Computational Studies on Water-Catalyzed Mechanisms for Stereoinversion of Glutarimide Intermediates Formed from Glutamic Acid Residues in Aqueous Phase

 ,
,
Abstract
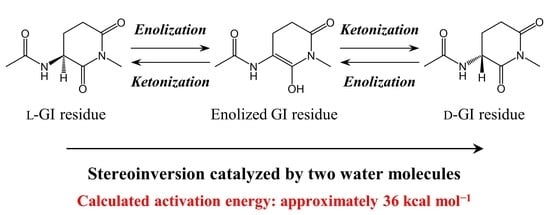
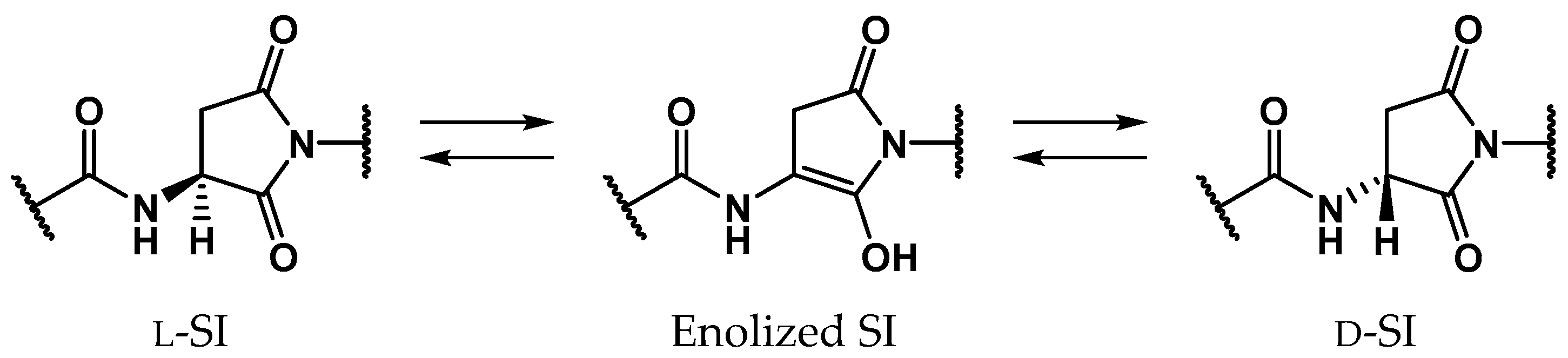
:
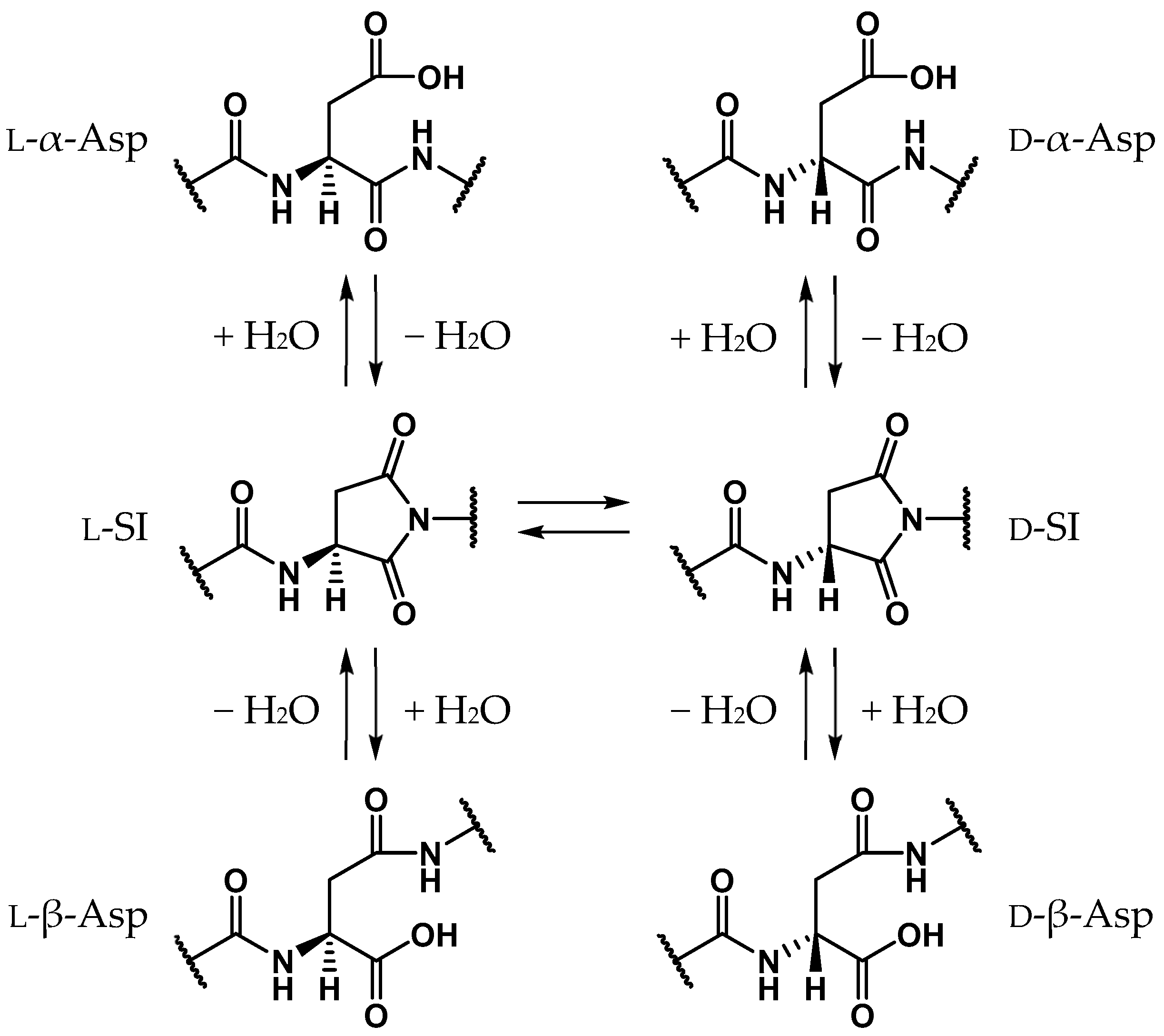
