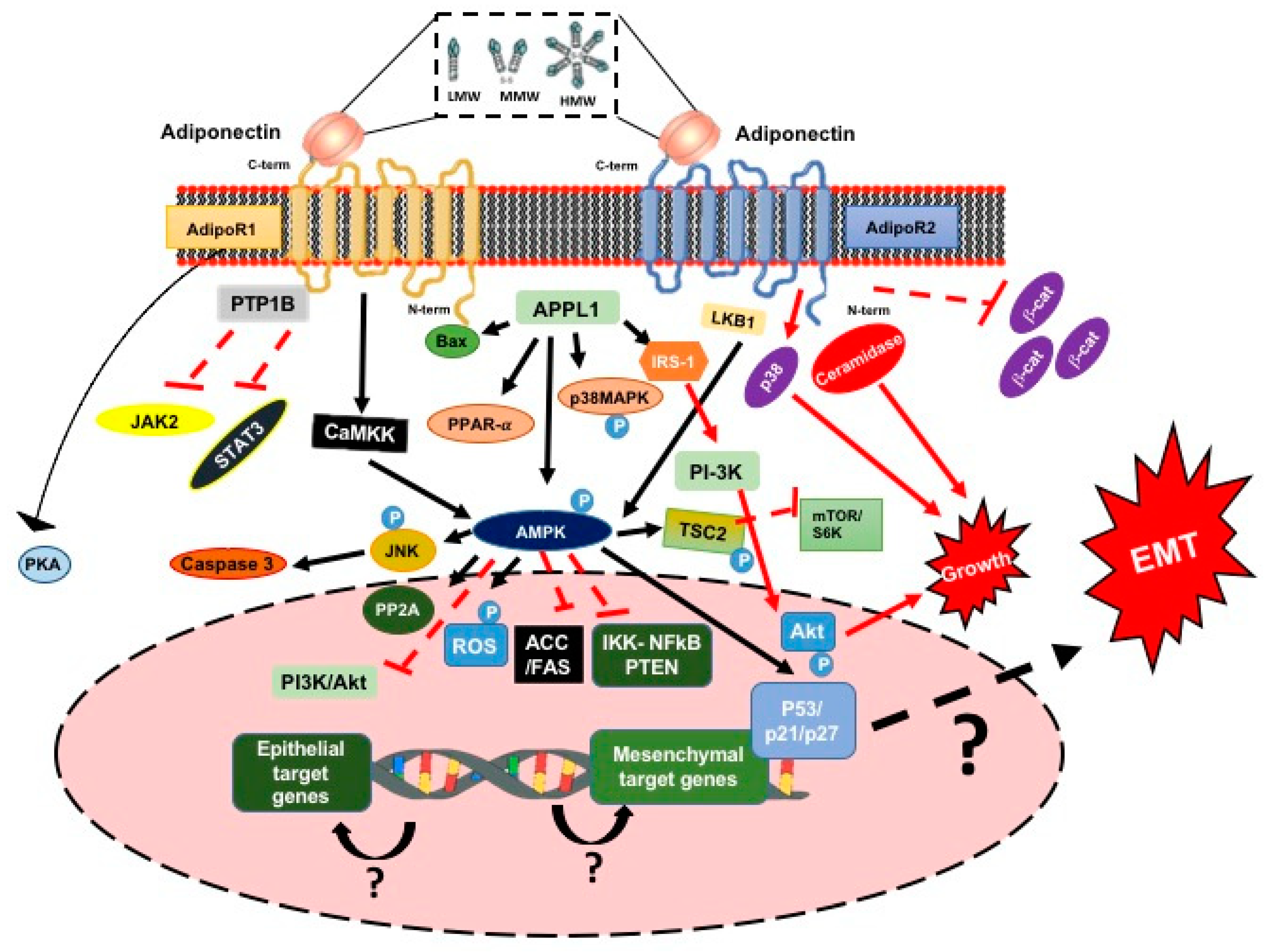
3.1.3. Adiponectin
3.1.4. Adiponectin and the Signaling Pathway
3.1.5. Adiponectin, the EMT Process and ETC
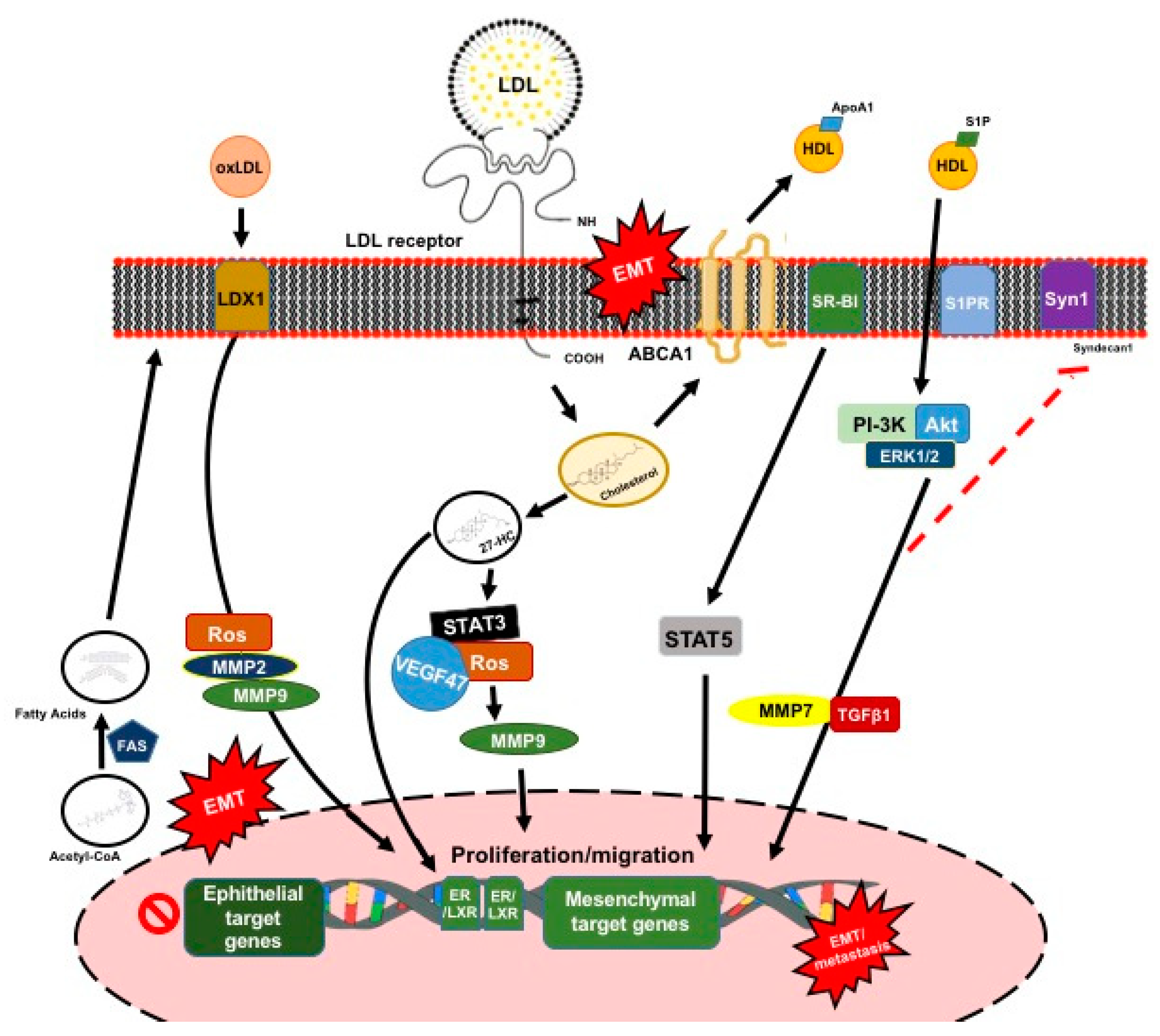
3.2. Plasma Lipoprotein Particles
3.2.1. Role of Low-Density Lipoproteins
3.2.2. Role of High-Density Lipoproteins
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABC | ATP binding cassette |
| AdipoR | Adiponectin receptors |
| AT | Adipose tissue |
| ATC | Anaplastic thyroid cancer |
| CSC | Cancer stem cells |
| CC | Cancer cell |
| CHR | Cytokine homology regions |
| CaMKK | Calcium-dependent kinases |
| DC | Dendritic cell |
| DM2 | Type 2 diabetes |
| ETC | Epithelial thyroid cancer |
| EMT | Epithelial- mesenchymal transition |
| FTC | Follicular thyroid cancer |
| HMW | High molecular weight |
| HDL | High-density lipoproteins |
| HSC | Hematopoietic stem cells |
| IFNα | interferon α |
| ILs | interleukins |
| LPS | lipopolysaccharide |
| LDL | Low-density lipoproteins |
| LMW | Low molecular weight |
| LOX-1 | Lecithin-like receptor 1 |
| MTA1 | Metastasis-associated protein 1 |
| MET | Mesenchymal-epithelial-transition |
| MMW | Medium molecular weight |
| MMP | Matrix metalloproteinase |
| oxLDL | Oxidized low-density lipoproteins |
| PDTC | Poorly differentiated thyroid cancer |
| PTC | Papillary thyroid cancer |
| PTEN | Phosphatase and tensin homolog |
| RAI | Radioactive iodine |
| ROS | Reactive oxygen species |
| SR-BI | Receptor class B type I |
| TAMs | Tumor-associated macrophagues |
| UTC | Undifferentiated thyroid carcinoma |
| VEGF | Vascular endothelial growth factor |
| WDTC | Well-Differentiated thyroid cancer |
| ZEB1 | Zinc finger E-box binding homeobox 1 |
References
- Shah, J.P. Thyroid Carcinoma: Epidemiology, Histology, and Diagnosis. Clin. Adv. Hematol. Oncol. 2015, 13, 3–6. [Google Scholar] [PubMed]
- AJCC Cancer Staging Form Supplement (Eight Edition). Available online: https://cancerstaging.org/references-tools/deskreferences/Documents/AJCC%20Cancer%20Staging%20Form%20Supplement.pdf (accessed on 5 May 2019).
