Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease



Abstract
1. Introduction
1.1. Glia: A Brief Overview of Diversity in Form and Function
1.2. AMPAR
1.3. AMPAR Involvement in Cellular Injury
1.4. AMPAR in Glial Cells
2. Astrocytes
2.1. Expression and Functional Properties of AMPAR in Astrocytes
2.2. Astrocyte AMPAR Functions Under Physiological Conditions
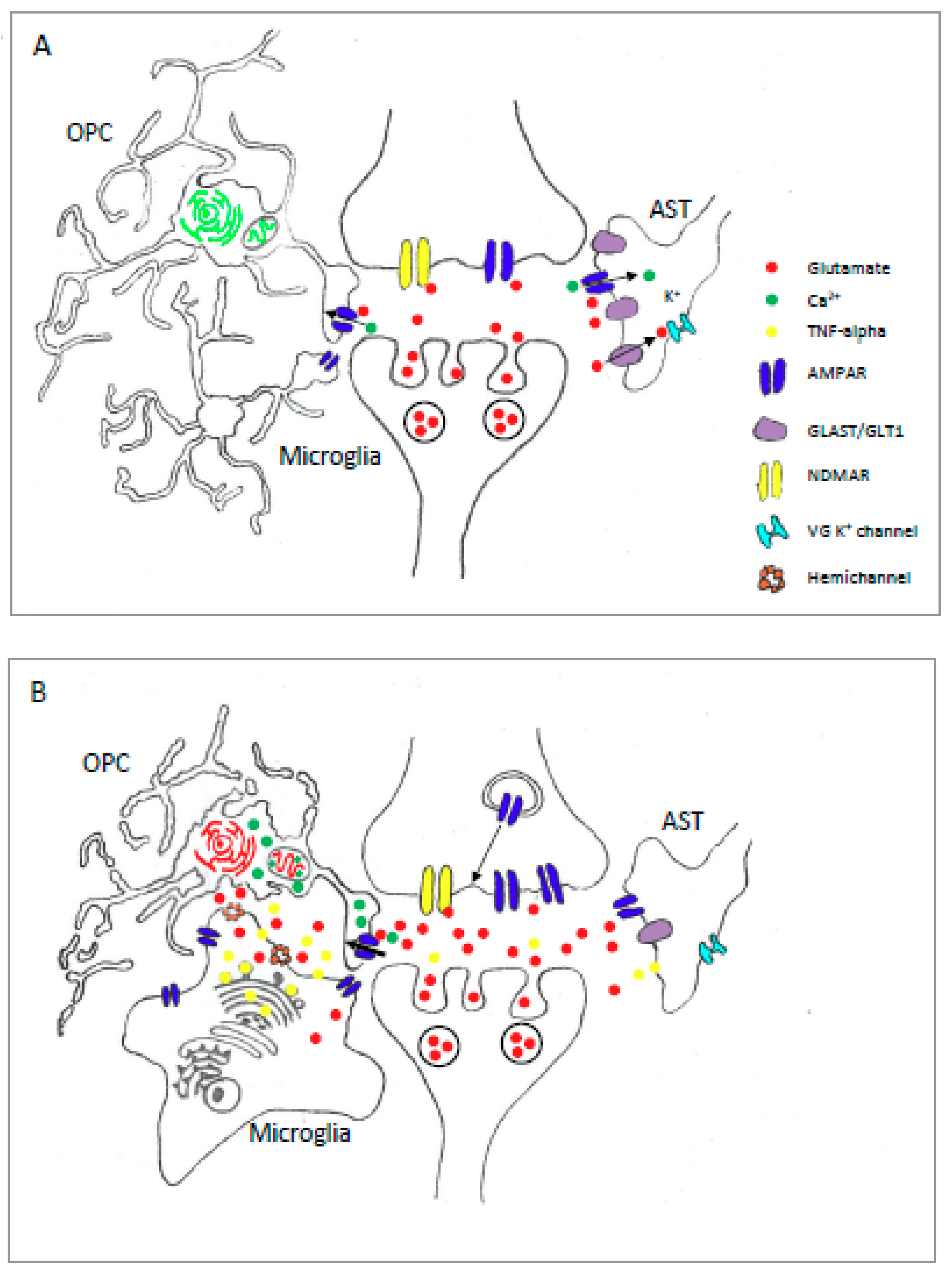
2.3. Astrocyte AMPAR in Pathology
3. Oligodendrocytes
3.1. Expression and Functional Properties of AMPAR in Oligodendrocytes
3.2. Oligodendrocyte AMPAR Functions Under Physiological Conditions
3.3. Oligodendrocyte AMPAR in Pathology
4. Microglia
4.1. Expression and Functional Properties of AMPAR in Microglia
4.2. Microglial AMPAR in Pathology
5. AMPAR in Other Glial Cells
5.1. Radial Glia
5.2. Schwann Cells
5.3. Satellite Glia
5.4. Enteric Glia
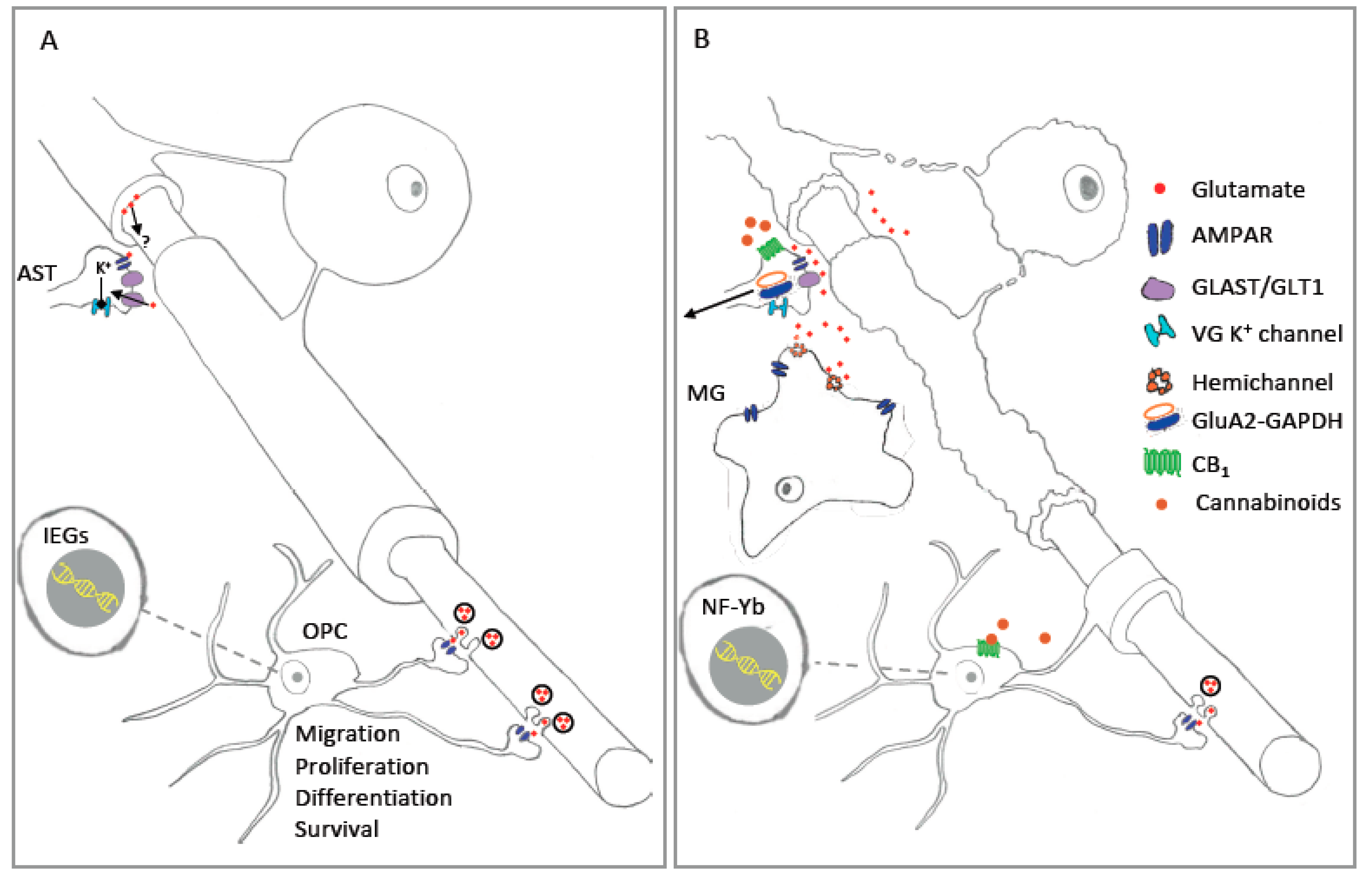
6. Cannabinoids and AMPA Receptor
7. AMPAR-Stimulated Gene Expression in Glial Cells: Contributions to Injury and Disease?
8. Summary
Acknowledgments
Conflicts of Interest
References
- Butt, A.M.; Fern, R.F.; Matute, C. Neurotransmitter signaling in white matter. Glia 2014, 62, 1762–1779. [Google Scholar] [CrossRef] [PubMed]
- Olmos, G.; Lladó, J. Tumor Necrosis Factor Alpha: A Link between Neuroinflammation and Excitotoxicity. Mediators Inflamm. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Fern, R.F.; Matute, C.; Stys, P.K. White matter injury: Ischemic and nonischemic. Glia 2014, 62, 1780–1789. [Google Scholar] [CrossRef]
- Levite, M. Glutamate, T cells and multiple sclerosis. J. Neural Transm. 2017, 124, 775–798. [Google Scholar] [CrossRef]
- Yamane, T. Mouse Yolk Sac Hematopoiesis. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef]
- Kimelberg, H.K.; Nedergaard, M. Functions of Astrocytes and their Potential As Therapeutic Targets. Neurotherapeutics 2010, 7, 338–353. [Google Scholar] [CrossRef]
- Baumann, N.; Pham-Dinh, D. Biology of Oligodendrocyte and Myelin in the Mammalian Central Nervous System. Physiol. Rev. 2001, 81, 871–910. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetanova, I.D.; Nave, K.A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin. Neurobiol. 2013, 23, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.D. A new mechanism of nervous system plasticity: Activity-dependent myelination. Nat. Rev. Neurosci. 2015, 16, 756–767. [Google Scholar] [CrossRef] [PubMed]
- de Faria, O.; Pama, E.A.C.; Evans, K.; Luzhynskaya, A.; Káradóttir, R.T. Neuroglial interactions underpinning myelin plasticity. Dev. Neurobiol. 2018, 78, 93–107. [Google Scholar] [CrossRef]
- Richardson, W.D.; Young, K.M.; Tripathi, R.B.; McKenzie, I. NG2-glia as Multipotent Neural Stem Cells: Fact or Fantasy? Neuron 2011, 70, 661–673. [Google Scholar] [CrossRef]
- Nishiyama, A.; Boshans, L.; Goncalves, C.M.; Wegrzyn, J.; Patel, K.D. Lineage, fate, and fate potential of NG2-glia. Brain Res. 2015, 1638, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Wigley, R.; Hamilton, N.; Nishiyama, A.; Kirchhoff, F.; Butt, A.M. Morphological and physiological interactions of NG2-glia with astrocytes and neurons. Proc. J. Anat. 2007, 210, 661–670. [Google Scholar]
- Hill, R.A.; Nishiyama, A. NG2 cells (polydendrocytes): Listeners to the neural network with diverse properties. Glia 2014, 62, 1195–1210. [Google Scholar] [CrossRef] [PubMed]
- Zuchero, J.B.; Barres, B.A. Glia in mammalian development and disease. Development 2015, 142, 3805–3809. [Google Scholar] [CrossRef] [PubMed]
- Spassky, N. Adult Ependymal Cells Are Postmitotic and Are Derived from Radial Glial Cells during Embryogenesis. J. Neurosci. 2005, 25, 10–18. [Google Scholar] [CrossRef]
- Götz, M.; Barde, Y.-A. Radial Glial Cells. Neuron 2005, 46, 369–372. [Google Scholar] [CrossRef]
- Parnavelas, J.G.; Nadarajah, B. Radial glial cells: Are they really glia? Neuron 2001, 31, 881–884. [Google Scholar] [CrossRef]
- Strazielle, N.; Ghersi-Egea, J.F. Choroid plexus in the central nervous system: Biology and physiopathology. J. Neuropathol. Exp. Neurol. 2000, 59, 561–574. [Google Scholar] [CrossRef]
- Bolborea, M.; Dale, N. Hypothalamic tanycytes: Potential roles in the control of feeding and energy balance. Trends Neurosci. 2013, 36, 91–100. [Google Scholar] [CrossRef]
- Kaur, C.; Rathnasamy, G.; Ling, E.A. Biology of microglia in the developing brain. J. Neuropathol. Exp. Neurol. 2017, 76, 736–753. [Google Scholar] [CrossRef]
- Kidd, G.J.; Ohno, N.; Trapp, B.D. Biology of Schwann cells. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 115, pp. 55–79. ISBN 9780444529022. [Google Scholar]
- Kim, J.; Lee, H.; Park, H. Two faces of Schwann cell dedifferentiation in peripheral neurodegenerative diseases: Pro-demyelinating and axon-preservative functions. Neural Regen. Res. 2014, 9, 1952. [Google Scholar] [CrossRef]
- Hanani, M. Satellite glial cells in sympathetic and parasympathetic ganglia: In search of function. Brain Res. Rev. 2010, 64, 304–327. [Google Scholar] [CrossRef]
- Volianskis, A.; France, G.; Jensen, M.S.; Bortolotto, Z.A.; Jane, D.E.; Collingridge, G.L. Long-term potentiation and the role of N-methyl-d-aspartate receptors. Brain Res. 2015, 1621, 5–16. [Google Scholar] [CrossRef]
- Lerma, J.; Marques, J.M. Kainate receptors in health and disease. Neuron 2013, 80, 292–311. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Kirchhoff, F. NMDA receptors in glia. Neuroscientist 2007, 13, 28–37. [Google Scholar] [CrossRef]
- Dzamba, D.; Honsa, P.; Anderova, M. NMDA Receptors in Glial Cells: Pending Questions. Curr. Neuropharmacol. 2013, 11, 250–262. [Google Scholar] [CrossRef]
- Swanson, G.T.; Kamboj, S.K.; Cull-Candy, S.G. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J. Neurosci. 1997, 17, 58–69. [Google Scholar] [CrossRef]
- Shepherd, J.D.; Huganir, R.L. The Cell Biology of Synaptic Plasticity: AMPA Receptor Trafficking. Annu. Rev. Cell Dev. Biol. 2007, 23, 613–643. [Google Scholar] [CrossRef]
- Geiger, J.R.P.; Melcher, T.; Koh, D.S.; Sakmann, B.; Seeburg, P.H.; Jonas, P.; Monyer, H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 1995, 15, 193–204. [Google Scholar] [CrossRef]
- Hollmann, M.; Hartley, M.; Heinemann, S. Ca2+ permeability of KA-AMPA - gated glutamate receptor channels depends on subunit composition. Science 1991, 252, 851–853. [Google Scholar] [CrossRef]
- Palacios, J.M.; Pazos, A.; Tomiyama, M.; Cortés, R.; Mengod, G.; Rodríguez-Puertas, R. Flip and flop splice variants of AMPA receptor subunits in the spinal cord of amyotrophic lateral sclerosis. Synapse 2002, 45, 245–249. [Google Scholar]
- Haering, S.C.; Tapken, D.; Pahl, S.; Hollmann, M. Auxiliary subunits: Shepherding AMPA receptors to the plasma membrane. Membranes 2014, 4, 469–490. [Google Scholar] [CrossRef]
- Bowie, D. Polyamine-mediated channel block of ionotropic glutamate receptors and its regulation by auxiliary proteins. J. Biol. Chem. 2018, 293, 18789–18802. [Google Scholar] [CrossRef]
- Ishiuchi, S.; Tsuzuki, K.; Yoshida, Y.; Yamada, N.; Hagimura, N.; Okado, H.; Miwa, A.; Kurihara, H.; Nakazato, Y.; Sasaki, T.; et al. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002, 8, 971–978. [Google Scholar] [CrossRef]
- Harlow, D.E.; Saul, K.E.; Komuro, H.; Macklin, W.B. Myelin Proteolipid Protein Complexes with v Integrin and AMPA Receptors In Vivo and Regulates AMPA-Dependent Oligodendrocyte Progenitor Cell Migration through the Modulation of Cell-Surface GluR2 Expression. J. Neurosci. 2015, 35, 12018–12032. [Google Scholar] [CrossRef]
- Iino, M.; Goto, K.; Kakegawa, W.; Okado, H.; Sudo, M.; Ishiuchi, S.; Miwa, A.; Takayasu, Y.; Saito, I.; Tsuzuki, K.; et al. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science 2001, 292, 926–929. [Google Scholar] [CrossRef] [PubMed]
- Fannon, J.; Tarmier, W.; Fulton, D. Neuronal activity and AMPA-type glutamate receptor activation regulates the morphological development of oligodendrocyte precursor cells. Glia 2015, 63, 1021–1035. [Google Scholar] [CrossRef]
- Gallo, V.; Zhou, J.; McBain, C.; Wright, P.; Knutson, P.; Armstrong, R. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. J. Neurosci. 1996, 16, 2659–2670. [Google Scholar] [CrossRef]
- Yuan, X.; Eisen, A.M.; McBain, C.J.; Gallo, V. A role for glutamate and its receptors in the regulation of oligodendrocyte development in cerebellar tissue slices. Development 1998, 125, 2901–2914. [Google Scholar]
- Pende, M.; Holtzclaw, L.A.; Curtis, J.L.; Russell, J.T.; Gallo, V. Glutamate regulates intracellular calcium and gene expression in oligodendrocyte progenitors through the activation of DL-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 3215–3219. [Google Scholar] [CrossRef]
- Begum, G.; Otsu, M.; Ahmed, U.; Ahmed, Z.; Stevens, A.; Fulton, D. NF-Y-dependent regulation of glutamate receptor 4 expression and cell survival in cells of the oligodendrocyte lineage. Glia 2018, 66, 1896–1914. [Google Scholar] [CrossRef] [PubMed]
- Kougioumtzidou, E.; Shimizu, T.; Hamilton, N.B.; Tohyama, K.; Sprengel, R.; Monyer, H.; Attwell, D.; Richardson, W.D. Signalling through AMPA receptors on oligodendrocyte precursors promotes myelination by enhancing oligodendrocyte survival. Elife 2017, 6, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Borges, K.; Kettenmann, H. Blockade of K+ channels induced by AMPA/kainate receptor activation in mouse oligodendrocyte precursor cells is mediated by NA+ entry. J. Neurosci. Res. 1995, 42, 579–593. [Google Scholar] [CrossRef]
- Robert, A.; Magistretti, P.J. AMPA/kainate receptor activation blocks K+ currents via internal Na+ increase in mouse cultured stellate astrocytes. Glia 1997, 20, 38–50. [Google Scholar] [CrossRef]