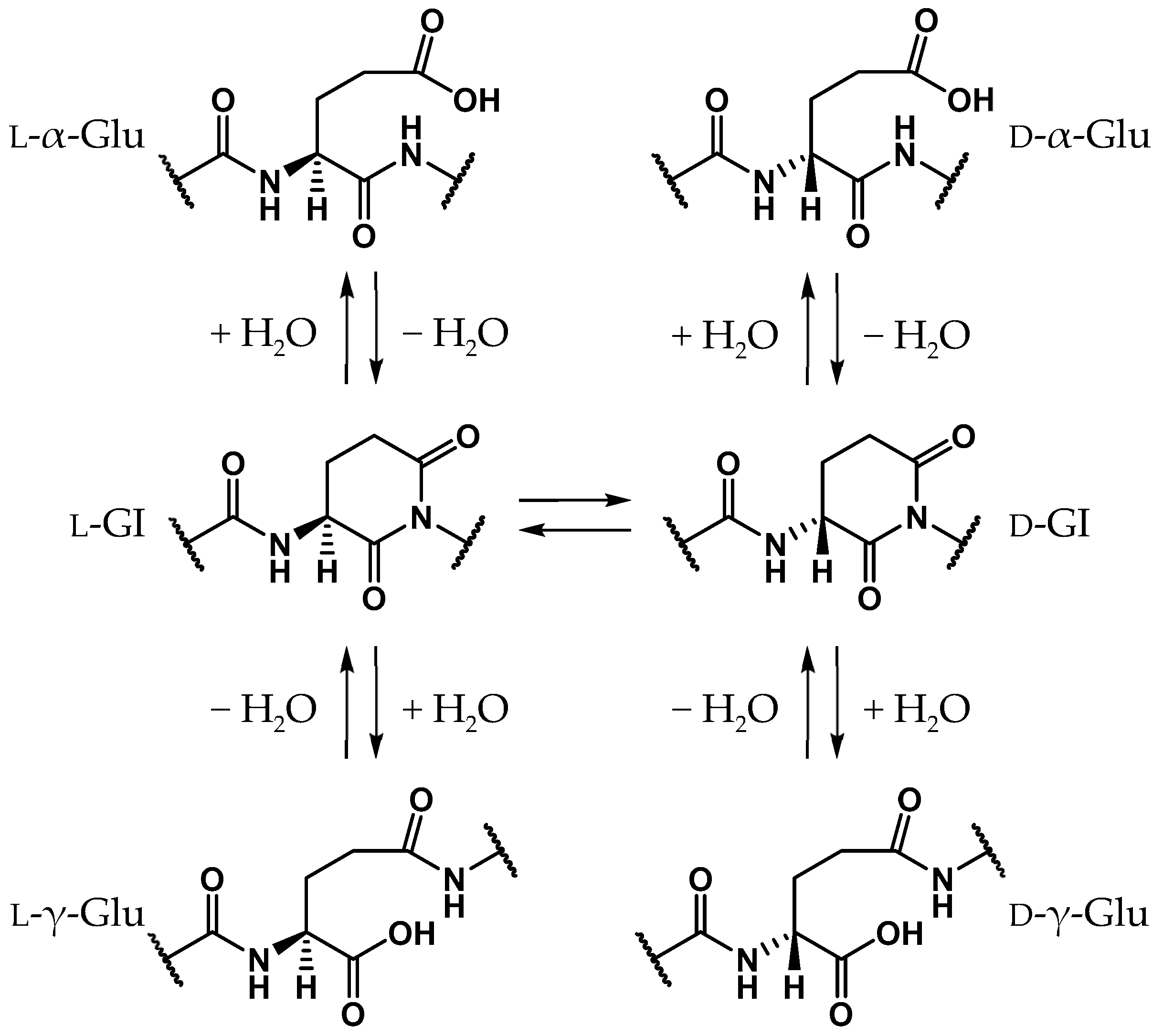
1. Introduction