- Amin, M.B.; Edge, S.; Greene, F.; Byrd, D.R.; Brookland, R.K.; Washington, M.K.; Gershenwald, J.E.; Compton, C.C.; Hess, K.R.; Sullivan, D.C.; et al. AJCC Cancer Staging Manual, 8th ed.; Springer: New York, NY, USA, 2017. [Google Scholar]
- Chao, T.C.; Lin, J.D.; Chen, M.F. Insular carcinoma: Infrequent subtype of thyroid cancer with aggressive clinical course. World J. Surg. 2004, 28, 393–396. [Google Scholar] [CrossRef]
- Cipriani, N.A.; Nagar, S.; Kaplan, S.P.; White, M.G.; Antic, T.; Sadow, P.M.; Aschebrook-Kilfoy, B.; Angelos, P.; Kaplan, E.L.; Grogan, R.H. Follicular Thyroid Carcinoma: How Have Histologic Diagnoses Changed in the Last Half-Century and What Are the Prognostic Implications? Thyroid 2015, 25, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Ganly, I.; Nixon, I.J.; Wang, L.Y.; Palmer, F.L.; Migliacci, J.C.; Aniss, A.; Sywak, M.; Eskander, A.E.; Freeman, J.L.; Campbell, M.J.; et al. Survival from Differentiated Thyroid Cancer: What Has Age Got to Do with It? Thyroid 2015, 25, 1106–1114. [Google Scholar] [CrossRef]
- Kakudo, K.; Bychkov, A.; Bai, Y.; Li, Y.; Liu, Z.; Jung, C.K. The new 4th edition World Health Organization classification for thyroid tumors, Asian perspectives. Pathol. Int. 2018, 68, 641–664. [Google Scholar] [CrossRef] [PubMed]
- Volante, M.; Collini, P.; Nikiforov, Y.E.; Sakamoto, A.; Kakudo, K.; Katoh, R.; Lloyd, R.V.; LiVolsi, V.A.; Papotti, M.; Sobrinho-Simoes, M.; et al. Poorly differentiated thyroid carcinoma: The Turin proposal for the use of uniform diagnostic criteria and algorithmic diagnostic approach. Am. J. Surg. Pathol. 2007, 31, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, S.M.; Loree, T.R.; Rigual, N.R.; Hicks, W.L., Jr.; Douglas, W.G.; Anferson, G.R.; Stoler, D.L. Anaplastic transformation of thyroid cancer. Review of clinical, pathologic, and molecular evidence provides new insights into disease biology and future therapy. Head Neck 2003, 25, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Luster, M.; Clarke, S.E.; Dietlein, M.; Lassmann, M.; Lind, P.; Oyen, W.J.G.; Tennvall, J.; Bombardieri, E.; European Association of Nuclear Medicine (EANM). Guidelines for radioiodine therapy of differentiated thyroid cancer. Eur. J. Nucl. Mol. Imaging 2008, 35, 1941–1959. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef]
- Pak, K.; Suh, S.; Kim, S.J.; Kim, I.J. Prognostic value of genetic mutations in thyroid cancer: A meta-analysis. Thyroid 2015, 25, 63–70. [Google Scholar] [CrossRef]
- Patel, K.N. Genetic mutations, molecular markers and future directions in research. Oral Oncol. 2013, 49, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Xing, M. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid 2010, 20, 697–706. [Google Scholar] [CrossRef]
- Brzezianska, E.; Pastuszak-Lewandoska, D.A. A minireview: The role of MAPK/ERK and PI3K/Akt pathways in thyroid follicular cell-derived neoplasm. Front Biosci. 2011, 1, 422–439. [Google Scholar] [CrossRef]
- Nakao, Y.; Mitsuyasu, T.; Kawano, S.; Nakamura, N.; Kanda, S.; Nakamura, S. Fibroblast growth factors 7 and 10 are involved in ameloblastoma proliferation via the mitogen-activated protein kinase pathway. Int. J. Oncol. 2013, 43, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, Y.; Lal, G.; Howe, J.R.; Weigel, R.J.; Komorowski, R.A.; Shilyansky, J.; Sugg, S.L. Invasion in follicular thyroid cancer cell lines is mediated by EphA2 and pAkt. Surgery 2012, 152, 1218–1224. [Google Scholar] [CrossRef]
- Palona, I.; Namba, H.; Mitsutake, N.; Starenki, D.; Podtcheko, A.; Sedliarou, I.; Ohtsuru, A.; Saenko, V.; Nagayama, Y.; Umezawa, K.; et al. BRAFv600E promotes invasiveness of thyroid cancer cells through nuclear factor kB activation. Endocrinology 2006, 147, 5699–5707. [Google Scholar] [CrossRef]
- Pyo, J.S.; Kang, G.; Kim, D.H.; Chae, S.W.; Park, C.; Kim, K.; Do, S.I.; Lee, H.J.; Kim, J.H.; Sohn, J.H. Activation of nuclear factor-kappaB contributes to growth and aggressiveness of papillary thyroid carcinoma. Pathol. Res. Pract. 2013, 209, 228–232. [Google Scholar] [CrossRef]
- Li, X.; Abdel-Mageed, A.B.; Mondal, D.; Kandil, E. The nuclear factor kappa-B signaling pathway as a therapeutic target against thyroid cancers. Thyroid 2013, 23, 209–218. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, M.H.; Kim, D.W.; Lee, S.; Huang, S.; Ryu, M.J.; Kim, Y.K.; Kim, S.J.; Kim, S.J.; Hwang, J.H.; et al. Cross-regulation between oncogenic BRAF(V600E) kinase and the MST1 pathway in papillary thyroid carcinoma. PLoS ONE 2011, 13, e16180. [Google Scholar] [CrossRef]