- Olloquequi, J.; Cornejo-Córdova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Li, N.; Guo, D.-Z.; Pan, S.-Y.; Li, H.; Yang, C. High Plasma Glutamate Levels are Associated with Poor Functional Outcome in Acute Ischemic Stroke. Cell. Mol. Neurobiol. 2014, 35, 159–165. [Google Scholar] [CrossRef]
- Castellanos, M.; Sobrino, T.; Pedraza, S.; Moldes, O.; Pumar, J.M.; Silva, Y.; Serena, J.; Garcia-Gil, M.; Castillo, J.; Davalos, A. High plasma glutamate concentrations are associated with infarct growth in acute ischemic stroke. Neurology 2008, 71, 1862–1868. [Google Scholar] [CrossRef]
- Martínez-Sánchez, P.; Gutiérrez-Fernández, M.; Fuentes, B.; Masjuán, J.; de Leciñana Cases, M.A.; Novillo-López, M.E.; Díez-Tejedor, E. Biochemical and inflammatory biomarkers in ischemic stroke: translational study between humans and two experimental rat models. J. Transl. Med. 2014, 12, 1–13. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Zhang, T.; Liren, C. Excitatory amino acids in cerebrospinal fluid of patients with acute head injuries. Clin. Chem. 2001, 47, 1458–1462. [Google Scholar]
- Kierans, A.S.; Kirov, I.I.; Gonen, O.; Haemer, G.; Nisenbaum, E.; Babb, J.S.; Grossman, R.I.; Lui, Y.W. Myoinositol and glutamate complex neurometabolite abnormality after mild traumatic brain injury. Neurology 2014, 82, 521–528. [Google Scholar] [CrossRef]
- Schroeter, M.; Jander, S. T-Cell Cytokines in Injury-Induced Neural Damage Michael Schroeter and Sebastian Jander *. NeuroMolecular Med. 2005, 7, 183–195. [Google Scholar] [CrossRef]
- Pazos, M.R.; Cinquina, V.; Gómez, A.; Layunta, R.; Santos, M.; Fernández-Ruiz, J.; Martínez-Orgado, J. Cannabidiol administration after hypoxia-ischemia to newborn rats reduces long-term brain injury and restores neurobehavioral function. Neuropharmacology 2012, 63, 776–783. [Google Scholar] [CrossRef]
- Dang, Y.X.; Shi, K.N.; Wang, X.M. Early changes in glutamate metabolism and perfusion in basal ganglia following hypoxia-ischemia in neonatal piglets: A multi-sequence 3.0t MR study. Front. Physiol. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Ceprián, M.; Jimenez-Sanchez, L.; Vargas, C.; Barata, L.; Hind, W.; Martínez-Orgado, J. Cannabidiol reduces brain damage and improves functional recovery in a neonatal rat model of arterial ischemic stroke. Neuropharmacology 2017, 116, 151–159. [Google Scholar] [CrossRef]
- Van Laar, V.S.; Roy, N.; Liu, A.; Rajprohat, S.; Arnold, B.; Dukes, A.A.; Holbein, C.D.; Berman, S.B. Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and, in the presence of N-acetyl cysteine, mitophagy. Neurobiol. Dis. 2015, 74, 180–193. [Google Scholar] [CrossRef]
- Ambrosi, G.; Cerri, S.; Blandini, F. A further update on the role of excitotoxicity in the pathogenesis of Parkinson’s disease. J. Neural Transm. 2014, 121, 849–859. [Google Scholar] [CrossRef]
- Zádori, D.; Veres, G.; Szalárdy, L.; Klivényi, P.; Vécsei, L. Alzheimer’s Disease: Recent Concepts on the Relation of Mitochondrial Disturbances, Excitotoxicity, Neuroinflammation, and Kynurenines. J. Alzheimer’s Dis. 2018, 62, 523–547. [Google Scholar] [CrossRef]
- Al Gawwam, G.; Sharquie, I.K. Serum Glutamate Is a Predictor for the Diagnosis of Multiple Sclerosis. Sci. World J. 2017, 2017. [Google Scholar] [CrossRef]
- Foran, E.; Trotti, D. Glutamate Transporters and the Excitotoxic Path to Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis. Antioxid. Redox Signal. 2009, 11, 1587–1602. [Google Scholar] [CrossRef] [PubMed]
- James, A.; Patel, V. Hypoxic ischaemic encephalopathy. Paediatr. Child Heal. (United Kingdom) 2014, 24, 385–389. [Google Scholar] [CrossRef]
- Alberdi, E.; Sánchez-Gómez, M.V.; Marino, A.; Matute, C. Ca2+ influx through AMPA or kainate receptors alone is sufficient to initiate excitotoxicity in cultured oligodendrocytes. Neurobiol. Dis. 2002, 9, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, S.; Wang, P.; Wang, W. Transient mitochondrial permeability transition mediates excitotoxicity in glutamate-sensitive NSC34D motor neuron-like cells. Exp. Neurol. 2015, 271, 122–130. [Google Scholar] [CrossRef]
- Randall, R.D.; Thayer, S.A. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J. Neurosci. 1992, 12, 1882–1895. [Google Scholar] [CrossRef]
- Thornton, C.; Baburamani, A.A.; Kichev, A.; Hagberg, H. Oxidative stress and endoplasmic reticulum (ER) stress in the development of neonatal hypoxic–ischaemic brain injury. Biochem. Soc. Trans. 2017, 45, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Prentice, H.; Modi, J.P.; Wu, J.-Y. Mechanisms of Neuronal Protection against Excitotoxicity, Endoplasmic Reticulum Stress, and Mitochondrial Dysfunction in Stroke and Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2015, 2015, 1–7. [Google Scholar] [CrossRef]
- Zhao, M.; Zhu, P.; Fujino, M.; Zhuang, J.; Guo, H.; Sheikh, I.; Zhao, L.; Li, X.-K. Oxidative Stress in Hypoxic-Ischemic Encephalopathy: Molecular Mechanisms and Therapeutic Strategies. Int. J. Mol. Sci. 2016, 17, 2078. [Google Scholar] [CrossRef]
- Lin, X.; Zhao, Y.; Li, S. Astaxanthin attenuates glutamate-induced apoptosis via inhibition of calcium influx and endoplasmic reticulum stress. Eur. J. Pharmacol. 2017, 806, 43–51. [Google Scholar] [CrossRef]
- Guemez-Gamboa, A.; Estrada-Sánchez, A.M.; Montiel, T.; Páramo, B.; Massieu, L.; Morán, J. Activation of NOX2 by the stimulation of ionotropic and metabotropic glutamate receptors contributes to glutamate neurotoxicity in vivo through the production of reactive oxygen species and calpain activation. J. Neuropathol. Exp. Neurol. 2011, 70, 1020–1035. [Google Scholar] [CrossRef]
- Blanco, S.; Hernández, R.; Franchelli, G.; Ramos-Álvarez, M.M.; Peinado, M.Á. Melatonin influences NO/NOS pathway and reduces oxidative and nitrosative stress in a model of hypoxic-ischemic brain damage. Nitric Oxide - Biol. Chem. 2017, 62, 32–43. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, F.; Li, L.; Tang, F.; Siedlak, S.L.; Fujioka, H.; Liu, Y.; Su, B.; Pi, Y.; Wang, X. MFN2 couples glutamate excitotoxicity and mitochondrial dysfunction in motor neurons. J. Biol. Chem. 2015, 290, 168–182. [Google Scholar] [CrossRef]
- Andreyev, A.; Tamrakar, P.; Rosenthal, R.E.; Fiskum, G. Calcium uptake and cytochrome c release from normal and ischemic brain mitochondria. Neurochem. Int. 2018, 117, 15–22. [Google Scholar] [CrossRef]
- Pottorf, W.J., II; Johanns, T.M.; Derrington, S.M.; Strehler, E.E.; Eneyde, A.; Thayer, S. Glutamate-induced protease-mediated loss of plasma membrane Ca2+ pump activity in rat hippocampal neurons. J. Neurochem. 2006, 98, 1646–1656. [Google Scholar] [CrossRef]
- Thornton, C.; Jones, A.; Nair, S.; Aabdien, A.; Mallard, C.; Hagberg, H. Mitochondrial dynamics, mitophagy and biogenesis in neonatal hypoxic-ischaemic brain injury. FEBS Lett. 2017, 1–19. [Google Scholar] [CrossRef]
- Brostrom, M.A.; Brostrom, C.O. Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: Implications for cell growth and adaptability. Cell Calcium 2003, 34, 345–363. [Google Scholar] [CrossRef]
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic reticulum stress and oxidative stress: A vicious nexus implicated in bowel disease pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef]
- Toral-Ojeda, I.; Aldanondo, G.; Lasa-Elgarresta, J.; Lasa-Fernández, H.; Fernández-Torrón, R.; López de Munain, A.; Vallejo-Illarramendi, A. Calpain 3 deficiency affects SERCA expression and function in the skeletal muscle. Expert Rev. Mol. Med. 2016, 18, e7. [Google Scholar] [CrossRef]
- French, J.P.; Quindry, J.C.; Falk, D.J.; Staib, J.L.; Lee, Y.; Wang, K.K.W.; Powers, S.K. Ischemia-reperfusion-induced calpain activation and SERCA2a degradation are attenuated by exercise training and calpain inhibition. Am. J. Physiol. Circ. Physiol. 2005, 290, H128–H136. [Google Scholar] [CrossRef]
- Pedrozo, Z.; Sánchez, G.; Torrealba, N.; Valenzuela, R.; Fernández, C.; Hidalgo, C.; Lavandero, S.; Donoso, P. Calpains and proteasomes mediate degradation of ryanodine receptors in a model of cardiac ischemic reperfusion. Biochim. Biophys. Acta - Mol. Basis Dis. 2010, 1802, 356–362. [Google Scholar] [CrossRef]
- Neginskaya, M.; Berezhnaya, E.; Uzdensky, A.B.; Abramov, A.Y. Reactive oxygen species produced by a photodynamic effect induced calcium signal in neurons and astrocytes. Mol. Neurobiol. 2018, 55, 96–102. [Google Scholar] [CrossRef]
- Ruiz, A.; Matute, C.; Alberdi, E. Endoplasmic reticulum Ca2+ release through ryanodine and IP3 receptors contributes to neuronal excitotoxicity. Cell Calcium 2009, 46, 273–281. [Google Scholar] [CrossRef]
- Mei, B.; Sun, Z.-W.; Zhang, L.; Zhu, S.-J.; Chen, W.-C. Excitotoxicity effects of glutamate on human neuroblastoma SH-SY5Y cells via oxidative damage. Neurosci. Bull. 2010, 26, 8–16. [Google Scholar]
- Martínez-Fábregas, J.; Díaz-Moreno, I.; González-Arzola, K.; Janocha, S.; Navarro, J.A.; Hervás, M.; Bernhardt, R.; Velázquez-Campoy, A.; Díaz-Quintana, A.; De la Rosa, M.A. Structural and Functional Analysis of Novel Human Cytochrome c Targets in Apoptosis. Mol. Cell. Proteomics 2014, 13, 1439–1456. [Google Scholar] [CrossRef]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAf-1 · cytochrome C multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef]
- Imbriano, C.; Gnesutta, N.; Mantovani, R. The NF-Y/p53 liaison: Well beyond repression. Biochim. Biophys. Acta - Rev. Cancer 2012, 1825, 131–139. [Google Scholar] [CrossRef]
- Filous, A.R.; Silver, J. Targeting astrocytes in CNS injury and disease: A translational research approach. Prog. Neurobiol. 2016, 144, 173–187. [Google Scholar] [CrossRef]
