Protein Structure Determination in Living Cells


Abstract

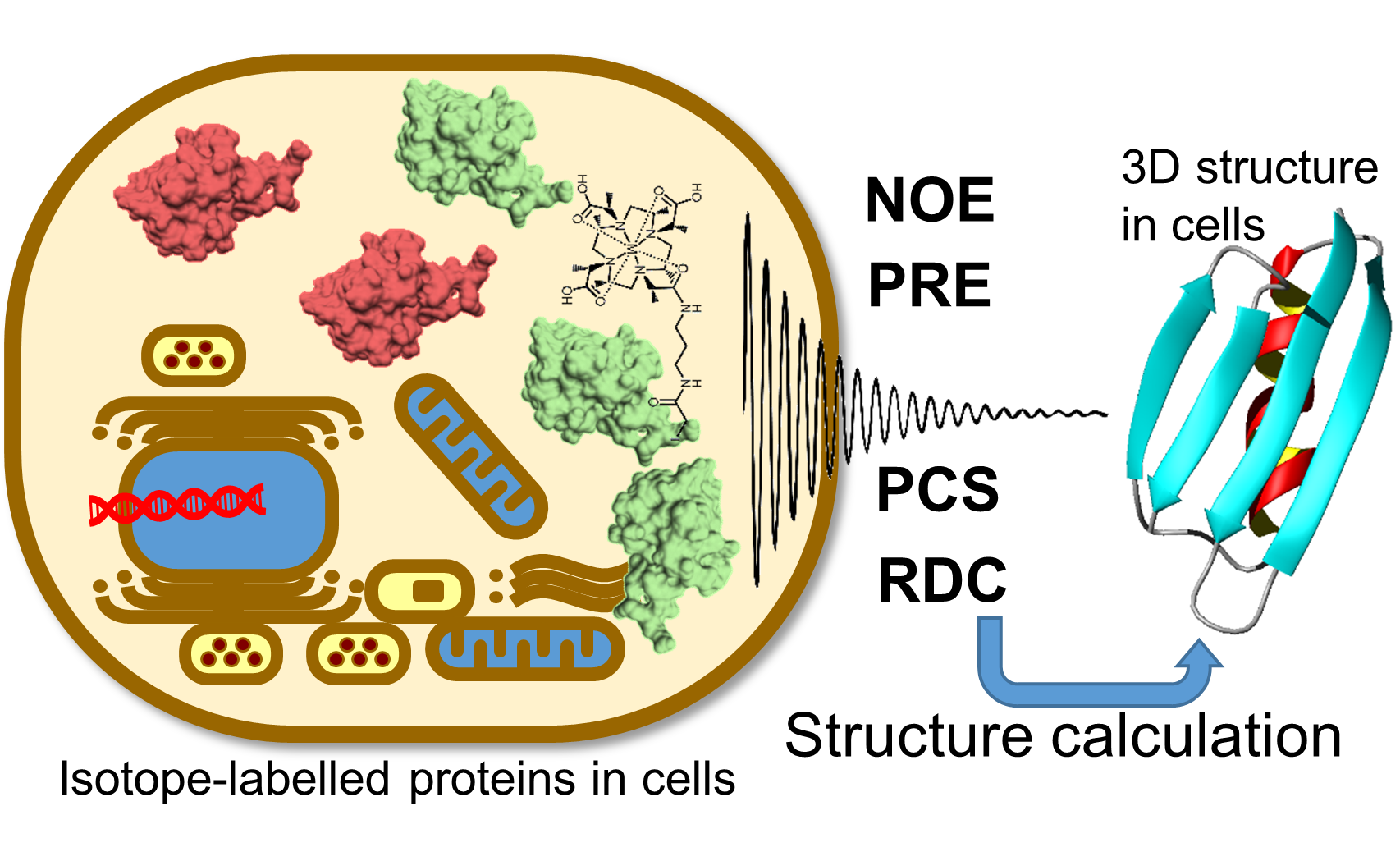
1. Introduction
2. Paramagnetic NMR
3. De Novo in-Cell Protein Structure Determination
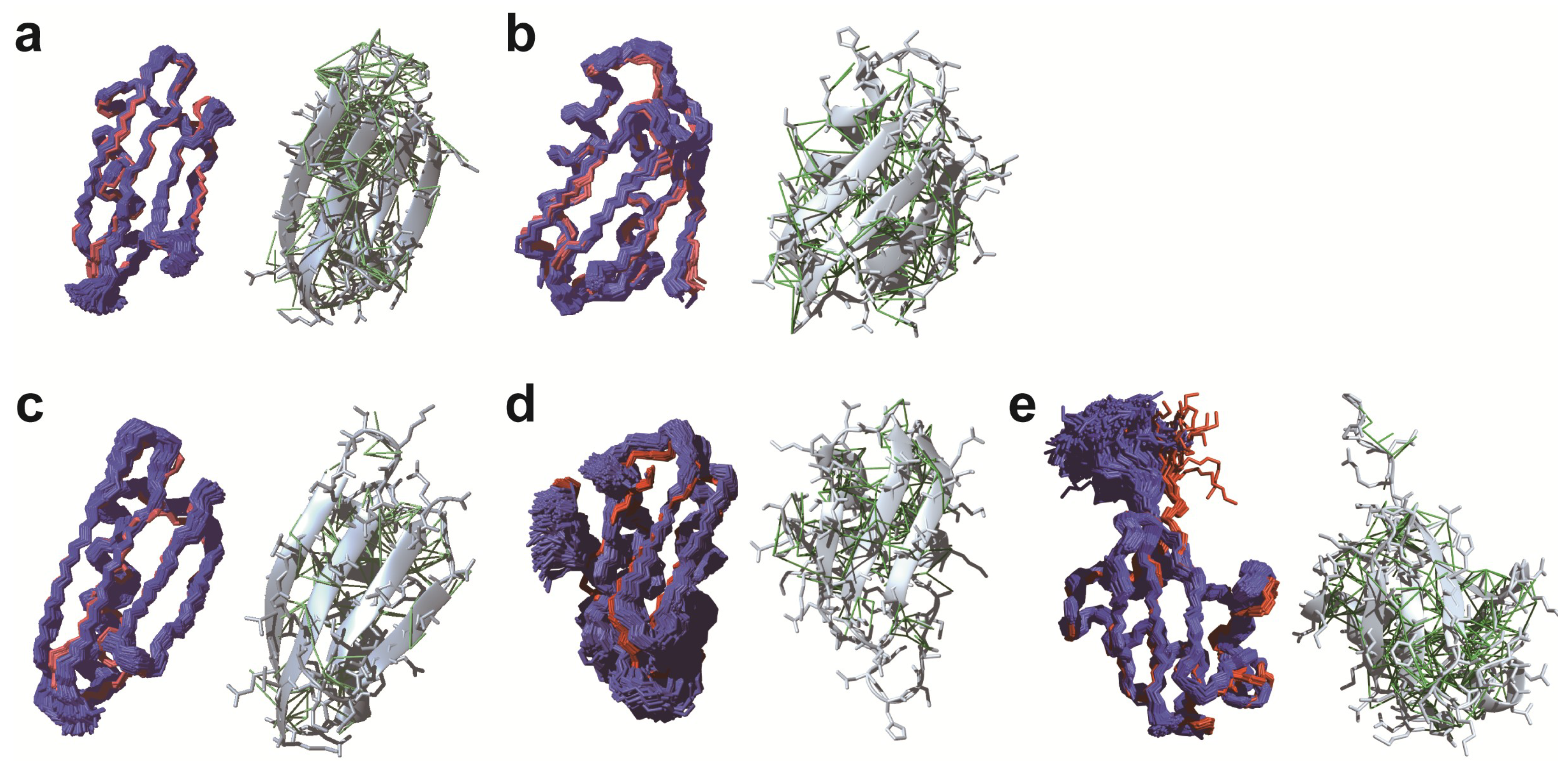
3.1. De novo Structure Determination in Prokaryotic Cells
3.2. De novo Structure Determination in Eukaryotic Cells
4. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HSQC | Heteronuclear Single Quantum Coherence |
| H/D | Hydrogen/Deuterium |
| NUS | Non-uniform Sampling |
| SOD | Superoxide Dismutase |
| PCS | Pseudo-Contact Shift |
| RDC | Residual Dipolar Coupling |
| IDP | Intrinsically Disordered Protein |
| LBT | Lanthanide-Binding Tag |
| NUS | Non-Uniform Sampling |
| NOE | Nuclear Overhauser Effect |
| GB1 | protein G B1 domain |
| PRE | Paramagnetic Relaxation Enhancement |
| PDB | Protein Data Bank |
References
- Serber, Z.; Keatinge-Clay, A.T.; Ledwidge, R.; Kelly, A.E.; Miller, S.M.; Dötsch, V. High-resolution macromolecular NMR spectroscopy inside living cells. J. Am. Chem. Soc. 2001, 123, 2446–2447. [Google Scholar] [CrossRef]
- Freedberg, D.I.; Selenko, P. Live cell NMR. Annu. Rev. Biophys. 2014, 43, 171–192. [Google Scholar] [CrossRef]
- Luchinat, E.; Banci, L. In-cell NMR: A topical review. IUCrJ 2017, 4, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Ikeya, T.; Ban, D.; Lee, D.; Ito, Y.; Kato, K.; Griesinger, C. Solution NMR views of dynamical ordering of biomacromolecules. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 287–306. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J. Macromolecular crowding: Obvious but underappreciated. Trends Biochem. Sci. 2001, 26, 597–604. [Google Scholar] [CrossRef]
- Majumder, S.; Xue, J.; DeMott, C.M.; Reverdatto, S.; Burz, D.S.; Shekhtman, A. Probing protein quinary interactions by in-cell nuclear magnetic resonance spectroscopy. Biochemistry 2015, 54, 2727–2738. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-X.; Rivas, G.; Minton, A.P. Macromolecular Crowding and Confinement: Biochemical, Biophysical, and Potential Physiological Consequences. Annu. Rev. Biophys. 2008, 37, 375–397. [Google Scholar] [CrossRef] [PubMed]
- Miklos, A.C.; Li, C.G.; Sharaf, N.G.; Pielak, G.J. Volume Exclusion and Soft Interaction Effects on Protein Stability under Crowded Conditions. Biochemistry 2010, 49, 6984–6991. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Li, C.G.; Pielak, G.J. Effects of Proteins on Protein Diffusion. J. Am. Chem. Soc. 2010, 132, 9392–9397. [Google Scholar] [CrossRef]