- Sastre-Perona, A.; Santisteban, P. Role of the wnt pathway in thyroid cancer. Front Endocrinol. 2012, 3, 31. [Google Scholar] [CrossRef]
- Enns, L.; Ladiges, W. Mitochondrial redox signaling and cancer invasiveness. J. Bioenerg. Biomembr. 2012, 44, 635–638. [Google Scholar] [CrossRef][Green Version]
- Dubey, R.; Levin, M.D.; Szabo, L.Z.; Laszlo, C.F.; Kushal, S.; Singh, J.B.; Oh, P.; Schnitzer, J.E.; Olenyuk, B.Z. Suppression of tumor growth by designed dimeric epidithiodiketopiperazine targeting hypoxia-inducible transcription factor complex. J. Am. Chem. Soc. 2013, 135, 4537–4549. [Google Scholar] [CrossRef] [PubMed]
- García-Jiménez, C.; Santisteban, P. TSH signalling and cancer. Arq. Bras. Endocrinol. Metabol. 2007, 51, 654–671. [Google Scholar] [CrossRef] [PubMed]
- Zafon, C.; Obiols, G. The mitogen-activated protein kinase (MAPK) signaling pathway in papillary thyroid cancer. From the molecular bases to clinical practice. Endocrinol. Nutr. 2009, 56, 176–186. [Google Scholar] [CrossRef]
- Romitti, M.; Wajner, S.M.; Zennig, N.; Goemann, I.M.; Bueno, A.L.; Meyer, E.L.; Maia, A.L. Increased type 3 deiodinase expression in papillary thyroid carcinoma. Thyroid 2012, 22, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Motti, M.L.; Califano, D.; Troncone, G.; De Marco, C.; Migliaccio, I.; Palmieri, E.; Pezzullo, L.; Palombini, L.; Fusco, A.; Viglietto, G. Complex regulation of the cyclin-dependent kinase inhibitor p27kip1 in thyroid cancer cells by the PI3K/Akt pathway: Regulation of p27kip1 expression and localization. Am. J. Pathol. 2005, 166, 737–749. [Google Scholar] [CrossRef]
- Serra, R.; Easter, S.L.; Jiang, W.; Baxley, S.E. Wnt5a as an effector of TGFβ in mammary development and cancer. J. Mammary Gland Biol. Neoplasia 2011, 16, 157–167. [Google Scholar] [CrossRef]
- Nieto, H.R.; Boelaert, K. Thyroid stimulating hormone in thyroid cancer: Does it matter? Endocr Relat Cancer 2016, 23, 109–121. [Google Scholar] [CrossRef]
- Xiaochen, X.; Xiaoguang, S.; Haixia, G.; Qiqiang, G.; Chenling, F.; Wenwu, D. P21-activated kinase 4 involves TSH induced papillary thyroid cancer cell proliferation. Oncotarget 2017, 8, 24882–24891. [Google Scholar] [CrossRef]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef]
- Rowe, C.W.; Paul, J.W.; Gedye, C.; Tolosa, J.M.; Bendinelli, C.; McGrath, S.; Smith, R. Targeting the TSH receptor in thyroid cancer. Endocr Relat Cancer 2017, 24, 191–202. [Google Scholar] [CrossRef]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their mMicroenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.C.; Zeniou, M. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Ma, R.; Minsky, N.; Morshed, S.A.; Davies, T.F. Stemness in human thyroid cancers and derived cell lines: The role of asymmetrically dividing cancer stem cells resistant to chemotherapy. J. Clin. Endocrinol. Metab. 2014, 99, E400–E409. [Google Scholar] [CrossRef] [PubMed]
- Garg, M. Emerging role of microRNAs in cancer stem cells: Implications in cancer therapy. World J. Stem Cells 2015, 7, 1078–1089. [Google Scholar] [CrossRef]
- Asadzadeh, Z.; Mansoori, B.; Mohammadi, A.; Aghajani, M.; Haji-Asgarzadeh, K.; Safarzadeh, E.; Mokhtarzadeh, A.; Duijf, P.H.G.; Baradaran, B. MicroRNAs in cancer stem cells: Biology, pathways, and therapeutic opportunities. J. Cell Physiol. 2019, 234, 10002–10017. [Google Scholar] [CrossRef]
- Goodell, M.A.; Brose, K.; Paradis, G.; Conner, A.S.; Mulligan, R.C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996, 183, 1797–1806. [Google Scholar] [CrossRef]
- Mato, E.; González, C.; Moral, A.; Pérez, J.I.; Bell, O.; Lerma, E.; de Leiva, A. ABCG2/BCRP gene expression is related to epithelial–mesenchymal transition inducer genes in a papillary thyroid carcinoma cell line (TPC-1). J. Mol. Endocrinol. 2014, 52, 289–300. [Google Scholar] [CrossRef][Green Version]
- Lan, L.; Luo, Y.; Cui, D.; Shi, B.Y.; Deng, W.; Huo, L.L.; Chen, H.L.; Zhang, G.Y.; Deng, L.L. Epithelial-mesenchymal transition triggers cancer stem cell generation in human thyroid cancer cells. Int. J. Oncol. 2013, 43, 113–120. [Google Scholar] [CrossRef]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef]
- Morandi, A.; Taddei, M.L.; Chiarugi, P.; Giannoni, E. Targeting the metabolic reprogramming that controls epithelial-to-mesenchymal transition in aggressive tumors. Front. Oncol. 2017, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brennan, J.P.; Slavin, J.L.; Blick, T.; Thompson, E.W.; Williams, E.D. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: Role of fibroblast growth factor receptor-2. Cancer Res. 2006, 66, 11271–11278. [Google Scholar] [CrossRef] [PubMed]