- Mishra, A. Binaural blood flow control by astrocytes: Listening to synapses and the vasculature. J. Physiol. 2017, 595, 1885–1902. [Google Scholar] [CrossRef] [PubMed]
- Kıray, H.; Lindsay, S.L.; Hosseinzadeh, S.; Barnett, S.C. The multifaceted role of astrocytes in regulating myelination. Exp. Neurol. 2016, 283, 541–549. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.R.; Robitaille, R.; Volterra, A. Gliotransmitters travel in time and space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef]
- Nedergaard, M.; Verkhratsky, A. Artifact versus reality-How astrocytes contribute to synaptic events. Glia 2012, 60, 1013–1023. [Google Scholar] [CrossRef]
- Bradley, S.J.; Challiss, R.A.J. G protein-coupled receptor signalling in astrocytes in health and disease: A focus on metabotropic glutamate receptors. Biochem. Pharmacol. 2012, 84, 249–259. [Google Scholar] [CrossRef]
- Burnashev, N.; Khodorova, A.; Jonas, P.; Helm, P.J.; Wisden, W.; Monyer, H.; Seeburg, P.H.; Sakmann, B. Calcium-permeable AMPA-kainate receptors in fusiform cerebellar glial cells. Science 1992, 256, 1566–1570. [Google Scholar] [CrossRef]
- Müller, T.; Möller, T.; Berger, T.; Schnitzer, J.; Kettenmann, H. Calcium entry through kainate receptors and resulting potassium-channel blockade in Bergmann glial cells. Science 1992, 256, 1563–1566. [Google Scholar] [CrossRef]
- Saab, A.S.; Neumeyer, A.; Jahn, H.M.; Cupido, A.; Le Meur, K.; Deitmer, J.W.; Monyer, H.; Boele, H.-J.; Scheller, A.; Gotz, M.; et al. Bergmann Glial AMPA Receptors Are Required for Fine Motor Coordination. Science 2012, 337, 749–753. [Google Scholar] [CrossRef]
- López-Bayghen, E.; Espinoza-Rojo, M.; Ortega, A. Glutamate down-regulates GLAST expression through AMPA receptors in Bergmann glial cells. Mol. Brain Res. 2003, 115, 1–9. [Google Scholar] [CrossRef]
- Mölders, A.; Koch, A.; Menke, R.; Klöcker, N. Heterogeneity of the astrocytic AMPA-receptor transcriptome. Glia 2018, 66, 2604–2616. [Google Scholar] [CrossRef]
- David, J.C.; Yamada, K.A.; Bagwe, M.R.; Goldberg, M.P. AMPA receptor activation is rapidly toxic to cortical astrocytes when desensitization is blocked. J. Neurosci. 1996, 16, 200–209. [Google Scholar] [CrossRef]
- Matthias, K.; Kirchhoff, F.; Seifert, G.; Hüttmann, K.; Matyash, M.; Kettenmann, H.; Steinhäuser, C. Segregated expression of AMPA-type glutamate receptors and glutamate transporters defines distinct astrocyte populations in the mouse hippocampus. J. Neurosci. 2003, 23, 1750–1758. [Google Scholar] [CrossRef]
- Droste, D.; Seifert, G.; Seddar, L.; Jädtke, O.; Steinhaüser, C.; Lohr, C. Ca2+-permeable AMPA receptors in mouse olfactory bulb astrocytes. Sci. Rep. 2017, 7, 44817. [Google Scholar] [CrossRef]
- Höft, S.; Griemsmann, S.; Seifert, G.; Steinhäuser, C. Heterogeneity in expression of functional ionotropic glutamate and GABA receptors in astrocytes across brain regions: Insights from the thalamus. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef]
- Lalo, U. NMDA Receptors Mediate Neuron-to-Glia Signaling in Mouse Cortical Astrocytes. J. Neurosci. 2006, 26, 2673–2683. [Google Scholar] [CrossRef]
- Li, S.; Stys, P.K. Mechanisms of Ionotropic Glutamate Receptor-Mediated Excitotoxicity in Isolated Spinal Cord White Matter. J. Neurosci. 2000, 20, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M.; Colquhoun, K.; Tutton, M.; Berry, M. Three-dimensional morphology of astrocytes and oligodendrocytes in the intact mouse optic nerve. J. Neurocytol. 1994, 23, 469–485. [Google Scholar] [CrossRef]
- Serwanski, D.R.; Jukkola, P.; Nishiyama, A. Heterogeneity of astrocyte and NG2 cell insertion at the node of ranvier. J. Comp. Neurol. 2017, 525, 535–552. [Google Scholar] [CrossRef]
- Micu, I.; Plemel, J.R.; Lachance, C.; Proft, J.; Jansen, A.J.; Cummins, K.; van Minnen, J.; Stys, P.K. The molecular physiology of the axo-myelinic synapse. Exp. Neurol. 2016, 276, 41–50. [Google Scholar] [CrossRef]
- Rosenbluth, J. Multiple functions of the paranodal junction of myelinated nerve fibers. J. Neurosci. Res. 2009, 87, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Marvin, J.S.; Borghuis, B.G.; Tian, L.; Cichon, J.; Harnett, M.T.; Akerboom, J.; Gordus, A.; Renninger, S.L.; Chen, T.W.; Bargmann, C.I.; et al. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 2013, 10, 162–170. [Google Scholar] [CrossRef]
- Savtchouk, I.; Carriero, G.; Volterra, A. Studying Axon-Astrocyte Functional Interactions by 3D Two-Photon Ca2+ Imaging: A Practical Guide to Experiments and “Big Data” Analysis. Front. Cell. Neurosci. 2018, 12, 98. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.G.; Lyons, D.A. On Myelinated Axon Plasticity and Neuronal Circuit Formation and Function. J. Neurosci. 2017, 37, 10023–10034. [Google Scholar] [CrossRef] [PubMed]
- Grosche, J.; Kettenmann, H.; Verkhratsky, A.; Matyash, V.; Möller, T.; Reichenbach, A. Microdomains for neuron–glia interaction: Parallel fiber signaling to Bergmann glial cells. Nat. Neurosci. 2002, 2, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Han, B.H.; Luo, N.L.; Chricton, C.A.; Xanthoudakis, S.; Tam, J.; Arvin, K.L.; Holtzman, D.M. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J. Neurosci. 2002, 22, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S. Synaptic release of excitatory amino acid neurotransmitter mediates anoxic neuronal death. J. Neurosci. 1984, 4, 1884–1891. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.J.; Nagarajah, R.; Banati, R.B.; Bennett, M.R. Glutamate induces directed chemotaxis of microglia. Eur. J. Neurosci. 2009, 29, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Salińska, E.; Danysz, W.; Łazarewicz, J.W. The role of excitotoxicity in neurodegeneration. Folia Neuropathol. 2005, 43, 322–339. [Google Scholar] [PubMed]
- Pitt, D.; Werner, P.; Raine, C.S. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 2000, 6, 67–70. [Google Scholar] [CrossRef]
- Kanwar, J.R.; Kanwar, R.K.; Krissansen, G.W. Simultaneous neuroprotection and blockade of inflammation reverses autoimmune encephalomyelitis. Brain 2004, 127, 1313–1331. [Google Scholar] [CrossRef]
- Groom, A.J.; Smith, T.; Turski, L. Multiple sclerosis and glutamate. Proc. Ann. N. Y. Acad. Sci. 2003, 993, 229–275. [Google Scholar]
- Kaito, M.; Ren, E.; Voskuhl, R.R.; Johnsonbaugh, H.; Tassoni, A.; Itoh, N.; Burda, J.; Ao, Y.; Farkhondeh, V.; Sofroniew, M.V.; et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc. Natl. Acad. Sci. USA 2017, 115, E302–E309. [Google Scholar]
- Lee, F.H.F.; Jia, Z.; Zhang, S.; Wong, A.H.C.; D’Souza, C.; Zhang, L.; Su, P.; Zhai, D.; Liu, F. Blocking GluR2-GAPDH ameliorates experimental autoimmune encephalomyelitis. Ann. Clin. Transl. Neurol. 2015, 2, 388–400. [Google Scholar]
- Wang, M.; Li, S.; Zhang, H.; Pei, L.; Zou, S.; Lee, F.J.S.; Wang, Y.T.; Liu, F. Direct interaction between GluR2 and GAPDH regulates AMPAR-mediated excitotoxicity. Mol. Brain 2012, 5, 13. [Google Scholar] [CrossRef]
- Zhai, D.; Chin, K.; Wang, M.; Liu, F. Disruption of the nuclear p53-GAPDH complex protects against ischemia-induced neuronal damage. Mol. Brain 2014, 7, 20. [Google Scholar] [CrossRef]
- Lee, F.H.F.; Zhang, H.; Jiang, A.; Zai, C.C.; Liu, F. Specific Alterations in Astrocyte Properties via the GluA2-GAPDH Complex Associated with Multiple Sclerosis. Sci. Rep. 2018, 8, 12856. [Google Scholar] [CrossRef]
- Pérez-Cerdá, F.; Sánchez-Gómez, M.V.; Matute, C. Pío del Río Hortega and the discovery of the oligodendrocytes. Front. Neuroanat. 2015, 9, 7–12. [Google Scholar] [CrossRef]
- Kessaris, N.; Fogarty, M.; Iannarelli, P.; Grist, M.; Wegner, M.; Richardson, W.D. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 2006, 9, 173–179. [Google Scholar] [CrossRef]
- Winkler, C.C.; Yabut, O.R.; Fregoso, S.P.; Gomez, H.G.; Dwyer, B.E.; Pleasure, S.J.; Franco, S.J. The Dorsal Wave of Neocortical Oligodendrogenesis Begins Embryonically and Requires Multiple Sources of Sonic Hedgehog. J. Neurosci. 2018, 38, 5237–5250. [Google Scholar] [CrossRef]
- Hashimoto, R.; Sakai, K.; Masuyama, N.; Akiyama, H.; Inoue, Y.U.; Hoshino, M.; Inoue, T.; Hayase, Y.; Kawaguchi, Y.; Koizumi, S.; et al. Origins of oligodendrocytes in the cerebellum, whose development is controlled by the transcription factor, Sox9. Mech. Dev. 2016, 140, 25–40. [Google Scholar] [CrossRef]
- Fogarty, M.; Richardson, W.D.; Kessaris, N. A subset of oligodendrocytes generated from radial glia in the dorsal spinal cord. Development 2005, 132, 1951–1959. [Google Scholar] [CrossRef]
- Pringle, N.P.; Yu, W.P.; Guthrie, S.; Roelink, H.; Lumsden, A.; Peterson, A.C.; Richardson, W.D. Determination of neuroepithelial cell fate: Induction of the oligodendrocyte lineage by ventral midline cells and Sonic hedgehog. Dev. Biol. 1996, 177, 30–42. [Google Scholar] [CrossRef]
- Barateiro, A.; Fernandes, A. Temporal oligodendrocyte lineage progression: In vitro models of proliferation, differentiation and myelination. Biochim. Biophys. Acta - Mol. Cell Res. 2014, 1843, 1917–1929. [Google Scholar] [CrossRef]
- Skaper, S.D.; Zusso, M.; Marinelli, C.; Giusti, P.; Bertalot, T. Systematic Review of Pharmacological Properties of the Oligodendrocyte Lineage. Front. Cell. Neurosci. 2016, 10, 1–19. [Google Scholar]
- Anna Cunningham, L.; Newville, J.; Jantzie, L.L. Embracing oligodendrocyte diversity in the context of perinatal injury. Neural Regen Res 2017, 12, 1575–1585. [Google Scholar] [CrossRef]
- Roy, N.S.; Wang, S.; Harrison-Restelli, C.; Benraiss, A.; Fraser, R.A.R.; Gravel, M.; Braun, P.E.; Goldman, S.A. Identification, Isolation, and Promoter-Defined Separation of Mitotic Oligodendrocyte Progenitor Cells from the Adult Human Subcortical White Matter. J. Neurosci. 1999, 19, 9986–9995. [Google Scholar] [CrossRef]