- Inomata, K.; Ohno, A.; Tochio, H.; Isogai, S.; Tenno, T.; Nakase, I.; Takeuchi, T.; Futaki, S.; Ito, Y.; Hiroaki, H.; et al. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature 2009, 458, 106–109. [Google Scholar] [CrossRef]
- Danielsson, J.; Mu, X.; Lang, L.; Wang, H.; Binolfi, A.; Theillet, F.X.; Bekei, B.; Logan, D.T.; Selenko, P.; Wennerstrom, H.; et al. Thermodynamics of protein destabilization in live cells. Proc. Natl. Acad. Sci. USA 2015, 112, 12402–12407. [Google Scholar] [CrossRef]
- Smith, A.E.; Zhou, L.Z.; Gorensek, A.H.; Senske, M.; Pielak, G.J. In-cell thermodynamics and a new role for protein surfaces. Proc. Natl. Acad. Sci. USA 2016, 113, 1725–1730. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.O.; Monteith, W.B.; Pielak, G.J. Internal and global protein motion assessed with a fusion construct and in-cell NMR spectroscopy. ChemBioChem 2011, 12, 390–391. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Zhou, L.Z.; Pielak, G.J. Hydrogen exchange of disordered proteins in Escherichia coli. Protein Sci. 2015, 24, 706–713. [Google Scholar] [CrossRef]
- Ikeya, T.; Sasaki, A.; Sakakibara, D.; Shigemitsu, Y.; Hamatsu, J.; Hanashima, T.; Mishima, M.; Yoshimasu, M.; Hayashi, N.; Mikawa, T.; et al. NMR protein structure determination in living E. coli cells using nonlinear sampling. Nat. Protoc. 2010, 5, 1051–1060. [Google Scholar] [CrossRef]
- Hamatsu, J.; O’Donovan, D.; Tanaka, T.; Shirai, T.; Hourai, Y.; Mikawa, T.; Ikeya, T.; Mishima, M.; Boucher, W.; Smith, B.O.; et al. High-resolution heteronuclear multidimensional NMR of proteins in living insect cells using a baculovirus protein expression system. J. Am. Chem. Soc. 2013, 135, 1688–1691. [Google Scholar] [CrossRef] [PubMed]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; Wiley-Interscience: New York, NY, USA, 1986. [Google Scholar]
- Clore, G.M.; Iwahara, J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem. Rev. 2009, 109, 4108–4139. [Google Scholar] [CrossRef] [PubMed]
- Otting, G. Protein NMR using paramagnetic ions. Annu. Rev. Biophys. 2010, 39, 387–405. [Google Scholar] [CrossRef]
- Saio, T.; Inagaki, F. Experimental Approaches of NMR Spectroscopy: Structural Study of Proteins by Paramagnetic Lanthanide Probe Methods; Springer: New York, NY, USA, 2018; pp. 227–252. [Google Scholar]
- Ye, Y.; Liu, X.; Xu, G.; Liu, M.; Li, C. Direct observation of Ca2+-induced calmodulin conformational transitions in intact Xenopus laevis oocytes by 19F NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2015, 54, 5328–5330. [Google Scholar] [CrossRef]