- Montemayor-Garcia, C.; Hardin, H.; Guo, Z.; Larrain, C.; Buehler, D.; Asioli, S.; Chen, H.; Lloyd, R.V. The role of epithelial mesenchymal transition markers in thyroid carcinoma progression. Endocr. Pathol. 2013, 24, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Olea-Flores, M.; Juárez-Cruz, J.C.; Mendoza-Catalán, M.A.; Padilla-Benavides, T.; Navarro-Tito, N. Signaling pathways induced by leptin during epithelial−mesenchymal transition in breast cancer. International journal of molecular sciences. Int. J. Mol. Sci. 2018, 19, 3493. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Bonnefond, S.; Morshed, S.A.; Latif, R.; Davies, T.F. Stemness is derived from thyroid cancer cells. Front. Endocrinol. 2014, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Wu, B.; Xiao, K.; Kang, J.; Xie, J.; Zhang, X.; Fan, Y. MiR-146b-5p promotes metastasis and induces epithelial-mesenchymal transition in thyroid cancer by targeting ZNRF3. Cell Physiol. Biochem. 2015, 35, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef]
- Tsuji, T.; Ibaragi, S.; Shima, K.; Hu, M.G.; Katsurano, M.; Sasaki, A.; Hu, G.F. Epithelial-mesenchymal transition induced by growth suppressor p12CDK2-AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res. 2008, 68, 10377–10386. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Ricci, C.; Behrens, G.; Leitzmann, M.F. Adiposity and risk of thyroid cancer: A systematic review and meta-analysis. Obes. Rev. 2015, 16, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Huang, M.; Wang, L.; Ye, W.; Tong, Y.; Wang, H. Obesity and Risk of Thyroid Cancer: Evidence from a Meta-Analysis of 21 Observational Studies. Med. Sci. Monit. 2015, 21, 283–291. [Google Scholar] [CrossRef]
- Shih, S.R.; Chiu, W.Y.; Chang, T.C.; Tseng, C.H. Diabetes and Thyroid Cancer Risk. Literature Review. Exp. Diabetes Res. 2012, 2012, 578285. [Google Scholar] [CrossRef] [PubMed]
- Yeo, Y.; Ma, S.H.; Hwang, Y.; Horn-Ross, P.L.; Hsing, A.; Lee, K.E.; Park, Y.J.; Park, D.J.; Yoo, K.Y.; Park, S.K. Diabetes Mellitus and Risk of Thyroid Cancer: A Meta-Analysis. PLoS ONE 2014, 9, e98135. [Google Scholar] [CrossRef]
- Zhang, W.; Bai, X.; Ge, H.; Cui, H.; Zhijiang, W.; Han, G. Meta-analysis in the association between obesity and risk of thyroid cancer. Int. J. Clin. Exp. Med. 2014, 7, 5268–5274. [Google Scholar] [PubMed]
- Rose, D.P.; Gracheck, P.J.; Vona-Davis, L. The Interactions of Obesity, Inflammation and Insulin Resistance in Breast Cancer. Cancers 2015, 27, 2147–2168. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Yang, Q.; Cao, J.; Xie, N.; Liu, K.; Shou, P.; Qian, F.; Wang, Y.; Shi, Y. Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death Dis. 2016, 7, e2167. [Google Scholar] [CrossRef]
- Nielsen, S.R.; Schmid, M.C. Macrophages as Key Drivers of Cancer Progression and Metastasis. Mediators Inflamm. 2017, 2017, 9624760. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Nannan, L.V. Inflammatory mediators, tumor necrosis factor- and interferon-, induce EMT in human PTC cell lines. Oncol Lett. 2015, 10, 2591–2597. [Google Scholar] [CrossRef]
- Park, J. Obesity and cancer—Mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 2014, 10, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Cui, E.; Guo, H.; Shen, M.; Yu, H.; Gu, D.; Mao, W.; Wang, X. Adiponectin inhibits migration and invasion by reversing epithelial-mesenchymal transition in non-small cell lung carcinoma. Oncol. Rep. 2018, 40, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Cignarelli, A.; Genchi, V.A.; Perrini, S.; Natalicchio, A.; Laviola, L.; Giorgino, F. Insulin and insulin receptors in adipose tissue development. Int. J. Mol. Sci. 2019, 20, 759. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Avtanski, D.; Saxena, N.K.; Sharma, D. Leptin-induced epithelial-mesenchymal transition in breast cancer cells requires-catenin activation via Akt/GSK3-and MTA1/Wnt1 protein-dependent pathways. J. Biol. Chem. 2012, 287, 8598–8612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.A.; Hou, S.; Han, S.; Zhou, J.; Wang, X.; Cui, W. Clinicopathological implications of leptin and leptin receptor expression in papillary thyroid cancer. Oncol. Lett. 2013, 5, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Hedayati, M.; Yaghmaei, P.; Pooyamanesh, Z.; Yeganeh, M.Z.; Rad, L.H. Leptin: A correlated peptide to papillary thyroid carcinoma? J. Thyroid Res. 2011, 2011, 832163. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.P.; Chi, C.W.; Tzen, C.Y.; Yang, T.L.; Lee, J.J.; Liu, T.P. Clinicopathologic significance of leptin and leptin receptor expressions in papillary thyroid carcinoma. Surgery 2010, 147, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Pappa, T.; Alevizaki, M. Obesity and Thyroid Cancer: A Clinical Update. Thyroid 2013, 24, 190–199. [Google Scholar] [CrossRef]