- Tanaka, K.; Nogawa, S.; Suzuki, S.; Dembo, T.; Kosakai, A. Upregulation of oligodendrocyte progenitor cells associated with restoration of mature oligodendrocytes and myelination in peri-infarct area in the rat brain. Brain Res. 2003, 989, 172–179. [Google Scholar] [CrossRef]
- Singh, J.; Sharma, K.; Frost, E.E.; Pillai, P.P. Role of PDGF-A-Activated ERK Signaling Mediated FAK-Paxillin Interaction in Oligodendrocyte Progenitor Cell Migration. J. Mol. Neurosci. 2019, 1, 10. [Google Scholar] [CrossRef]
- Biname, F.; Sakry, D.; Dimou, L.; Jolivel, V.; Trotter, J. NG2 Regulates Directional Migration of Oligodendrocyte Precursor Cells via Rho GTPases and Polarity Complex Proteins. J. Neurosci. 2013, 33, 10858–10874. [Google Scholar] [CrossRef]
- Hamilton, N.; Vayro, S.; Wigley, R.; Butt, A.M. Axons and astrocytes release ATP and glutamate to evoke calcium signals in NG2-glia. Glia 2010, 58, 66–79. [Google Scholar] [CrossRef]
- Larson, V.A.; Zhang, Y.; Bergles, D.E. Electrophysiological properties of NG2+cells: Matching physiological studies with gene expression profiles. Brain Res. 2016, 1638, 138–160. [Google Scholar] [CrossRef]
- Gard, A.L.; Williams, C., II; Burrell, M.R. Oligodendroblasts Distinguished from O-2A Glial Progenitors by Surface Phenotype (O4+GalC-) and Response to Cytokines Using Signal Transducer LIFRB. Dev. Biol. 1995, 167, 596–608. [Google Scholar] [CrossRef]
- Niu, J.; Mei, F.; Wang, L.; Tian, Y.; Mo, W.; Li, H.; Lu, Q.R.; Xiao, L. Phosphorylated Olig1 Localizes to the Cytosol of Oligodendrocytes and Promotes Membrane Expansion and Maturation. Glia 2012, 60, 1427–1436. [Google Scholar] [CrossRef]
- Rivkin, M.J.; Flax, J.; Mozell, R.; Osathanondh, R.; Volpe, J.J.; Villa-Komaroff, L. Oligodendroglial development in human fetal cerebrum. Ann. Neurol. 2005, 38, 92–101. [Google Scholar] [CrossRef]
- Liu, S.; Qu, Y.; Stewart, T.J.; Howard, M.J.; Chakrabortty, S.; Holekamp, T.F.; McDonald, J.W. Embryonic stem cells differentiate into oligodendrocytes and myelinate in culture and after spinal cord transplantation. Proc. Natl. Acad. Sci. USA 2000, 97, 6126–6131. [Google Scholar] [CrossRef]
- Ziemka-Nalecz, M.; Janowska, J.; Strojek, L.; Jaworska, J. Impact of neonatal hypoxia-ischaemia on oligodendrocyte survival, maturation and myelinating potential. J. Cell. Mol. Med. 2018, 22, 207–222. [Google Scholar] [CrossRef]
- Stiefel, K.M.; Torben-Nielsen, B.; Coggan, J.S. Proposed evolutionary changes in the role of myelin. Front. Neurosci. 2013, 7, 1–9. [Google Scholar] [CrossRef]
- Gautier, H.O.B.; Franklin, R.J.M.; Reynolds, R.; Lao-Peregrin, C.; Lundgaard, I.; Evans, K.A.; Sitnikov, S.; James, R.; Volbracht, K.; James, F.; et al. Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat. Commun. 2015, 6, 8518. [Google Scholar] [CrossRef]
- Drobyshevsky, A.; Jiang, R.; Lin, L.; Derrick, M.; Luo, K.; Back, S.A.; Tan, S. Unmyelinated axon loss with postnatal hypertonia after fetal hypoxia. Ann. Neurol. 2014, 75, 533–541. [Google Scholar] [CrossRef]
- Mitew, S.; Lulu, Y.; Merson, T.D. Axonal activity-dependent myelination in development: Insights for myelin repair. J. Chem. Neuroanat. 2016, 76, 2–8. [Google Scholar] [CrossRef]
- Stedehouder, J.; Brizee, D.; Shpak, G.; Kushner, S.A. Activity-Dependent Myelination of Parvalbumin Interneurons Mediated by Axonal Morphological Plasticity. J. Neurosci. 2018, 38, 3631–3642. [Google Scholar] [CrossRef]
- Nave, K.-A. Myelination and support of axonal integrity by glia. Nature 2010, 468, 244–252. [Google Scholar] [CrossRef]
- Lee, K.Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Hossain, S.; Liu, H.N.; Fragoso, G.; Almazan, G. Agonist-induced down-regulation of AMPA receptors in oligodendrocyte progenitors. Neuropharmacology 2014, 79, 506–514. [Google Scholar] [CrossRef]
- Itoh, T.; Beesley, J.; Itoh, A.; Cohen, A.S.; Kavanaugh, B.; Coulter, D.A.; Grinspan, J.B.; Pleasure, D. AMPA glutamate receptor-mediated calcium signaling is transiently enhanced during development of oligodendrocytes. J. Neurochem. 2002, 81, 390–402. [Google Scholar] [CrossRef]
- Gallo, V.; Patneau, D.K.; Mayer, M.L.; Vaccarino, F.M. Excitatory amino acid receptors in glial progenitor cells: Molecular and functional properties. Glia 1994, 11, 94–101. [Google Scholar] [CrossRef]
- Seifert, G.; Rehn, L.; Weber, M.; Steinhäuser, C. AMPA receptor subunits expressed by single astrocytes in the juvenile mouse hippocampus. Mol. Brain Res. 1997, 47, 286–294. [Google Scholar] [CrossRef]
- Wright, P.; Chew, L.-J.; Fleck, M.W.; Gallo, V.; Scherer, S.E.; Mayer, M.L. Growth Factor-Induced Transcription of GluR1 Increases Functional AMPA Receptor Density in Glial Progenitor Cells. J. Neurosci. 2018, 17, 227–240. [Google Scholar]
- Bergles, D.E.; Roberts, J.D.B.; Somogyl, P.; Jahr, C.E. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 2000, 405, 187–191. [Google Scholar] [CrossRef]
- Zhou, W.; Jan, Y.N.; Luo, Q.; Ge, W.-P.; Jan, L.Y. Dividing glial cells maintain differentiated properties including complex morphology and functional synapses. Proc. Natl. Acad. Sci. USA 2008, 106, 328–333. [Google Scholar]
- Haberlandt, C.; Derouiche, A.; Wyczynski, A.; Haseleu, J.; Pohle, J.; Karram, K.; Trotter, J.; Seifert, G.; Frotscher, M.; Steinhäuser, C.; et al. Gray Matter NG2 Cells Display Multiple Ca2+-Signaling Pathways and Highly Motile Processes. PLoS ONE 2011, 6, e17575. [Google Scholar] [CrossRef]
- Seifert, G.; Zhou, M.; Steinhäuser, C. Analysis of AMPA Receptor Properties During Postnatal Development of Mouse Hippocampal Astrocytes. J. Neurophysiol. 1997, 78, 2916–2923. [Google Scholar] [CrossRef]
- Yoshioka, A.; Bacskai, B.; Pleasure, D. Pathophysiology of oligodendroglial excitotoxicity. J. Neurosci. Res. 1996, 46, 427–437. [Google Scholar] [CrossRef]
- Zhang, Y.; Bennett, M.L.; Maniatis, T.; Wu, J.Q.; Deng, S.; O’Keeffe, S.; Guarnieri, P.; Scholze, A.R.; Chen, K.; Zhang, C.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.G.; Fern, R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 2005, 438, 1167–1171. [Google Scholar] [CrossRef] [PubMed]
- Renzel, R.; Sadek, A.R.; Chang, C.H.; Gray, W.P.; Seifert, G.; Steinhäuser, C. Polarized distribution of AMPA, but not GABAA, receptors in radial glia-like cells of the adult dentate gyrus. Glia 2013, 61, 1146–1154. [Google Scholar] [CrossRef]
- Christensen, P.C.; Samadi-Bahrami, Z.; Pavlov, V.; Stys, P.K.; Moore, G.R.W. Ionotropic glutamate receptor expression in human white matter. Neurosci. Lett. 2016, 630, 1–8. [Google Scholar] [CrossRef]
- Talos, D.M.; Fishman, R.E.; Park, H.; Folkerth, R.D.; Follett, P.L.; Volpe, J.J.; Jensen, F.E. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. I. Rodent cerebral white matter and cortex. J. Comp. Neurol. 2006, 497, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Ness, J.K.; Scaduto, R.C.; Wood, T.L. IGF-I Prevents Glutamate-Mediated Bax Translocation and Cytochrome C Release in O4+ Oligodendrocyte Progenitors. Glia 2004, 46, 183–194. [Google Scholar] [CrossRef]
- Ness, J.K.; Wood, T.L. Insulin-like growth factor I, but not neurotrophin-3, sustains Akt activation and provides long-term protection of immature oligodendrocytes from glutamate-mediated apoptosis. Mol. Cell. Neurosci. 2002, 20, 476–488. [Google Scholar] [CrossRef]
- Deng, W.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc. Natl. Acad. Sci. USA 2003, 100, 6801–6806. [Google Scholar] [CrossRef] [PubMed]
- Follett, P.L.; Deng, W.; Dai, W.; Talos, D.M.; Massillon, L.J.; Rosenberg, P.A.; Volpe, J.J.; Jensen, J.B. Glutamate Receptor-Mediated Oligodendrocyte Toxicity in Periventricular Leukomalacia: A Protective Role for Topiramate. J. Neurosci. 2004, 24, 4412–4420. [Google Scholar] [CrossRef]
- Deng, W.; Neve, R.L.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. α-Amino-3-hydroxy-5-methyl-4-isoxazole Propionate Receptor Subunit Composition and cAMP-response Element-binding Protein Regulate Oligodendrocyte Excitotoxicity. J. Biol. Chem. 2006, 281, 36004–36011. [Google Scholar] [CrossRef]
- Jabs, R.; Kirchhoff, F.; Kettenmann, H.; Steinhäuser, C. Kainate activates Ca2+-permeable glutamate receptors and blocks voltage-gated K+ currents in glial cells of mouse hippocampal slices. Pflügers Arch. Eur. J. Physiol. 1994, 426, 310–319. [Google Scholar] [CrossRef]
- Ge, W.P.; Yang, X.J.; Zhang, Z.; Wang, H.K.; Shen, W.; Deng, Q.D.; Duan, S. Long-term potentiation of neuron-glia synapses mediated by Ca 2+-permeable AMPA receptors. Science 2006, 312, 1533–1537. [Google Scholar] [CrossRef]
- Sahel, A.; Ortiz, F.C.; Kerninon, C.; Maldonado, P.P.; Angulo, M.C.; Nait-Oumesmar, B. Alteration of synaptic connectivity of oligodendrocyte precursor cells following demyelination. Front. Cell. Neurosci. 2015, 9, 1–12. [Google Scholar] [CrossRef]
- Matute, C.; Alberdi, E.; Domercq, M.; Sánchez-Gómez, M.V.; Pérez-Samartín, A.; Rodríguez-Antigüedad, A.; Pérez-Cerdá, F. Excitotoxic damage to white matter. Proc. J. Anat. 2007, 210, 693–702. [Google Scholar]
- Follett, P.L.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. NBQX attenuates excitotoxic injury in developing white matter. J. Neurosci. 2000, 20, 9235–9241. [Google Scholar] [CrossRef]
- Rosenberg, L.J.; Teng, Y.D.; Wrathall, J.R. 2,3-Dihydroxy-6-Nitro-7-Sulfamoyl-Benzo(f)Quinoxaline Reduces Glial Loss and Acute White Matter Pathology after Experimental Spinal Cord Contusion. J. Neurosci. 1999, 19, 464–475. [Google Scholar] [CrossRef]
- Chen, T.J.; Kula, B.; Nagy, B.; Barzan, R.; Gall, A.; Ehrlich, I.; Kukley, M. In Vivo Regulation of Oligodendrocyte Precursor Cell Proliferation and Differentiation by the AMPA-Receptor Subunit GluA2. Cell Rep. 2018, 25, 852–861.e7. [Google Scholar] [CrossRef]