- Müntener, T.; Häussinger, D.; Selenko, P.; Theillet, F.X. In-Cell Protein Structures from 2D NMR Experiments. J. Phys. Chem. Lett. 2016, 7, 2821–2825. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.B.; Yang, F.; Ye, Y.; Wu, Q.; Li, C.; Huber, T.; Su, X.C. 3D structure determination of a protein in living cells using paramagnetic NMR spectroscopy. Chem. Commun. (Camb.) 2016, 52, 10237–10240. [Google Scholar] [CrossRef]
- Hikone, Y.; Hirai, G.; Mishima, M.; Inomata, K.; Ikeya, T.; Arai, S.; Shirakawa, M.; Sodeoka, M.; Ito, Y. A new carbamidemethyl-linked lanthanoid chelating tag for PCS NMR spectroscopy of proteins in living HeLa cells. J. Biomol. NMR 2016, 66, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Müntener, T.; Kottelat, J.; Huber, A.; Häussinger, D. New Lanthanide Chelating Tags for PCS NMR Spectroscopy with Reduction Stable, Rigid Linkers for Fast and Irreversible Conjugation to Proteins. Bioconjug. Chem. 2018, 29, 3344–3351. [Google Scholar] [CrossRef] [PubMed]
- Häussinger, D.; Huang, J.R.; Grzesiek, S. DOTA-M8: An extremely rigid, high-affinity lanthanide chelating tag for PCS NMR spectroscopy. J. Am. Chem. Soc. 2009, 131, 14761–14767. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, J.T.; Pei, Y.Y.; Su, X.C. Site-specific tagging proteins via a rigid, stable and short thiolether tether for paramagnetic spectroscopic analysis. Chem. Commun. (Camb.) 2015, 51, 2824–2827. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.X.; Binolfi, A.; Liokatis, S.; Verzini, S.; Selenko, P. Paramagnetic relaxation enhancement to improve sensitivity of fast NMR methods: Application to intrinsically disordered proteins. J. Biomol. NMR 2011, 51, 487–495. [Google Scholar] [CrossRef]
- Bertini, I.; Luchinat, C.; Parigi, G. Magnetic susceptibility in paramagnetic NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2002, 40, 249–273. [Google Scholar] [CrossRef]
- Koehler, J.; Meiler, J. Expanding the utility of NMR restraints with paramagnetic compounds: Background and practical aspects. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 59, 360–389. [Google Scholar] [CrossRef]
- Schmitz, C.; Stanton-Cook, M.J.; Su, X.C.; Otting, G.; Huber, T. Numbat: An interactive software tool for fitting Deltachi-tensors to molecular coordinates using pseudocontact shifts. J. Biomol. NMR 2008, 41, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Suturina, E.A.; Kuprov, I. Pseudocontact shifts from mobile spin labels. Phys. Chem. Chem. Phys. 2016, 18, 26412–26422. [Google Scholar] [CrossRef] [PubMed]