- Uddin, S.; Bavi, P.; Siraj, A.K.; Ahmed, M.; Al-Rasheed, M.; Hussain, A.R.; Ahmed, M.; Amin, T.; Alzahrani, A.; Al-Dayel, F.; et al. Leptin-R and its association with PI3K/Akt signaling pathway in papillary thyroid carcinoma. Endocr. Relat. Cancer 2010, 17, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Wu, M.J.; Yang, J.Y.; Camarillo, I.G.; Chang, C.J. Leptin-STAT3-G9a signaling promotes obesity-mediated breast cancer progression. Cancer Res. 2015, 75, 2375–2386. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Belfiore, A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front. Endocrinol. 2014, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.P.; Yin, P.H.; Chang, Y.C.; Lee, C.H.; Huang, S.Y.; Chi, C.W. Differential roles of leptin in regulating cell migration in thyroid cancer cells. Oncol. Rep. 2010, 23, 1721–1727. [Google Scholar] [PubMed]
- Dossus, L.; Franceschi, S.; Biessy, C.; Navionis, A.S.; Travis, R.C.; Weiderpass, E.; Scalbert, A.; Romieu, I.; Tjønneland, A.; Olsen, A.; et al. Adipokines and inflammation markers and risk of differentiated thyroid carcinoma: The EPIC study. Int. J. Cancer 2017, 142, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The role of adiponectin in cancer: A review of current evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.H.; Lu, M.E. Adiponectin increases motility of human prostate cancer cells via AdipoR, p38, AMPK, and NF-κB pathways. Prostate 2009, 69, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Schettino, P.; Polito, R.; Scudiero, O.; Monaco, M.L.; De Palma, G.D. Adiponectin and colon cancer: Evidence for inhibitory effects on viability and migration of human colorectal cell lines. Mol. Cell Biochem. 2018, 448, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Achari, A.E.; Jain, S.K. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, F.; Niu, R. Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci. Rep. 2015, 5, 17663. [Google Scholar] [CrossRef]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 2011, 17, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Kelesidis, I.; Kelesidis, T.; Mantzoros, C.S. Adiponectin and cancer: A systematic review. Br. J. Cancer 2006, 94, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Barchuk, M.; Miksztowicz, V. Behavior of metalloproteinases in adipose tissue, liver and arterial wall: An update of extracellular matrix remodeling. Cells 2019, 8, 158. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Straight, A.M.; Mann, H.; Duffy, E.; Fenton, C.; Dinauer, C.; Tuttle, R.M.; Francis, G.L. Matrix metalloproteinase (MMP) expression by differentiated thyroid carcinoma of children and adolescents. J. Endocrinol. Invest. 2002, 25, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Roth, L.; Kalev-Altman, R.; Monsonego-Ornan, E.; Sela-Donenfeld, D. A new role of the membrane-type matrix metalloproteinase 16 (MMP16/MT3-MMP) in neural crest cell migration. Int. J. Dev. Biol. 2017, 61, 245–256. [Google Scholar] [CrossRef]
- Bai, X.; Li, Y.Y.; Zhang, H.Y.; Wang, F.; He, H.L.; Yao, J.C.; Liu, L.; Li, S.S. Role of metalloproteinase-9 in transforming growth factor—β1-induced epithelial-mesenchymal transition in esophageal squamous cell carcinoma. Onco Targets Ther. 2017, 10, 2837–2847. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lodish, H.F. Adiponectin deficiency promotes tumor growth in mice by reducing macrophage infiltration. PLoS ONE 2010, 5, e11987. [Google Scholar] [CrossRef]
- Cheng, S.P.; Liu, C.L.; Hsu, Y.C.; Chang, Y.C.; Huang, S.Y.; Lee, J.J. Expression and biologic significance of adiponectin receptors in papillary thyroid carcinoma. Cell Biochem. Biophys. 2013, 65, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Pazaitou-Panayiotou, K.; Aronis, K.N.; Moon, H.S.; Chamberland, J.P.; Liu, X.; Diakopoulos, K.N.; Kyttaris, V.; Panagiotou, V.; Mylvaganam, G.; et al. Circulating adiponectin is inversely associated with risk of thyroid cancer: In vivo and in vitro studies. J. Clin. Endocrinol. Metab. 2011, 96, E2023–E2028. [Google Scholar] [CrossRef] [PubMed]