- Hines, J.H.; Ravanelli, A.M.; Schwindt, R.; Scott, E.K.; Appel, B. Neuronal activity biases axon selection for myelination in vivo. Nat. Neurosci. 2015, 18, 683–689. [Google Scholar] [CrossRef]
- Etxeberria, A.; Mei, F.; Ullian, E.M.; Dao, D.Q.; Redmond, S.A.; Mayoral, S.R.; Hokanson, K.C.; Chan, J.R. Dynamic Modulation of Myelination in Response to Visual Stimuli Alters Optic Nerve Conduction Velocity. J. Neurosci. 2016, 36, 6937–6948. [Google Scholar] [CrossRef]
- Mensch, S.; Baraban, M.; Almeida, R.; Czopka, T.; Ausborn, J.; El Manira, A.; Lyons, D.A. Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat. Neurosci. 2015, 18, 628–630. [Google Scholar] [CrossRef]
- Káradóttir, R.; Cavelier, P.; Bergersen, L.H.; Attwell, D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005, 438, 1162–1166. [Google Scholar] [CrossRef]
- Lundgaard, I.; Luzhynskaya, A.; Stockley, J.H.; Wang, Z.; Evans, K.A.; Swire, M.; Volbracht, K.; Gautier, H.O.B.; Franklin, R.J.M.; Ffrench-Constant, C.; et al. Neuregulin and BDNF Induce a Switch to NMDA Receptor-Dependent Myelination by Oligodendrocytes. PLoS Biol. 2013, 11, e1001743. [Google Scholar] [CrossRef]
- De Biase, L.M.; Pucak, M.L.; Mishina, M.; Kang, S.H.; Calabresi, P.A.; Baxi, E.G.; Fukaya, M.; Bergles, D.E. NMDA Receptor Signaling in Oligodendrocyte Progenitors Is Not Required for Oligodendrogenesis and Myelination. J. Neurosci. 2011, 31, 12650–12662. [Google Scholar] [CrossRef]
- Kukley, M.; Capetillo-Zarate, E.; Dietrich, D. Vesicular glutamate release from axons in white matter. Nat. Neurosci. 2007, 10, 311–320. [Google Scholar] [CrossRef]
- Ziskin, J.L.; Nishiyama, A.; Rubio, M.; Fukaya, M.; Bergles, D.E. Vesicular release of glutamate from unmyelinated axons in white matter. Nat. Neurosci. 2007, 10, 321–330. [Google Scholar] [CrossRef]
- De Biase, L.M.; Nishiyama, A.; Bergles, D.E. Excitability and Synaptic Communication within the Oligodendrocyte Lineage. J. Neurosci. 2010, 30, 3600–3611. [Google Scholar] [CrossRef]
- Gallo, V.; Mangin, J.M.; Kukley, M.; Dietrich, D. Synapses on NG2-expressing progenitors in the brain: Multiple functions? J. Physiol. 2008, 586, 3767–3781. [Google Scholar] [CrossRef]
- Kukley, M.; Nishiyama, A.; Dietrich, D. The Fate of Synaptic Input to NG2 Glial Cells: Neurons Specifically Downregulate Transmitter Release onto Differentiating Oligodendroglial Cells. J. Neurosci. 2010, 30, 8320–8331. [Google Scholar] [CrossRef]
- Butt, A.M.; Hamilton, N.; Hubbard, P.; Pugh, M.; Ibrahim, M. Synantocytes: The fifth element. J. Anat. 2005, 207, 695–706. [Google Scholar] [CrossRef]
- Sakry, D.; Neitz, A.; Singh, J.; Frischknecht, R.; Marongiu, D.; Binamé, F.; Perera, S.S.; Endres, K.; Lutz, B.; Radyushkin, K.; et al. Oligodendrocyte Precursor Cells Modulate the Neuronal Network by Activity-Dependent Ectodomain Cleavage of Glial NG2. PLoS Biol. 2014, 12, e1001993. [Google Scholar] [CrossRef]
- Passlick, S.; Trotter, J.; Seifert, G.; Steinhäuser, C.; Jabs, R. The NG2 Protein Is Not Required for Glutamatergic Neuron-NG2 Cell Synaptic Signaling. Cereb. Cortex 2016, 26, 51–57. [Google Scholar] [CrossRef][Green Version]
- Micu, I.; Plemel, J.R.; Caprariello, A.V.; Nave, K.A.; Stys, P.K. Axo-myelinic neurotransmission: A novel mode of cell signalling in the central nervous system. Nat. Rev. Neurosci. 2018, 19, 49–57. [Google Scholar] [CrossRef]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef]
- McDonald, J.W.; Althomsons, S.P.; Hyrc, K.L.; Choi, D.W.; Goldberg, M.P. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat. Med. 1998, 4, 291–297. [Google Scholar] [CrossRef]
- Zonouzi, M.; Renzi, M.; Farrant, M.; Cull-Candy, S.G. Bidirectional plasticity of calcium-permeable AMPA receptors in oligodendrocyte lineage cells. Nat. Neurosci. 2011, 14, 1430–1438. [Google Scholar] [CrossRef]
- Back, S.A.; Rosenberg, P.A. Pathophysiology of Glia in Perinatal White Matter Injury. Glia 2014, 62, 1790–1815. [Google Scholar] [CrossRef]
- Khwaja, O.; Volpe, J.J. Pathogenesis of cerebral white matter injury of prematurity. Arch Dis Child Fetal Neonatal Ed. 2008, 93, F153–F161. [Google Scholar] [CrossRef]
- Volpe, J.J. Confusions in Nomenclature: “Periventricular Leukomalacia” and “White Matter Injury”—Identical, Distinct, or Overlapping? Pediatr. Neurol. 2017, 73, 3–6. [Google Scholar] [CrossRef]
- Back, S.A.; Riddle, A.; McClure, M.M. Maturation-dependent vulnerability of perinatal white matter in premature birth. Stroke 2007, 38, 724–730. [Google Scholar] [CrossRef]
- Buser, J.R.; Segovia, K.N.; Dean, J.M.; Nelson, K.; Beardsley, D.; Gong, X.; Luo, N.L.; Ren, J.; Wan, Y.; Riddle, A.; et al. Timing of appearance of late oligodendrocyte progenitors coincides with enhanced susceptibility of preterm rabbit cerebral white matter to hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 1053–1065. [Google Scholar] [CrossRef]
- Riddle, A.; Luo, N.L.; Manese, M.; Beardsley, D.; Green, L.; Rorvik, D.A.; Kelly, K.A.; Barlow, C.H.; Kelly, J.J.; Hohimer, A.R.; et al. Spatial Heterogeneity in Oligodendrocyte Lineage Maturation and Not Cerebral Blood Flow Predicts Fetal Ovine Periventricular White Matter Injury. J. Neurosci. 2006, 26, 3045–3055. [Google Scholar] [CrossRef]
- Segovia, K.N.; Mcclure, M.; Moravec, M.; Luo, N.L.; Wan, Y.; Gong, X.; Riddle, A.; Craig, A.; Struve, J.; Sherman, L.S.; et al. Arrested Oligodendrocyte Lineage Maturation in Chronic Perinatal White Matter Injury. Ann. Neurol. 2008, 63, 520–530. [Google Scholar] [CrossRef]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Volpe, J.J.; Kinney, H.C. Arrested oligodendrocyte lineage progression during human cerebral white matter development: dissociation between the timing of progenitor differentiation and myelinogenesis. J. Neuropathol. Exp. Neurol. 2002, 61, 197–211. [Google Scholar] [CrossRef]
- Talos, D.M.; Follett, P.L.; Folkerth, R.D.; Fishman, R.E.; Trachtenberg, F.L.; Volpe, J.J.; Jensen, F.E. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. II. Human cerebral white matter and cortex. J. Comp. Neurol. 2006, 497, 61–77. [Google Scholar] [CrossRef]
- Page, K.J.; Everitt, B.J. The Distribution of Neurons Coexpressing Immunoreactivity to AMPA-sensitive Glutamate Receptor Subtypes (GluR1-4) and Nerve Growth Factor Receptor in the Rat Basal Forebrain. Eur. J. Neurosci. 1995, 7, 1022–1033. [Google Scholar] [CrossRef]
- Deng, Y.; Lu, J.; Sivakumar, V.; Ling, E.A.; Kaur, C. Amoeboid microglia in the periventricular white matter induce oligodendrocyte damage through expression of proinflammatory cytokines via MAP kinase signaling pathway in hypoxic neonatal rats. Brain Pathol. 2008, 18, 387–400. [Google Scholar] [CrossRef]
- Su, Z.; Yuan, Y.; Chen, J.; Zhu, Y.; Qiu, Y.; Zhu, F.; Huang, A.; He, C. Reactive astrocytes inhibit the survival and differentiation of oligodendrocyte precursor cells by secreted TNF-α. J. Neurotrauma 2011, 28, 1089–1100. [Google Scholar] [CrossRef]
- Rathnasamy, G.; Ling, E.; Kaur, C. Iron and Iron Regulatory Proteins in Amoeboid Microglial Cells Are Linked to Oligodendrocyte Death in Hypoxic Neonatal Rat Periventricular White Matter through Production of Proinflammatory Cytokines and Reactive Oxygen/Nitrogen Species. J. Neurosci. 2011, 31, 17982–17995. [Google Scholar] [CrossRef]
- Villapol, S.; Fau, S.; Renolleau, S.; Biran, V.; Charriaut-Marlangue, C.; Baud, O. Melatonin promotes myelination by decreasing white matter inflammation after neonatal stroke. Pediatr. Res. 2011, 69, 51–55. [Google Scholar] [CrossRef]
- Wada, T.; Sawano, T.; Tanaka, T.; Furuyama, T.; Fukumoto, M.; Yamaguchi, W.; Saino, O.; Takeda, Y.; Kogo, M.; Matsuyama, T.; et al. Absence of Sema4D improves oligodendrocyte recovery after cerebral ischemia/reperfusion injury in mice. Neurosci. Res. 2016, 108, 6–11. [Google Scholar] [CrossRef]
- Bonfanti, E.; Gelosa, P.; Fumagalli, M.; Dimou, L.; Viganò, F.; Tremoli, E.; Cimino, M.; Sironi, L.; Abbracchio, M.P. The role of oligodendrocyte precursor cells expressing the GPR17 receptor in brain remodeling after stroke. Cell Death Dis. 2017, 8, e2871. [Google Scholar] [CrossRef]
- Rebai, O.; Amri, M. Chlorogenic Acid Prevents AMPA-Mediated Excitotoxicity in Optic Nerve Oligodendrocytes Through a PKC and Caspase-Dependent Pathways. Neurotox. Res. 2018, 34, 559–573. [Google Scholar] [CrossRef]
- McCracken, E.; Fowler, J.H.; Dewar, D.; Morrison, S.; McCulloch, J. Grey matter and white matter ischemic damage is reduced by the competitive AMPA receptor antagonist, SPD 502. J. Cereb. Blood Flow Metab. 2002, 22, 1090–1097. [Google Scholar] [CrossRef]
- Viganò, F.; Möbius, W.; Götz, M.; Dimou, L. Transplantation reveals regional differences in oligodendrocyte differentiation in the adult brain. Nat. Neurosci. 2013, 16, 1370–1372. [Google Scholar] [CrossRef]
- Lentferink, D.H.; Jongsma, J.M.; Werkman, I.; Baron, W. Grey matter OPCs are less mature and less sensitive to IFNγ than white matter OPCs: Consequences for remyelination. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef]
- Newcombe, J.; Uddin, A.; Dove, R.; Patel, B.; Turski, L.; Nishizawa, Y.; Smith, T. Glutamate receptor expression in multiple sclerosis lesions. Brain Pathol. 2008, 18, 52–61. [Google Scholar] [CrossRef]
- Bannerman, P.; Horiuchi, M.; Feldman, D.; Hahn, A.; Itoh, A.; See, J.; Jia, Z.P.; Itoh, T.; Pleasure, D. GluR2-free alfa-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors intensify demyelination in experimental autoimmune encephalomyelitis. J. Neurochem. 2007, 102, 1064–1070. [Google Scholar] [CrossRef]