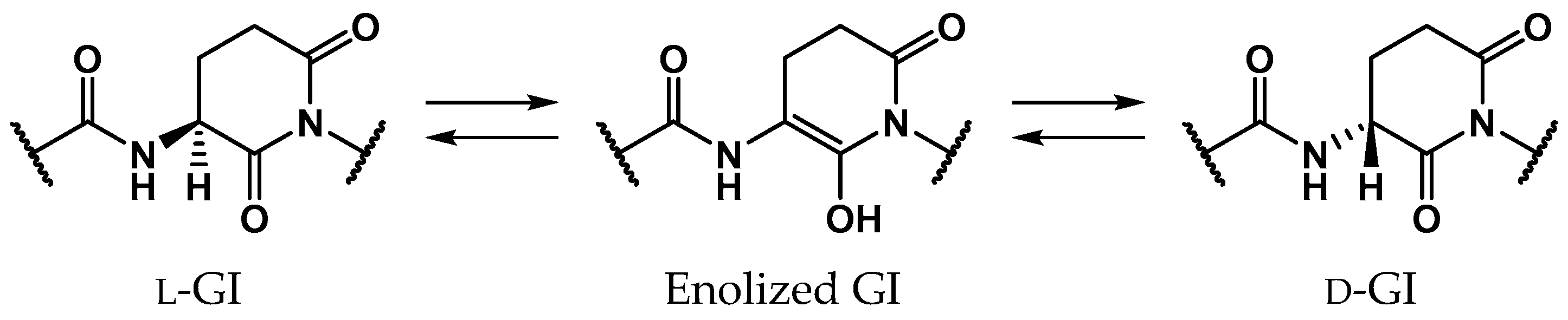
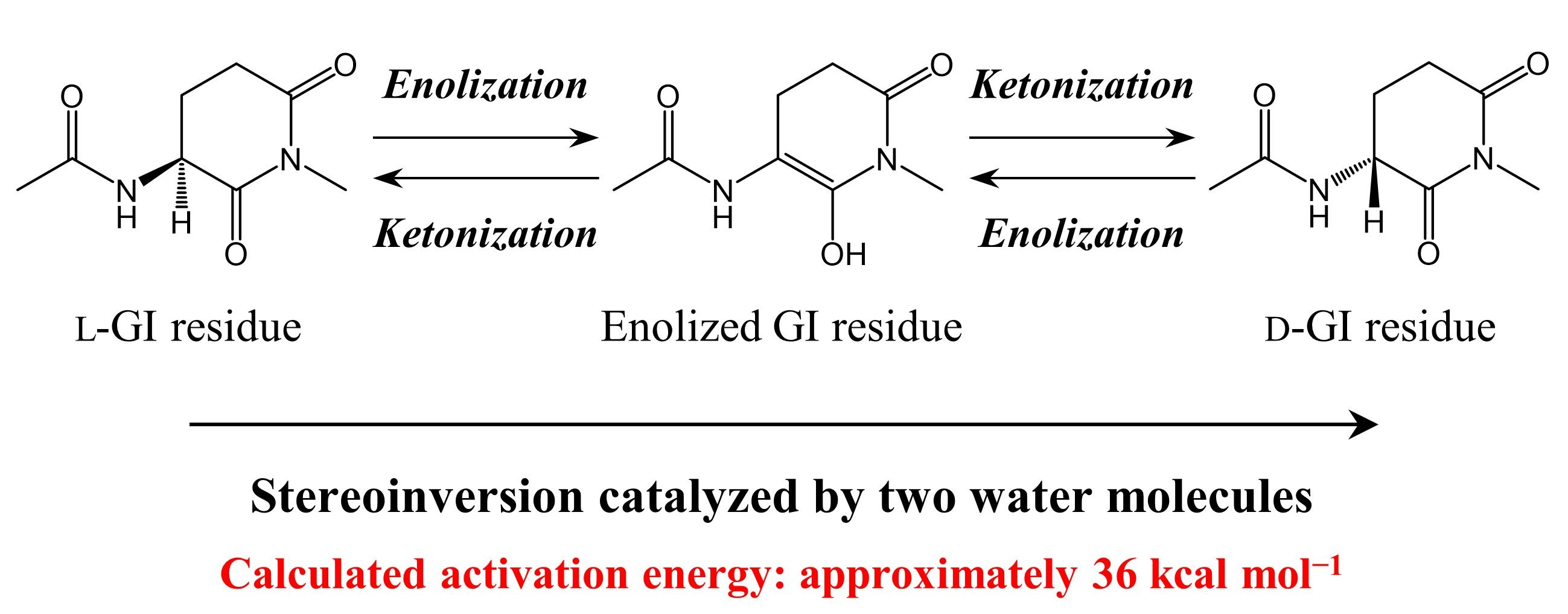
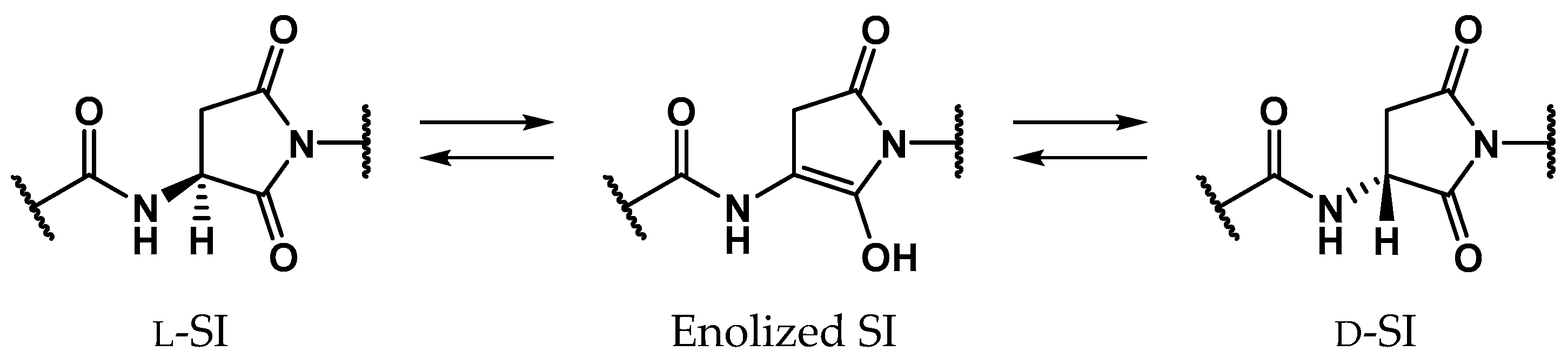