- Cruz, P.M.; Mo, H.; McConathy, W.J.; Sabnis, N.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013, 4, 119. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, D.; Simon, M.C. Hypoxia, lipids, and cancer: Surviving the harsh tumor microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Botas, J.; Ferruelo, A.J.; Suárez, Y.; Fernández, C.; Gómez-Coronado, D.; Lasunción, M.A. Dose-dependent effects of lovastatin on cell cycle progression. Distinct requirement of cholesterol and non-sterol mevalonate derivatives. Biochim. Biophys. Acta 2001, 1532, 185–194. [Google Scholar] [CrossRef]
- Fernández, C.; Val T. Lobo, M.d.; Gómez-Coronado, D.; Lasunción, M.A. Cholesterol is essential for mitosis progression and its deficiency induces polyploid cell formation. Exp. Cell Res. 2004, 300, 109–120. [Google Scholar] [CrossRef]
- Jameel, F.; Phang, M.; Wood, L.G.; Garg, M.L. Acute effects of feeding fructose, glucose and sucrose on blood lipid levels and systemic inflammation. Lipids Health Dis. 2014, 13, 195. [Google Scholar] [CrossRef] [PubMed]
- Furuya, Y.; Sekine, Y.; Kato, H.; Miyazawa, Y.; Koike, H.; Suzuki, K. Low-density lipoprotein receptors play an important role in the inhibition of prostate cancer cell proliferation by statins. Prostate Int. 2016, 4, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.R. The significance of cholesterol and its metabolite, 27-hydroxycholesterol in breast cancer. Mol. Cell Endocrinol. 2018, 466, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, J.L.; Gerl, M.J.; Klose, C.; Ejsing, C.S.; Beug, H.; Simons, K.; Shevchenko, A. Membrane lipidome of an epithelial cell line. Proc. Natl. Acad. Sci. USA 2011, 108, 1903–1907. [Google Scholar] [CrossRef] [PubMed]
- Warita, K.; Warita, T.; Beckwitt, C.H.; Schurdak, M.E.; Vazquez, A.; Wells, A.; Oltvai, Z.N. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci. Rep. 2014, 4, 7593. [Google Scholar] [CrossRef]
- Li, J.; Dong, L.; Wei, D.; Wang, X.; Zhang, S.; Li, H. Fatty acid synthase mediates the epithelial-mesenchymal transition of breast cancer cells. Int. J. Biol. Sci. 2014, 10, 10171–10180. [Google Scholar] [CrossRef]
- Hung, C.M.; Kuo, D.H.; Chou, C.H.; Su, Y.C.; Ho, C.T.; Way, T.D. Osthole suppresses hepatocyte growth factor (HGF)-induced epithelial-mesenchymal transition via repression of the c-Met/Akt/mTOR pathway in human breast cancer cells. J. Agric. Food Chem. 2011, 59, 9683–9690. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Xiao, L.; Sugiura, H.; Huang, X.; Ali, A.; Kuro-o, M.; Deberardinis, R.J.; Boothman, D.A. Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene 2015, 34, 3908–3916. [Google Scholar] [CrossRef] [PubMed]
- Sudhof, T.C.; Goldstein, J.L.; Brown, M.S.; Russell, D.W. The LDL receptor gene: A mosaic of exons shared with different proteins. Science 1985, 228, 815–822. [Google Scholar] [CrossRef]
- Rotheneder, M.; Kostner, G.M. Effects of low- and high-density lipoproteins on the proliferation of human breast cancer cells in vitro: Differences between hormone-dependent and hormone-independent cell lines. Int. J. Cancer 1989, 43, 875–879. [Google Scholar] [CrossRef] [PubMed]
- De Gonzalo-Calvo, D.; López-Vilaró, L.; Nasarre, L.; Perez-Olabarria, M.; Vázquez, T.; Escuin, D.; Badimon, L.; Barnadas, A.; Lerma, E.; Llorente-Cortés, V. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: A molecular and clinicopathological study. BMC Cancer 2015, 15, 460. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.; Zelenko, Z.; Neel, B.A.; Antoniou, I.M.; Rajan, L.; Kase, N.; LeRoith, D. Elevated tumor LDLR expression accelerates LDL cholesterol-mediated breast cancer growth in mouse models of hyperlipidemia. Oncogene 2017, 36, 6462–6471. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, C.R.; Domingues, G.; Matias, I.; Matos, J.; Fonseca, I.; de Almeida, J.M.; Dias, S. LDL-cholesterol signaling induces breast cancer proliferation and invasion. Lipids Health Dis. 2014, 13, 16. [Google Scholar] [CrossRef]
- Cruz, P.; Epuñán, M.J.; Ramírez, M.E.; Torres, C.G.; Valladares, L.E.; Sierralta, W.D. 27-hydroxycholesterol and the expression of three estrogen-sensitive proteins in MCF7 cells. Oncol. Rep. 2012, 28, 992–998. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Ma, K.L.; Liu, J.; Wu, Y.; Hu, Z.B.; Liu, L.; Lu, J.; Zhang, X.L.; Liu, B.C. Inflammatory stress exacerbates lipid accumulation and podocyte injuries in diabetic nephropathy. Acta Diabetol. 2015, 52, 1045–1056. [Google Scholar] [CrossRef]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313, 17–29. [Google Scholar] [CrossRef]