- Docagne, F.; Muñetón, V.; Clemente, D.; Ali, C.; Loría, F.; Correa, F.; Hernangómez, M.; Mestre, L.; Vivien, D.; Guaza, C. Excitotoxicity in a chronic model of multiple sclerosis: Neuroprotective effects of cannabinoids through CB1 and CB2 receptor activation. Mol. Cell. Neurosci. 2007, 34, 551–561. [Google Scholar] [CrossRef]
- Nakajima, M.; Suda, S.; Sowa, K.; Sakamoto, Y.; Nito, C.; Nishiyama, Y.; Aoki, J.; Ueda, M.; Yokobori, S.; Yamada, M.; et al. AMPA Receptor Antagonist Perampanel Ameliorates Post-Stroke Functional and Cognitive Impairments. Neuroscience 2018, 386, 256–264. [Google Scholar] [CrossRef]
- Elting, J.-W.; Diener, H.C.; Teelken, A.W.; Lees, K.R.; Sulter, G.A.; Hommel, M.; Kaste, M.; Versavel, M.; De Keyser, J. AMPA Antagonist ZK200775 in Patients With Acute Ischemic Stroke. Stroke 2002, 33, 2813–2818. [Google Scholar] [CrossRef]
- Walters, M.R.; Kaste, M.; Lees, K.R.; Diener, H.C.; Hommel, M.; De Keyser, J.; Steiner, H.; Versavel, M. The AMPA antagonist ZK 200775 in patients with acute ischaemic stroke: A double-blind, multicentre, placebo-controlled safety and tolerability study. Cerebrovasc. Dis. 2005, 20, 304–309. [Google Scholar] [CrossRef]
- Gibson, E.M.; Barres, B.A.; Bieri, G.; Lin, G.L.; Woo, P.J.; Wood, L.S.; Vogel, H.; Zuchero, J.B.; Mount, C.W.; Purger, D.; et al. Neuronal Activity Promotes Oligodendrogenesis and Adaptive Myelination in the Mammalian Brain. Science 2014, 344, 1252304. [Google Scholar] [CrossRef]
- Barres, B.A.; Hart, I.K.; Coles, H.S.R.; Burne, J.F.; Voyvodic, J.T.; Richardson, W.D.; Raff, M.C. Cell death and control of cell survival in the oligodendrocyte lineage. Cell 1992, 70, 31–46. [Google Scholar] [CrossRef]
- Wake, H.; Lee, P.R.; Fields, R.D. Control of local protein synthesis and initial events in myelination by action potentials. Science 2011, 333, 1647–1651. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Gan, W.B. Microglia dynamics and function in the CNS. Curr. Opin. Neurobiol. 2010, 20, 595–600. [Google Scholar] [CrossRef]
- Yang, I.; Han, S.J.; Kaur, G.; Crane, C.; Parsa, A.T. The role of microglia in central nervous system immunity and glioma immunology. J. Clin. Neurosci. 2010, 17, 6–10. [Google Scholar] [CrossRef]
- Miron, V.E.; Franklin, R.J.M. Macrophages and CNS remyelination. J. Neurochem. 2014, 130, 165–171. [Google Scholar] [CrossRef]
- Jin, X.; Yamashita, T. Microglia in central nervous system repair after injury. J. Biochem. 2016, 159, 491–496. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist. Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Noda, M.; Nakanishi, H.; Nabekura, J.; Akaike, N. AMPA-kainate subtypes of glutamate receptor in rat cerebral microglia. J. Neurosci. 2000, 20, 251. [Google Scholar] [CrossRef]
- Hagino, Y.; Kariura, Y.; Manago, Y.; Amano, T.; Wang, B.; Sekiguchi, M.; Nishikawa, K.; Aoki, S.; Wada, K.; Noda, M. Heterogeneity and potentiation of AMPA type of glutamate receptors in rat cultured microglia. Glia 2004, 47, 68–77. [Google Scholar] [CrossRef]
- Christensen, R.N.; Ha, B.K.; Sun, F.; Bresnahan, J.C.; Beattie, M.S. Kainate induces rapid redistribution of the actin cytoskeleton in ameboid microglia. J. Neurosci. Res. 2006, 84, 170–181. [Google Scholar] [CrossRef]
- Sivakumar, V.; Ling, E.A.; Lu, J.; Kaur, C. Role of glutamate and its receptors and insulin-like growth factors in hypoxia induced periventricular white matter injury. Glia 2010, 58, 507–523. [Google Scholar] [CrossRef]
- Wong, W.T.; Wang, M.; Li, W. Regulation of microglia by ionotropic glutamatergic and GABAergic neurotransmission. Neuron Glia Biol. 2012, 7, 41–46. [Google Scholar] [CrossRef]
- Gottlieb, M.; Matute, C. Expression of ionotropic glutamate receptor subunits in glial cells of the hippocampal CA1 area following transient forebrain ischemia. J. Cereb. Blood Flow Metab. 1997, 17, 290–300. [Google Scholar] [CrossRef]
- Schilling, T.; Eder, C. Ion channel expression in resting and activated microglia of hippocampal slices from juvenile mice. Brain Res. 2007, 1186, 21–28. [Google Scholar] [CrossRef]
- Guan, J.; Bennet, L.; Gluckman, P.D.; Gunn, A.J. Insulin-like growth factor-1 and post-ischemic brain injury. Prog. Neurobiol. 2003, 70, 443–462. [Google Scholar] [CrossRef]
- Takeuchi, H.; Suzumura, A. Gap junctions and hemichannels composed of connexins: Potential therapeutic targets for neurodegenerative diseases. Front. Cell. Neurosci. 2014, 8, 189. [Google Scholar] [CrossRef]
- Leonoudakis, D.; Braithwaite, S.P.; Beattie, M.S.; Beattie, E.C.; Leonoudakis, D.D. TNFα-induced AMPA-receptor trafficking in CNS neurons; relevance to excitotoxicity? Neuron Glia Biol. 2004, 1, 263–273. [Google Scholar] [CrossRef]
- Zhu, W.; Zheng, H.; Li, Z.; Wang, W.; Shao, X.; Yao, Q. Excitotoxicity of TNFα derived from KA activated microglia on hippocampal neurons in vitro and in vivo. J. Neurochem. 2010, 114, 386–396. [Google Scholar] [CrossRef]
- Mandolesi, G.; De Vito, F.; Fresegna, D.; Gentile, A.; Marfia, G.A.; Bullitta, S.; Sepman, H.; Musella, A.; Centonze, D. Synaptopathy connects inflammation and neurodegeneration in multiple sclerosis. Nat. Rev. Neurol. 2015, 11, 711–724. [Google Scholar] [CrossRef]
- Centonze, D.; Ecconi, F.; Battistini, L.; Bernardi, G.; Bergamaschi, A.; Furlan, R.; Muzio, L.; Cencioni, M.T.; Cavallucci, V.; De Chiara, V.; et al. Inflammation Triggers Synaptic Alteration and Degeneration in Experimental Autoimmune Encephalomyelitis. J. Neurosci. 2009, 29, 3442–3452. [Google Scholar] [CrossRef]
- Rizzo, F.R.; Musella, A.; De Vito, F.; Fresegna, D.; Bullitta, S.; Vanni, V.; Guadalupi, L.; Stampanoni Bassi, M.; Buttari, F.; Mandolesi, G.; et al. Tumor Necrosis Factor and Interleukin-1β Modulate Synaptic Plasticity during Neuroinflammation. Neural Plast. 2018, 2018, 8430123. [Google Scholar] [CrossRef]
- Beppu, K.; Kosai, Y.; Kido, M.A.; Akimoto, N.; Mori, Y.; Kojima, Y.; Fujita, K.; Okuno, Y.; Yamakawa, Y.; Ifuku, M.; et al. Expression, subunit composition, and function of AMPA-type glutamate receptors are changed in activated microglia; possible contribution of GluA2 (GluR-B)-deficiency under pathological conditions. Glia 2013, 61, 881–891. [Google Scholar] [CrossRef]
- Carter, T.L.; Rissman, R.A.; Mishizen-Eberz, A.J.; Wolfe, B.B.; Hamilton, R.L.; Gandy, S.; Armstrong, D.M. Differential preservation of AMPA receptor subunits in the hippocampi of Alzheimer’s disease patients according to Braak stage. Exp. Neurol. 2004, 187, 299–309. [Google Scholar] [CrossRef]
- Noda, M.; Beppu, K. Possible Contribution of Microglial Glutamate Receptors to Inflammatory Response upon Neurodegenerative Diseases. J. Neurol. Disord. 2013, 1. [Google Scholar] [CrossRef]
- Jansson, L.C.; Wigren, H.K.; Nordström, T.; Åkerman, K.E. Functional α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors in differentiating embryonic neural progenitor cells. Neuroreport 2011, 22, 282–287. [Google Scholar] [CrossRef]
- Muth-Köhne, E.; Pachernegg, S.; Karus, M.; Faissner, A.; Hollmann, M. Expression of NMDA receptors and Ca-impermeable AMPA receptors requires neuronal differentiation and allows discrimination between two different types of neural stem cells. Cell. Physiol. Biochem. 2010, 26, 935–946. [Google Scholar] [CrossRef]
- Shtaya, A.; Sadek, A.R.; Zaben, M.; Seifert, G.; Pringle, A.; Steinhäuser, C.; Gray, W.P. AMPA receptors and seizures mediate hippocampal radial glia-like stem cell proliferation. Glia 2018, 66, 2397–2413. [Google Scholar] [CrossRef]
- Kawakami, S.I. Glial and neuronal localization of ionotropic glutamate receptor subunit-immunoreactivities in the median eminence of female rats: GluR2/3 and GluR6/7 colocalize with vimentin, not with glial fibrillary acidic protein (GFAP). Brain Res. 2000, 858, 198–204. [Google Scholar] [CrossRef]
- Demêmes, D.; Lleixa, A.; Dechesne, C.J. Cellular and subcellular localization of AMPA-selective glutamate receptors in the mammalian peripheral vestibular system. Brain Res. 1995, 671, 83–94. [Google Scholar] [CrossRef]
- Chen, T.-J.; Fröhlich, N.; Kula, B.; Barzan, R.; Kukley, M. Glutamate Activates AMPA Receptor Conductance in the Developing Schwann Cells of the Mammalian Peripheral Nerves. J. Neurosci. 2017, 37, 11818–11834. [Google Scholar] [CrossRef]
- Fink, T.; Davey, D.F.; Ansselin, A.D. Glutaminergic and adrenergic receptors expressed on adult guinea pig Schwann cells in vitro. Can. J. Physiol. Pharmacol. 1999, 77, 204–210. [Google Scholar] [CrossRef]
- Liu, G.J.; Bennett, M.R. ATP secretion from nerve trunks and Schwann cells mediated by glutamate. Neuroreport 2006, 14, 2079. [Google Scholar] [CrossRef]
- Kung, L.H.; Gong, K.; Adedoyin, M.; Ng, J.; Bhargava, A.; Ohara, P.T.; Jasmin, L. Evidence for Glutamate as a Neuroglial Transmitter within Sensory Ganglia. PLoS ONE 2013, 8, e68312. [Google Scholar] [CrossRef]
- Fernández-Montoya, J.; Avendaño, C.; Negredo, P. The glutamatergic system in primary somatosensory neurons and its involvement in sensory input-dependent plasticity. Int. J. Mol. Sci. 2018, 19, 69. [Google Scholar] [CrossRef]
- Von Boyen, G.; Steinkamp, M.; Adler, G.; Kirsch, J. Glutamate receptor subunit expression in primary enteric glia cultures. J. Recept. Signal Transduct. 2006, 26, 329–336. [Google Scholar] [CrossRef]
- Goffredo, D.; Conti, L.; Di Febo, F.; Biella, G.; Tosoni, A.; Vago, G.; Biunno, I.; Moiana, A.; Bolognini, D.; Toselli, M.; et al. Setting the conditions for efficient, robust and reproducible generation of functionally active neurons from adult subventricular zone-derived neural stem cells. Cell Death Differ. 2008, 15, 1847–1856. [Google Scholar] [CrossRef]
- Buffo, A.; Rossi, F. Origin, lineage and function of cerebellar glia. Prog. Neurobiol. 2013, 109, 1–22. [Google Scholar] [CrossRef]