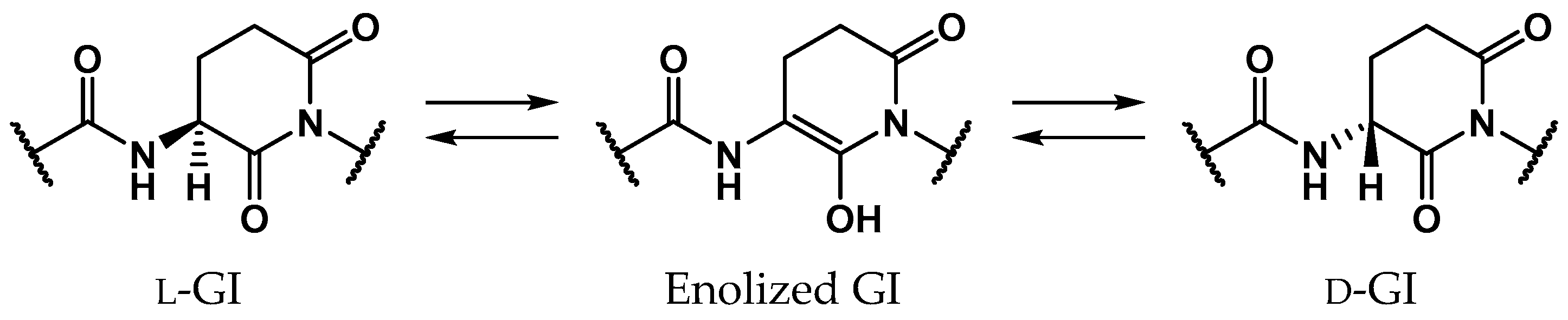
2. Results
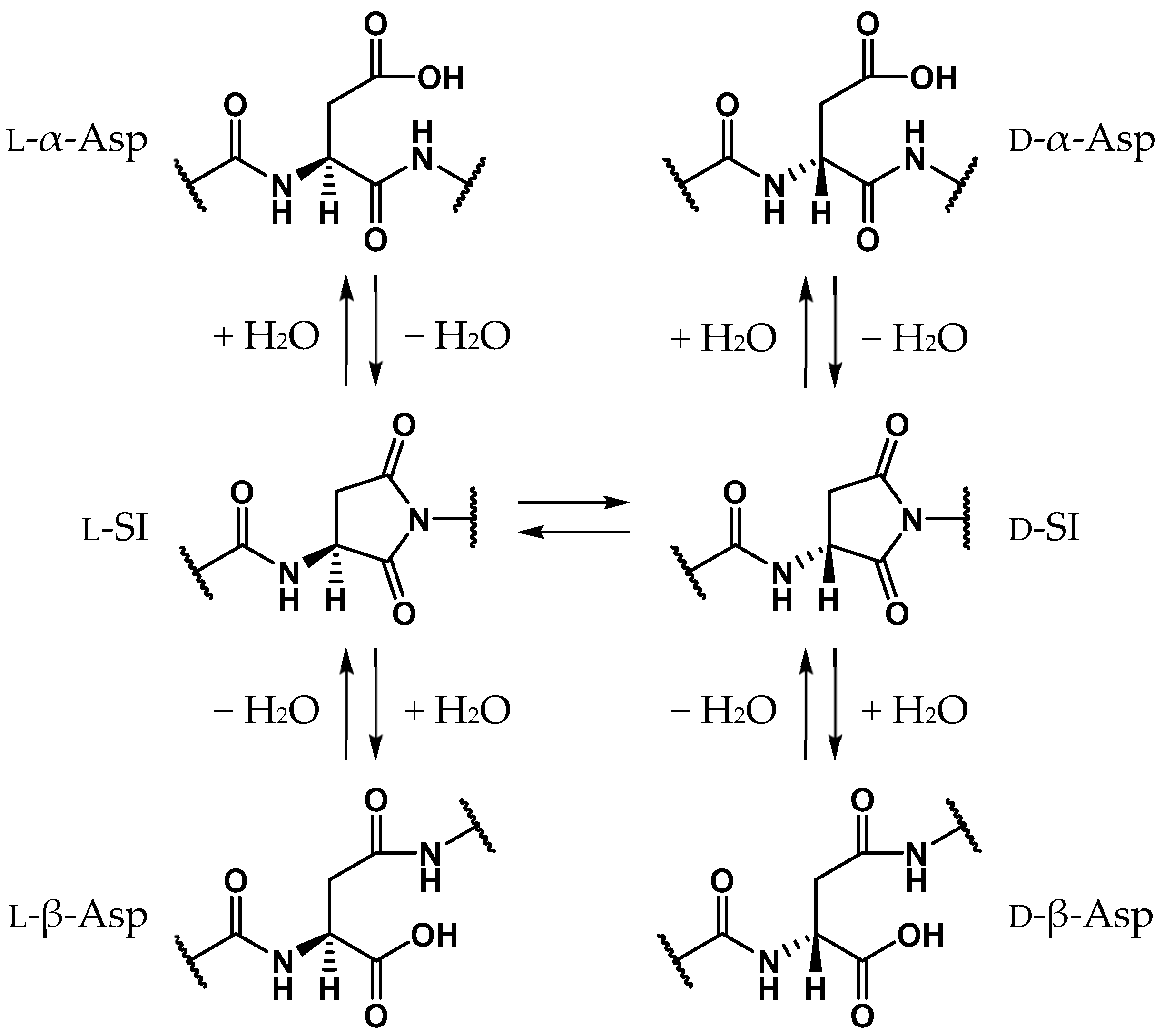