- Bradley, P.; Malmstrom, L.; Qian, B.; Schonbrun, J.; Chivian, D.; Kim, D.E.; Meiler, J.; Misura, K.M.; Baker, D. Free modeling with Rosetta in CASP6. Proteins 2005, 61, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, S.; Park, H.; Kim, D.E.; DiMaio, F.; Baker, D. Protein structure prediction using Rosetta in CASP12. Proteins 2018, 86, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M. Practical Aspects of Paramagnetic Relaxation Enhancement in Biological Macromolecules. Methods Enzymol. 2015, 564, 485–497. [Google Scholar]
- Battiste, J.L.; Wagner, G. Utilization of Site-Directed Spin Labeling and High-Resolution Heteronuclear Nuclear Magnetic Resonance for Global Fold Determination of Large Proteins with Limited Nuclear Overhauser Effect Data. Biochemistry 2000, 39, 5355–5365. [Google Scholar] [CrossRef]
- Bertrand, K.; Reverdatto, S.; Burz, D.S.; Zitomer, R.; Shekhtman, A. Structure of proteins in eukaryotic compartments. J. Am. Chem. Soc. 2012, 134, 12798–12806. [Google Scholar] [CrossRef]
- Banci, L.; Barbieri, L.; Bertini, I.; Luchinat, E.; Secci, E.; Zhao, Y.; Aricescu, A.R. Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nat. Chem. Biol. 2013, 9, 297–299. [Google Scholar] [CrossRef]
- Ogino, S.; Kubo, S.; Umemoto, R.; Huang, S.; Nishida, N.; Shimada, I. Observation of NMR signals from proteins introduced into living Mammalian cells by reversible membrane permeabilization using a pore-forming toxin, streptolysin o. J. Am. Chem. Soc. 2009, 131, 10834–10835. [Google Scholar] [CrossRef]
- Sakakibara, D.; Sasaki, A.; Ikeya, T.; Hamatsu, J.; Hanashima, T.; Mishima, M.; Yoshimasu, M.; Hayashi, N.; Mikawa, T.; Waälchli, M.; et al. Protein structure determination in living cells by in-cell NMR spectroscopy. Nature 2009, 458, 102–105. [Google Scholar] [CrossRef]
- Mobli, M.; Stern, A.S.; Hoch, J.C. Spectral reconstruction methods in fast NMR: Reduced dimensionality, random sampling and maximum entropy. J. Magn. Reson. 2006, 182, 96–105. [Google Scholar] [CrossRef]
- Laue, E.D.; Mayger, M.R.; Skilling, J.; Staunton, J. Reconstruction of phase sensitive 2D NMR spectra by maximum entropy. J. Magn. Reson. 1986, 68, 14–29. [Google Scholar]
- Hoch, J.C.; Maciejewski, M.W.; Mobli, M.; Schuyler, A.D.; Stern, A.S. Nonuniform Sampling and Maximum Entropy Reconstruction in Multidimensional NMR. Acc. Chem. Res. 2014, 47, 708–717. [Google Scholar] [CrossRef]