- Ko, C.; Robitaille, R. Perisynaptic Schwann Cells at the Neuromuscular Synapse: Adaptable, Multitasking Glial Cells. Cold Spring Harb. Perspect. Biol. 2015, 7, a020503. [Google Scholar] [CrossRef]
- Jansson, L.C.; Louhivuori, L.; Wigren, H.K.; Nordström, T.; Louhivuori, V.; Castrén, M.L.; Åkerman, K.E. Effect of glutamate receptor antagonists on migrating neural progenitor cells. Eur. J. Neurosci. 2013, 37, 1369–1382. [Google Scholar] [CrossRef]
- Herrera, A.A.; Qiang, H.; Ko, C.P. The role of perisynaptic Schwann cells in development of neuromuscular junctions in the frog (Xenopus laevis). J. Neurobiol. 2000, 45, 237–254. [Google Scholar] [CrossRef]
- Todd, K.J.; Darabid, H.; Robitaille, R. Perisynaptic Glia Discriminate Patterns of Motor Nerve Activity and Influence Plasticity at the Neuromuscular Junction. J. Neurosci. 2010, 30, 11870–11882. [Google Scholar] [CrossRef]
- Campana, W.M.; Mantuano, E.; Azmoon, P.; Henry, K.; Banki, M.A.; Kim, J.H.; Pizzo, D.P.; Gonias, S.L. Ionotropic glutamate receptors activate cell signaling in response to glutamate in Schwann cells. FASEB J. 2017, 31, 1744–1755. [Google Scholar] [CrossRef]
- Mantuano, E.; Lam, M.S.; Shibayama, M.; Campana, W.M.; Gonias, S.L. The NMDA receptor functions independently and as an LRP1 co-receptor to promote Schwann cell survival and migration. J. Cell Sci. 2015, 128, 3478–3488. [Google Scholar] [CrossRef]
- Saitoh, F.; Wakatsuki, S.; Tokunaga, S.; Fujieda, H.; Araki, T. Glutamate signals through mGluR2 to control Schwann cell differentiation and proliferation. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Christensen, P.C.; Welch, N.C.; Brideau, C.; Stys, P.K. Functional ionotropic glutamate receptors on peripheral axons and myelin. Muscle&Nerve 2016, 54, 451–459. [Google Scholar]
- Rebillard, G.; Ruel, J.; Nouvian, R.; Saleh, H.; Pujol, R.; Dehnes, Y.; Raymond, J.; Puel, J.L.; Devau, G. Glutamate transporters in the guinea-pig cochlea: Partial mRNA sequences, cellular expression and functional implications. Eur. J. Neurosci. 2003, 17, 83–92. [Google Scholar] [CrossRef]
- Tachibana, M.; Wenthold, R.J.; Morioka, H.; Petralia, R.S. Light and electron microscopic immunocytochemical localization of AMPA-selective glutamate receptors in the rat spinal cord. J. Comp. Neurol. 1994, 344, 431–454. [Google Scholar] [CrossRef]
- Ferrari, L.F.; Lotufo, C.M.; Araldi, D.; Rodrigues, M.A.; Macedo, L.P.; Ferreira, S.H.; Parada, C.A. Inflammatory sensitization of nociceptors depends on activation of NMDA receptors in DRG satellite cells. Proc. Natl. Acad. Sci. USA 2014, 111, 18363–18368. [Google Scholar] [CrossRef]
- Grubišić, V.; Gulbransen, B.D. Enteric glia: The most alimentary of all glia. J. Physiol. 2017, 595, 557–570. [Google Scholar] [CrossRef]
- Rao, M.; Nelms, B.D.; Dong, L.; Salinas-Rios, V.; Rutlin, M.; Gershon, M.D.; Corfas, G. Enteric glia express Proteolipid Protein 1 and are a transcriptionally unique population of glia in the mammalian nervous system. Glia 2015, 63, 2040–2057. [Google Scholar] [CrossRef]
- Carpanese, E.; Moretto, P.; Filpa, V.; Marchet, S.; Moro, E.; Crema, F.; Frigo, G.; Giaroni, C. Antagonism of ionotropic glutamate receptors attenuates chemical ischemia-induced injury in rat primary cultured myenteric ganglia. PLoS ONE 2014, 9, 1–17. [Google Scholar] [CrossRef]
- Kirchgessner, A.L.; Liu, M.-T.; Alcantara, F. Excitotoxicity in the Enteric Nervous System. J. Neurosci. 1997, 17, 8804–8816. [Google Scholar] [CrossRef] [PubMed]
- Shouman, B.; Fontaine, R.H.; Baud, O.; Schwendimann, L.; Keller, M.; Spedding, M.; Lelièvre, V.; Gressens, P. Endocannabinoids potently protect the newborn brain against AMPA-kainate receptor-mediated excitotoxic damage. Br. J. Pharmacol. 2006, 148, 442–451. [Google Scholar] [CrossRef]
- Gonzalez-Islas, C.; Garcia-Bereguiain, M.A.; Wenner, P. Tonic and Transient Endocannabinoid Regulation of AMPAergic Miniature Postsynaptic Currents and Homeostatic Plasticity in Embryonic Motor Networks. J. Neurosci. 2012, 32, 13597–13607. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Colizzi, M.; McGuire, P.; Pertwee, R.G.; Bhattacharyya, S. Effect of cannabis on glutamate signalling in the brain: A systematic review of human and animal evidence. Neurosci. Biobehav. Rev. 2016, 64, 359–381. [Google Scholar] [CrossRef]
- Ligresti, A.; De Petrocellis, L.; Di Marzo, V. From Phytocannabinoids to Cannabinoid Receptors and Endocannabinoids: Pleiotropic Physiological and Pathological Roles Through Complex Pharmacology. Physiol. Rev. 2016, 96, 1593–1659. [Google Scholar] [CrossRef]
- Gaoni, Y.; Mechoulam, R. Isolation, structure and partial synthesis of an active constituent of hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Hillard, C.J. Circulating Endocannabinoids: From Whence Do They Come and Where are They Going? Nature Publishing Group: London, UK, 2018; Volume 43, ISBN 4149558493. [Google Scholar]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-gómez, E.; Matias, I.; Delamarre, A.; Metna-laurent, M.; Cannich, A.; et al. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Shosaku, T.; Kano, M. Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr. Opin. Neurobiol. 2014, 29, 1–898. [Google Scholar] [CrossRef]
- Augustin, S.M.; Lovinger, D.M. Functional Relevance of Endocannabinoid-Dependent Synaptic Plasticity in the Central Nervous System. ACS Chem. Neurosci. 2018, 9, 2146–2161. [Google Scholar] [CrossRef]
- Hashimotodani, Y.; Ohno-Shosaku, T.; Tanimura, A.; Kita, Y.; Sano, Y.; Shimizu, T.; Di Marzo, V.; Kano, M. Acute inhibition of diacylglycerol lipase blocks endocannabinoid-mediated retrograde signalling: Evidence for on-demand biosynthesis of 2-arachidonoylglycerol. J. Physiol. 2013, 591, 4765–4776. [Google Scholar] [CrossRef]
- Ilyasov, A.A.; Milligan, C.E.; Pharr, E.P.; Howlett, A.C. The Endocannabinoid System and Oligodendrocytes in Health and Disease. Front. Neurosci. 2018, 12, 1–10. [Google Scholar] [CrossRef]
- Molina-Holgado, E.; Vela, J.M.; Arévalo-Martín, A.; Almazán, G.; Molina-holgado, F.; Borrel, J.; Guaza, C. Cannabinoids Promote Oligodendrocyte Progenitor Survival: Involvement of Cannabinoid Receptors and Phosphatidylinositol-3 Kinase/Akt Signaling. J. Neurosci. 2002, 22, 9742–9753. [Google Scholar] [CrossRef]
- Gomez, O.; Arevalo-Martin, A.; Garcia-Ovejero, D.; Ortega-Gutierrez, S.; Cisneros, J.A.; Almazan, G.; Sánnchez-Rodriguez, M.A.; Molina-Holgado, F.; Molina-Holgado, E. The constitutive production of the endocannabinoid 2-arachidonoylglycerol participates in oligodendrocyte differentiation. Glia 2010, 58, 1913–1927. [Google Scholar] [CrossRef]
- Gomez, O.; Sanchez-Rodriguez, M.A.; Ortega-Gutierrez, S.; Vazquez-Villa, H.; Guaza, C.; Molina-Holgado, F.; Molina-Holgado, E. A Basal Tone of 2-Arachidonoylglycerol Contributes to Early Oligodendrocyte Progenitor Proliferation by Activating Phosphatidylinositol 3-Kinase (PI3K)/AKT and the Mammalian Target of Rapamycin (MTOR) Pathways. J. Neuroimmune Pharmacol. 2015, 10, 309–317. [Google Scholar] [CrossRef]
- Tomas-Roig, J.; Wirths, O.; Salinas-Riester, G.; Havemann-Reinecke, U. The Cannabinoid CB1/CB2 Agonist WIN55212.2 Promotes Oligodendrocyte Differentiation In Vitro and Neuroprotection During the Cuprizone-Induced Central Nervous System Demyelination. CNS Neurosci. Ther. 2016, 22, 387–395. [Google Scholar] [CrossRef]
- Gomez, O.; Sanchez-Rodriguez, A.; Le, M.Q.U.; Sanchez-Caro, C.; Molina-Holgado, F.; Molina-Holgado, E. Cannabinoid receptor agonists modulate oligodendrocyte differentiation by activating PI3K/Akt and the mammalian target of rapamycin (mTOR) pathways. Br. J. Pharmacol. 2011, 163, 1520–1532. [Google Scholar] [CrossRef]
- Sanchez-Rodriguez, M.A.; Gomez, O.; Esteban, P.F.; Garcia-Ovejero, D.; Molina-Holgado, E. The endocannabinoid 2-arachidonoylglycerol regulates oligodendrocyte progenitor cell migration. Biochem. Pharmacol. 2018, 157, 180–188. [Google Scholar] [CrossRef]
- Arevalo-Martin, A.; Garcia-Ovejero, D.; Rubio-Araiz, A.; Gomez, O.; Molina-Holgado, F.; Molina-Holgado, E. Cannabinoids modulate Olig2 and polysialylated neural cell adhesion molecule expression in the subventricular zone of post-natal rats through cannabinoid receptor 1 and cannabinoid receptor 2. Eur. J. Neurosci. 2007, 26, 1548–1559. [Google Scholar] [CrossRef]
- Giacoppo, S.; Bramanti, P.; Mazzon, E. Sativex in the management of multiple sclerosis-related spasticity: An overview of the last decade of clinical evaluation. Mult. Scler. Relat. Disord. 2017, 17, 22–31. [Google Scholar] [CrossRef]
- Russo, M.; Naro, A.; Leo, A.; Sessa, E.; D’Aleo, G.; Bramanti, P.; Calabrò, R.S. Evaluating sativex® in neuropathic pain management: A clinical and neurophysiological assessment in multiple sclerosis. Pain Med. (United States) 2016, 17, 1145–1154. [Google Scholar] [CrossRef]
- Feliú, A.; Moreno-Martet, M.; Mecha, M.; Carrillo-Salinas, F.J.; De Lago, E.; Fernández-Ruiz, J.; Guaza, C. A Sativex®-like combination of phytocannabinoids as a disease-modifying therapy in a viral model of multiple sclerosis. Br. J. Pharmacol. 2015, 172, 3579–3595. [Google Scholar] [CrossRef]
- Giacoppo, S.; Rajan, T.S.; Galuppo, M.; Pollastro, F.; Grassi, G.; Bramanti, P.; Mazzon, E. Purified Cannabidiol, the main non-psychotropic component of Cannabis sativa, alone, counteracts neuronal apoptosis in experimental multiple sclerosis. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4906–4919. [Google Scholar]
- Mecha, M.; Feliú, A.; Iñigo, P.M.; Mestre, L.; Carrillo-Salinas, F.J.; Guaza, C. Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: A role for A2A receptors. Neurobiol. Dis. 2013, 59, 141–150. [Google Scholar] [CrossRef]