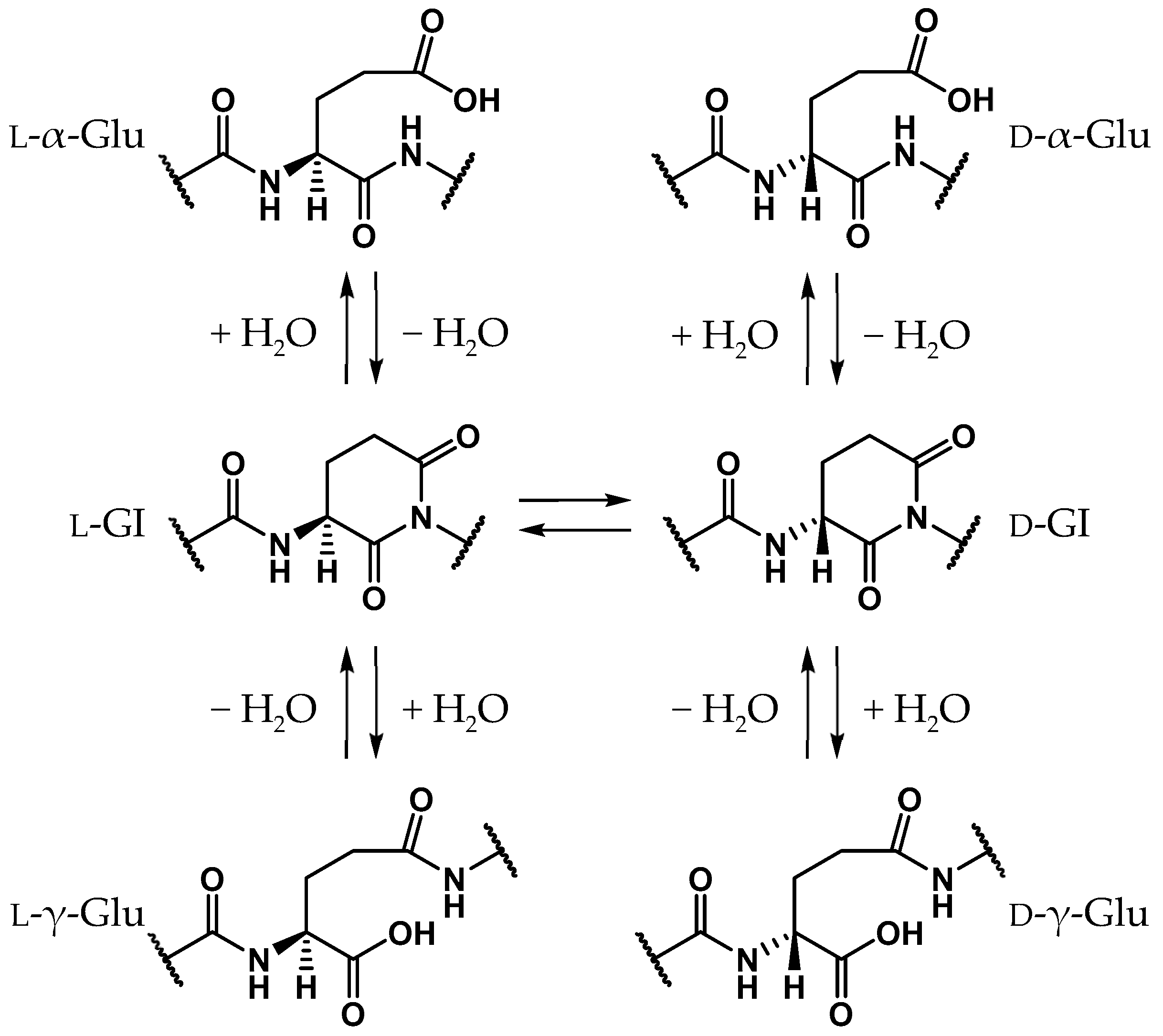
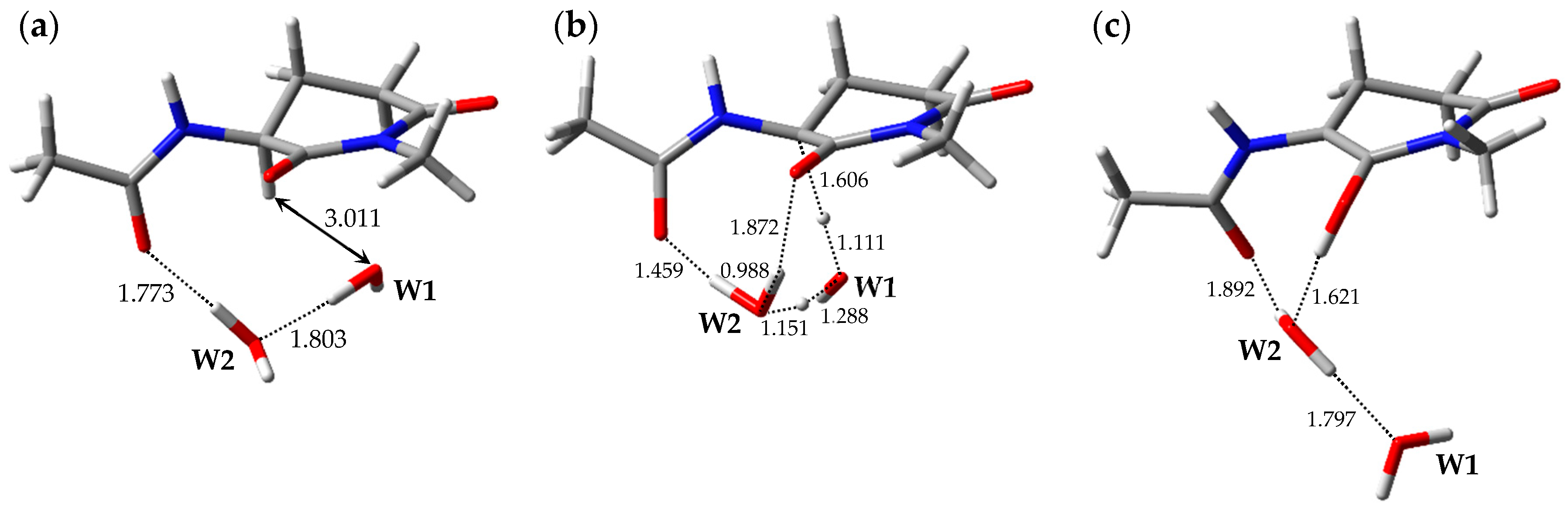