- Ikeya, T.; Hanashima, T.; Hosoya, S.; Shimazaki, M.; Ikeda, S.; Mishima, M.; Güntert, P.; Ito, Y. Improved in-cell structure determination of proteins at near-physiological concentration. Sci. Rep. 2016, 6, 38312. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.D. Polymerization of the bacterial elongation factor for protein synthesis, EF-Tu. Eur. J. Biochem. 1979, 97, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Schmidt, A.; Malmstroem, J.; Claassen, M.; Ori, A.; Szymborska, A.; Herzog, F.; Rinner, O.; Ellenberg, J.; Aebersold, R. The quantitative proteome of a human cell line. Mol. Syst. Biol. 2011, 7, 549. [Google Scholar] [CrossRef]
- Boucher, W. Azara, V2.0; Department of Biochemistry, University of Cambridge: Cambridge, UK, 1996. [Google Scholar]
- Schmidt, E.; Güntert, P. A new algorithm for reliable and general NMR resonance assignment. J. Am. Chem. Soc. 2012, 134, 12817–12829. [Google Scholar] [CrossRef] [PubMed]
- Ikeya, T.; Jee, J.G.; Shigemitsu, Y.; Hamatsu, J.; Mishima, M.; Ito, Y.; Kainosho, M.; Güntert, P. Exclusively NOESY-based automated NMR assignment and structure determination of proteins. J. Biomol. NMR 2011, 50, 137–146. [Google Scholar] [CrossRef]
- Schmidt, E.; Güntert, P. Reliability of exclusively NOESY-based automated resonance assignment and structure determination of proteins. J. Biomol. NMR 2013, 57, 193–204. [Google Scholar] [CrossRef]
- Pritišanac, I.; Würz, J.M.; Alderson, T.R.; Güntert, P. Automatic structure-based NMR methyl resonance assignment in large proteins. bioRxiv 2019, 1, 538272. [Google Scholar]
- Ikeya, T.; Ikeda, S.; Kigawa, T.; Ito, Y.; Güntert, P. Protein NMR Structure Refinement based on Bayesian Inference. J. Phys. Conf. Ser. 2016, 699, 012005. [Google Scholar] [CrossRef]
- Tanaka, T.; Ikeya, T.; Kamoshida, H.; Suemoto, Y.; Mishima, M.; Shirakawa, M.; Güntert, P.; Ito, Y. High Resolution Protein 3D Structure Determination in Living Eukaryotic Cells. Angew. Chem. Int. Ed. Engl. 2019, 58. [Google Scholar] [CrossRef]
- Sharaf, N.G.; Barnes, C.O.; Charlton, L.M.; Young, G.B.; Pielak, G.J. A bioreactor for in-cell protein NMR. J. Magn. Reson. 2010, 202, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.; Nishida, N.; Udagawa, Y.; Takarada, O.; Ogino, S.; Shimada, I. A gel-encapsulated bioreactor system for NMR studies of protein-protein interactions in living mammalian cells. Angew. Chem. Int. Ed. Engl. 2013, 52, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Inomata, K.; Kamoshida, H.; Ikari, M.; Ito, Y.; Kigawa, T. Impact of cellular health conditions on the protein folding state in mammalian cells. Chem. Commun. (Camb.) 2017, 53, 11245–11248. [Google Scholar] [CrossRef]