- Bernal-Chico, A.; Canedo, M.; Manterola, A.; Victoria Sánchez-Gómez, M.; Pérez-Samartín, A.; Rodríguez-Puertas, R.; Matute, C.; Mato, S. Blockade of monoacylglycerol lipase inhibits oligodendrocyte excitotoxicity and prevents demyelination in vivo. Glia 2015, 63, 163–176. [Google Scholar] [CrossRef]
- Manterola, A.; Bernal-Chico, A.; Cipriani, R.; Canedo-Antelo, M.; Moreno-García, Á.; Martín-Fontecha, M.; Pérez-Cerdá, F.; Sánchez-Gómez, M.V.; Ortega-Gutiérrez, S.; Brown, J.M.; et al. Deregulation of the endocannabinoid system and therapeutic potential of ABHD6 blockade in the cuprizone model of demyelination. Biochem. Pharmacol. 2018, 157, 189–201. [Google Scholar] [CrossRef]
- Mato, S.; Alberdi, E.; Ledent, C.; Watanabe, M.; Matute, C. CB1 cannabinoid receptor-dependent and -independent inhibition of depolarization-induced calcium influx in oligodendrocytes. Glia 2009, 57, 295–306. [Google Scholar] [CrossRef]
- Lozovaya, N.; Min, R.; Tsintsadze, V.; Burnashev, N. Dual modulation of CNS voltage-gated calcium channels by cannabinoids: Focus on CB1 receptor-independent effects. Cell Calcium 2009, 46, 154–162. [Google Scholar] [CrossRef]
- Mato, S.; Victoria Sánchez-Gómez, M.; Matute, C. Cannabidiol induces intracellular calcium elevation and cytotoxicity in oligodendrocytes. Glia 2010, 58, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Mecha, M.; Torrao, A.S.; Mestre, L.; Carrillo-Salinas, F.J.; Mechoulam, R.; Guaza, C. Cannabidiol protects oligodendrocyte progenitor cells from inflammation-induced apoptosis by attenuating endoplasmic reticulum stress. Cell Death Dis. 2012, 3, e331. [Google Scholar] [CrossRef] [PubMed]
- Pazos, M.R.; Mohammed, N.; Lafuente, H.; Santos, M.; Martínez-Pinilla, E.; Moreno, E.; Valdizan, E.; Romero, J.; Pazos, A.; Franco, R.; et al. Mechanisms of cannabidiol neuroprotection in hypoxic-ischemic newborn pigs: Role of 5HT1A and CB2 receptors. Neuropharmacology 2013, 71, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Khaksar, S.; Bigdeli, M.R. Anti-excitotoxic effects of cannabidiol are partly mediated by enhancement of NCX2 and NCX3 expression in animal model of cerebral ischemia. Eur. J. Pharmacol. 2016, 794, 270–279. [Google Scholar] [CrossRef]
- El-Remessy, A.B.; Khalil, I.E.; Matragoon, S.; Abou-Mohamed, G.; Tsai, N.J.; Roon, P.; Caldwell, R.B.; Caldwell, R.W.; Green, K.; Liou, G.I. Neuroprotective Effect of(-)Δ9-Tetrahydrocannabinol and Cannabidiol in N-Methyl-D-Aspartate-Induced Retinal Neurotoxicity: Involvement of Peroxynitrite. Am. J. Pathol. 2003, 163, 1997–2008. [Google Scholar] [CrossRef]
- Loría, F.; Petrosino, S.; Hernangómez, M.; Mestre, L.; Spagnolo, A.; Correa, F.; Di Marzo, V.; Docagne, F.; Guaza, C. An endocannabinoid tone limits excitotoxicity in vitro and in a model of multiple sclerosis. Neurobiol. Dis. 2010, 37, 166–176. [Google Scholar] [CrossRef]
- De Lago, E.; Moreno-Martet, M.; Cabranes, A.; Ramos, J.A.; Fernández-Ruiz, J. Cannabinoids ameliorate disease progression in a model of multiple sclerosis in mice, acting preferentially through CB1 receptor-mediated anti-inflammatory effects. Neuropharmacology 2012, 62, 2299–2308. [Google Scholar] [CrossRef]
- Cabranes, A.; Venderova, K.; De Lago, E.; Fezza, F.; Sánchez, A.; Mestre, L.; Valenti, M.; García-Merino, A.; Ramos, J.A.; Di Marzo, V.; et al. Decreased endocannabinoid levels in the brain and beneficial effects of agents activating cannabinoid and/or vanilloid receptors in a rat model of multiple sclerosis. Neurobiol. Dis. 2005, 20, 207–217. [Google Scholar] [CrossRef]
- Good, C.H.; Lupica, C.R. Afferent-specific AMPA receptor subunit composition and regulation of synaptic plasticity in midbrain dopamine neurons by abused drugs. J. Neurosci. 2010, 30, 7900–7909. [Google Scholar] [CrossRef]
- Luján, M.Á.; Castro-Zavala, A.; Alegre-Zurano, L.; Valverde, O. Repeated Cannabidiol treatment reduces cocaine intake and modulates neural proliferation and CB1R expression in the mouse hippocampus. Neuropharmacology 2018, 143, 163–175. [Google Scholar] [CrossRef]
- Pérez-Cadahía, B.; Drobic, B.; Davie, J.R. Activation and function of immediate-early genes in the nervous system. Biochem. Cell Biol. 2011, 89, 61–73. [Google Scholar] [CrossRef]
- Minatohara, K.; Akiyoshi, M.; Okuno, H. Role of Immediate-Early Genes in Synaptic Plasticity and Neuronal Ensembles Underlying the Memory Trace. Front. Mol. Neurosci. 2016, 8, 78. [Google Scholar] [CrossRef]
- Hasel, P.; Dando, O.; Jiwaji, Z.; Baxter, P.; Todd, A.C.; Heron, S.; Márkus, N.M.; McQueen, J.; Hampton, D.W.; Torvell, M.; et al. Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat. Commun. 2017, 8, 15132. [Google Scholar] [CrossRef]
- Molnar, G.; Crozat, A.; Pardee, A.B. The immediate-early gene Egr-1 regulates the activity of the thymidine kinase promoter at the G0-to-G1 transition of the cell cycle. Mol. Cell. Biol. 2015, 14, 5242–5248. [Google Scholar] [CrossRef]
- Sukhatme, V.P.; Cao, X.; Chang, L.C.; Tsai-Morris, C.H.; Stamenkovich, D.; Ferreira, P.C.P.; Cohen, D.R.; Edwards, S.A.; Shows, T.B.; Curran, T.; et al. A zinc finger-encoding gene coregulated with c-fos during growth and differentiation, and after cellular depolarization. Cell 1988, 53, 37–43. [Google Scholar] [CrossRef]
- Sanchez-Gomez, M.V.; Perez-Navarro, E.; Alberdi, E.; Alberch, J.; Matute, C. Bax and Calpain Mediate Excitotoxic Oligodendrocyte Death Induced by Activation of Both AMPA and Kainate Receptors. J. Neurosci. 2011, 31, 2996–3006. [Google Scholar] [CrossRef]
- Kumar, S.; Cakouros, D. Transcriptional control of the core cell-death machinery. Trends Biochem. Sci. 2004, 29, 193–199. [Google Scholar] [CrossRef]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Benatti, P.; Basile, V.; Merico, D.; Fantoni, L.I.; Tagliafico, E.; Imbriano, C. A balance between NF-Y and p53 governs the pro- and anti-apoptotic transcriptional response. Nucleic Acids Res. 2008, 36, 1415–1428. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Region | GluA Expression | Ca2+ Permeability | Functions | ||
|---|---|---|---|---|---|
| Degree | Evidence | Physiological | Pathophysiological | ||
| Cb | GluA1, GluA4 [96] | ++ | ePhys [96] | Synapse modulation [40] Blockade of K+ currents [97] | Altered motor control [98] GLAST downregulation [99] |
| Ctx | GluA2 dominant (qPCR) [100] | +/− | Ci [101] | Regulate K+ currents [48] | Excitotoxicity [101] |
| Hp | GluA2 dominant (qPCR) [100] No expression (ephys) [102] | No AMPA current | ePhys [102] | N.D. | N.D. |
| OB | GluA1, 2, 4 (IHC) [103] | + | ci, ePhys [103] | N.D. | N.D. |
| Th | GluA1-4 (RT-PCR) [104] | +/− | ePhys [104] | N.D. | N.D. |
| Preparation | GluA Expression | Ca2+ Permeability | Functions | ||
|---|---|---|---|---|---|
| Degree | Evidence | Physiological | Pathophysiological | ||
| OPC in vitro | GluA2, 3, 4 (qPCR, WB) [154,155] | ++ | Ci [155,169] ePhys [155] | IEG expression [44] | Excitotoxicity [170] OGD injury [171,172,173] NF-Y-dependent apoptosis [45] |
| OPC ex vivo | N.D. | + | Ci, ePhys [161] | Proliferation, Differentiation [41,43] Migration [39] Blockade of K+ currents [43,174] OPC LTP [175] | OGD injury [165] |
| OPC in vivo | GluA2-4 (FISH) [46] | N.D. | N.D. | Survival [165] | H-I injury [172] Remyelination [148,176] |
| OL in vitro | GluA2, 3, 4 (qPCR, WB) [155] | + | Ci [155,177] ePhys [155] | N.D. | Excitotoxicity [65] |
| OL ex vivo | GluA4 (IHC) [106] | + | Ci [109] | Activate NMDAR [109] | Excitotoxicity [106] |
| OL in vivo | GluA2/3 (IHC) [177] GluA4 IHC [167] | N.D. | N.D. | N.D. | Demyelination [119] H-I injury [178] Spinal cord injury [179] |
| Preparation | GluA Expression | Ca2+ Permeability | Functions | ||
|---|---|---|---|---|---|
| Degree | Evidence | Physiological | Pathophysiological | ||
| In vitro | GluA2,3,4 (RT-PCR) [233] GluA1,2,3 (qPCR) [234] GluA1 (ICC) [236] | - | ePhys [225,226] | Chemotaxis [117] | Hypoxia induced increase in GluA2-4 [237] Release of IL-1b and TNF-a [234,237] TNF-a release [234] |
| In vivo | GluA2/3, 4 (IHC) [237] No AMPAR (ePhys) [238] | No AMPAR | ePhys [238] | N.D. | Hypoxia/Ischemia increased GluA2-4 [237,239] |
| Tissue Age | GluA Expression | Ca2+ Permeability | Functions | ||
|---|---|---|---|---|---|
| Degree | Evidence | Physiological | Pathophysiological | ||
| RG Developing Tissue | GluA1, 2, 3 (qPCR, ICC) [251] GluA3, 4 (qPCR) [252] GluA3,4 [252] | + | Ci [251] | N.D. | N.D. |
| RG Adult tissue | GluA2, 3 and 4 (qPCR) [166] | N.D. | N.D. | Proliferation, survival [253] | N.D. |
| Tanycte | GluA2/3 (IHC) [254] | N.D. | N.D. | N.D. | N.D. |
| Schwann cell | GluA2/3, 4 (IHC, I-EM) [255] Functional AMPAR in developing cells [256] | + | ePhys [256,257] | ATP release [258] | N.D. |
| Satellite glia | GluA4 (IHC) [259,260] | + | Ci [259,260] | N.D. | N.D. |
| Enteric glia | GluA1, 3 (qPCR, WB) [261] | N.D. | N.D. | N.D. | N.D. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceprian, M.; Fulton, D. Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease. Int. J. Mol. Sci. 2019, 20, 2450. https://doi.org/10.3390/ijms20102450
Ceprian M, Fulton D. Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease. International Journal of Molecular Sciences. 2019; 20(10):2450. https://doi.org/10.3390/ijms20102450
Chicago/Turabian StyleCeprian, Maria, and Daniel Fulton. 2019. "Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease" International Journal of Molecular Sciences 20, no. 10: 2450. https://doi.org/10.3390/ijms20102450
APA StyleCeprian, M., & Fulton, D. (2019). Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease. International Journal of Molecular Sciences, 20(10), 2450. https://doi.org/10.3390/ijms20102450