- Delimaris, I.; Faviou, E.; Antonakos, G.; Stathopoulou, E.; Zachari, A.; Dionyssiou-Asteriou, A. Oxidized LDL, serum oxidizability and serum lipid levels in patients with breast or ovarian cancer. Clin. Biochem. 2007, 15, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Khaidakov, M.; Mitra, S.; Kang, B.Y.; Wang, X.; Kadlubar, S.; Novelli, G.; Raj, V.; Winters, M.; Carter, W.C.; Mehta, J.L. Oxidized LDL Receptor 1 (OLR1) as a possible link between obesity, dyslipidemia and cancer. PLoS ONE 2011, 6, e20277. [Google Scholar] [CrossRef] [PubMed]
- Kume, N.; Kita, T. Roles of lectin-like oxidized LDL receptor-1 and its soluble forms in atherogenesis. Curr. Opin. Lipidol. 2001, 12, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ding, G.; Liang, W.; Chen, C.; Yang, H. Role of LOX-1 and ROS in oxidized low-density lipoprotein induced epithelial-mesenchymal transition of NRK52E. Lipids Health Dis. 2010, 9, 120. [Google Scholar] [CrossRef] [PubMed]
- Parathath, S.; Darlington, Y.F.; de la Llera Moya, M.; Drazul-Schrader, D.; Williams, D.L.; Phillips, M.C.; Rothblat, G.H.; Connelly, M.A. Effects of amino acid substitutions at glycine 420 on SR-BI cholesterol transport function. J. Lipid Res. 2007, 48, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Kinslechner, K.; Schörghofer, D.; Schütz, B.; Vallianou, M.; Wingelhofer, B.; Mikulits, W.; Röhrl, C.; Hengstschläger, M.; Moriggl, R.; Stangl, H.; et al. Malignant phenotypes in metastatic melanoma are governed by SR-BI and its association with glycosylation and STAT5 activation. Mol. Cancer Res. 2018, 16, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Luu, W.; Sharpe, L.J.; Gelissen, I.C.; Brown, A.J. The role of signalling in cellular cholesterol homeostasis. IUBMB Life 2013, 65, 675–684. [Google Scholar] [CrossRef]
- Zhao, W.; Prijic, S.; Urban, B.C.; Tisza, M.J.; Zuo, Y.; Li, L.; Tan, Z.; Chen, X.; Mani, S.A.; Chang, J.T. Candidate antimetastasis drugs suppress the metastatic capacity of breast cancer cells by reducing membrane fluidity. Cancer Res. 2016, 76, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Portolés, C.; Feliu, J.; Reglero, G.; Ramírez de Molina, A. ABCA1 overexpression worsens colorectal cancer prognosis by facilitating tumour growth and caveolin-1-dependent invasiveness and these effects can be ameliorated using the BET inhibitor apabetalone. Mol. Oncol. 2018, 12, 1735–1752. [Google Scholar] [CrossRef]
- Smith, B.; Land, H. Anticancer activity of the cholesterol exporter ABCA1 gene. Cell Rep. 2012, 2, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Yao, X.; Chen, L.; Yan, Z.; Liu, J.; Zhang, Y.; Feng, T.; Wu, J.; Liu, X. Sphingosine-1-phosphate induced epithelial-mesenchymal transition of hepatocellular carcinoma via an MMP-7/syndecan-1/TGF-β autocrine loop. Oncotarget 2016, 7, 63324–63337. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signaling Pathways Affected | Mutations | Types of Thyroid Tumors | Refs |
|---|---|---|---|
| MAPK | BRAF V600E, RAS, RET/PTC, RTK, ALK | PTC | [11,14] |
| PI3K/Akt1, PTEN | FTC | [11] | |
| TGF-β1 | PTC, FTC, ATC, PDTC | [16] | |
| PI3/AKT | PTEN | FTA, FTC | [17] |
| Akt1, Akt2 | FTC | [17] | |
| NF-KB | RET-PTC, RAS, BRAF-V600E | PTC, FTC, ATC | [18,19,20] |
| RASSF1/MST1/FOXO3 | MEK/MAPK, RASSF1/MST1/FOXO3, NF-κB, BRAF-V600E, ERK 1/2, Akt | PTC, FTC, ATC, PDTC | [21] |
| WNT/β-CATENIN | CTNNB1 | ATC, PDTC | [22] |
| HIF1A | HIF1, VEGFA, MET | PDTC, FCT, ATC | [23,24] |
| HIF1α | ATC | [19] | |
| TSHR | TSH-TSHR, NIS | PDTC, FCT, ATC, PTC | [25] |
| PAK4 | PTC | [25] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Revilla, G.; Corcoy, R.; Moral, A.; Escolà-Gil, J.C.; Mato, E. Cross-Talk between Inflammatory Mediators and the Epithelial Mesenchymal Transition Process in the Development of Thyroid Carcinoma. Int. J. Mol. Sci. 2019, 20, 2466. https://doi.org/10.3390/ijms20102466
Revilla G, Corcoy R, Moral A, Escolà-Gil JC, Mato E. Cross-Talk between Inflammatory Mediators and the Epithelial Mesenchymal Transition Process in the Development of Thyroid Carcinoma. International Journal of Molecular Sciences. 2019; 20(10):2466. https://doi.org/10.3390/ijms20102466
Chicago/Turabian StyleRevilla, Giovanna, Rosa Corcoy, Antonio Moral, Joan Carles Escolà-Gil, and Eugenia Mato. 2019. "Cross-Talk between Inflammatory Mediators and the Epithelial Mesenchymal Transition Process in the Development of Thyroid Carcinoma" International Journal of Molecular Sciences 20, no. 10: 2466. https://doi.org/10.3390/ijms20102466
APA StyleRevilla, G., Corcoy, R., Moral, A., Escolà-Gil, J. C., & Mato, E. (2019). Cross-Talk between Inflammatory Mediators and the Epithelial Mesenchymal Transition Process in the Development of Thyroid Carcinoma. International Journal of Molecular Sciences, 20(10), 2466. https://doi.org/10.3390/ijms20102466