- Hyberts, S.G.; Takeuchi, K.; Wagner, G. Poisson-gap sampling and forward maximum entropy reconstruction for enhancing the resolution and sensitivity of protein NMR data. J. Am. Chem. Soc. 2010, 132, 2145–2147. [Google Scholar] [CrossRef]
- Harada, R.; Tochio, N.; Kigawa, T.; Sugita, Y.; Feig, M. Reduced native state stability in crowded cellular environment due to protein-protein interactions. J. Am. Chem. Soc. 2013, 135, 3696–3701. [Google Scholar] [CrossRef] [PubMed]
- Hembram, D.S.; Haremaki, T.; Hamatsu, J.; Inoue, J.; Kamoshida, H.; Ikeya, T.; Mishima, M.; Mikawa, T.; Hayashi, N.; Shirakawa, M.; et al. An in-cell NMR study of monitoring stress-induced increase of cytosolic Ca2+ concentration in HeLa cells. Biochem. Biophys. Res. Commun. 2013, 438, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Kainosho, M.; Torizawa, T.; Iwashita, Y.; Terauchi, T.; Mei Ono, A.; Güntert, P. Optimal isotope labelling for NMR protein structure determinations. Nature 2006, 440, 52–57. [Google Scholar] [CrossRef]
- Kainosho, M.; Miyanoiri, Y.; Terauchi, T.; Takeda, M. Perspective: Next generation isotope-aided methods for protein NMR spectroscopy. J. Biomol. NMR 2018, 71, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xu, R.; Cowburn, D. Chapter 8 Segmental Isotopic Labeling of Proteins for Nuclear Magnetic Resonance. Methods Enzymol. 2009, 462, 151–175. [Google Scholar]
- Xue, J.; Burz, D.S.; Shekhtman, A. Segmental labeling to study multidomain proteins. Adv. Exp. Med. Biol. 2012, 992, 17–33. [Google Scholar]
- Minato, Y.; Ueda, T.; Machiyama, A.; Shimada, I.; Iwai, H. Segmental isotopic labeling of a 140 kDa dimeric multi-domain protein CheA from Escherichia coli by expressed protein ligation and protein trans-splicing. J. Biomol. NMR 2012, 53, 191–207. [Google Scholar] [CrossRef]
- Freiburger, L.; Sonntag, M.; Hennig, J.; Li, J.; Zou, P.; Sattler, M. Efficient segmental isotope labeling of multi-domain proteins using Sortase A. J. Biomol. NMR 2015, 63, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mikula, K.M.; Tascon, I.; Tommila, J.J.; Iwai, H. Segmental isotopic labeling of a single-domain globular protein without any refolding step by an asparaginyl endopeptidase. FEBS Lett. 2017, 591, 1285–1294. [Google Scholar] [CrossRef]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical assessment of methods of protein structure prediction (CASP)-Round XII. Proteins 2018, 86, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Holland, D.J.; Bostock, M.J.; Gladden, L.F.; Nietlispach, D. Fast multidimensional NMR spectroscopy using compressed sensing. Angew. Chem. Int. Ed. Engl. 2011, 50, 6548–6551. [Google Scholar] [CrossRef] [PubMed]
- Kazimierczuk, K.; Orekhov, V.Y. Accelerated NMR spectroscopy by using compressed sensing. Angew. Chem. Int. Ed. Engl. 2011, 50, 5556–5559. [Google Scholar] [CrossRef]
- Hyberts, S.G.; Milbradt, A.G.; Wagner, A.B.; Arthanari, H.; Wagner, G. Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson Gap scheduling. J. Biomol. NMR 2012, 52, 315–327. [Google Scholar] [CrossRef]
- Ying, J.; Delaglio, F.; Torchia, D.A.; Bax, A. Sparse multidimensional iterative lineshape-enhanced (SMILE) reconstruction of both non-uniformly sampled and conventional NMR data. J. Biomol. NMR 2017, 68, 101–118. [Google Scholar] [CrossRef]
- Kobayashi, N.; Hattori, Y.; Nagata, T.; Shinya, S.; Güntert, P.; Kojima, C.; Fujiwara, T. Noise peak filtering in multi-dimensional NMR spectra using convolutional neural networks. Bioinformatics 2018, 34, 4300–4301. [Google Scholar] [CrossRef]
- Yu, I.; Mori, T.; Ando, T.; Harada, R.; Jung, J.; Sugita, Y.; Feig, M. Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. Elife 2016, 5, e19274. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Name | Chemical Structure 1 | Reported Paramagnetic Effects | Linker | Reference | Commercially Available [CAS] 2 |
|---|---|---|---|---|---|
| DOTA-M8-CAM-I |  | PCS | N-propylene-acetamide | [24] | no |
| 4PhSO2-PyMTA |  | PCS/PRE | pyridine | [23,27] | yes |
| DOTA-maleimide |  | PRE | N-ethylene-maleimide | [28] | yes [1006711-09-5] |
| DOTA-M7Py |  | PCS | pyridine | [22] | no |
| DO2A |  | solvent PRE | ― | [29] | yes [112193-75-6] |
| M7PyThiazole-SO2Me-DOTA |  | PCS/RDC | pyridine thiazole | [25] | no |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikeya, T.; Güntert, P.; Ito, Y. Protein Structure Determination in Living Cells. Int. J. Mol. Sci. 2019, 20, 2442. https://doi.org/10.3390/ijms20102442
Ikeya T, Güntert P, Ito Y. Protein Structure Determination in Living Cells. International Journal of Molecular Sciences. 2019; 20(10):2442. https://doi.org/10.3390/ijms20102442
Chicago/Turabian StyleIkeya, Teppei, Peter Güntert, and Yutaka Ito. 2019. "Protein Structure Determination in Living Cells" International Journal of Molecular Sciences 20, no. 10: 2442. https://doi.org/10.3390/ijms20102442
APA StyleIkeya, T., Güntert, P., & Ito, Y. (2019). Protein Structure Determination in Living Cells. International Journal of Molecular Sciences, 20(10), 2442. https://doi.org/10.3390/ijms20102442