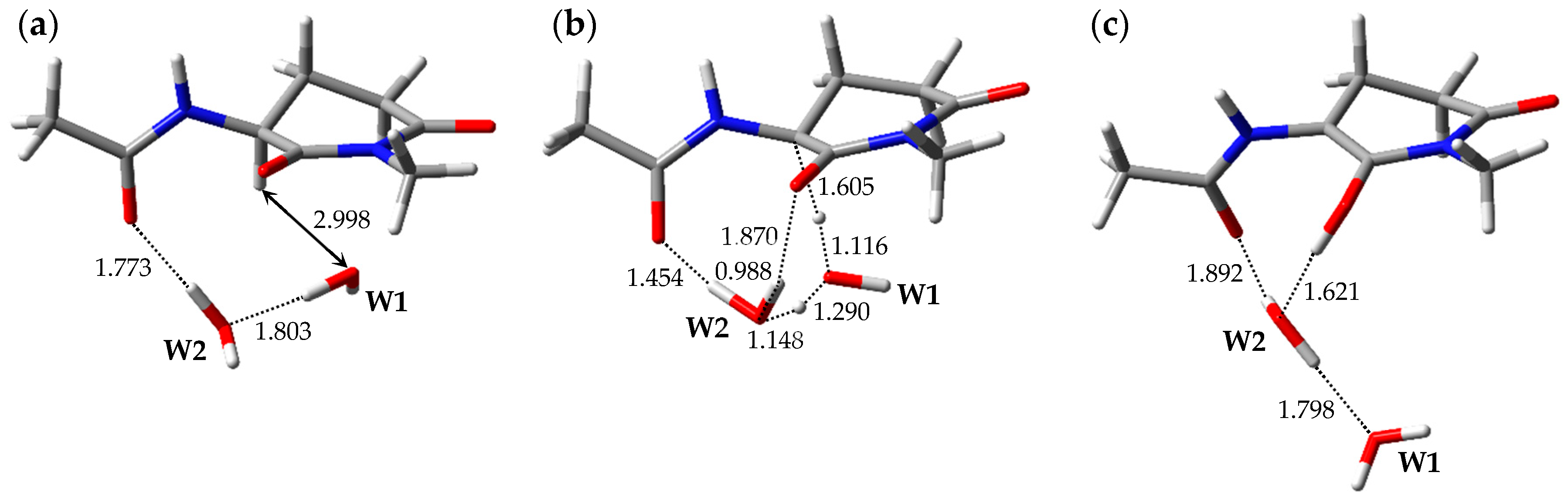
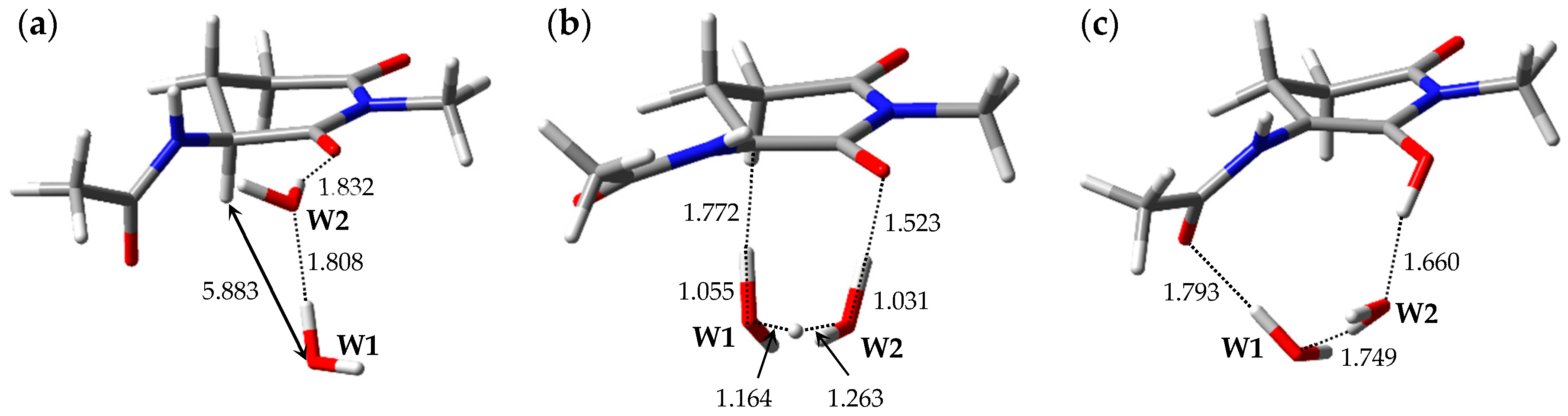
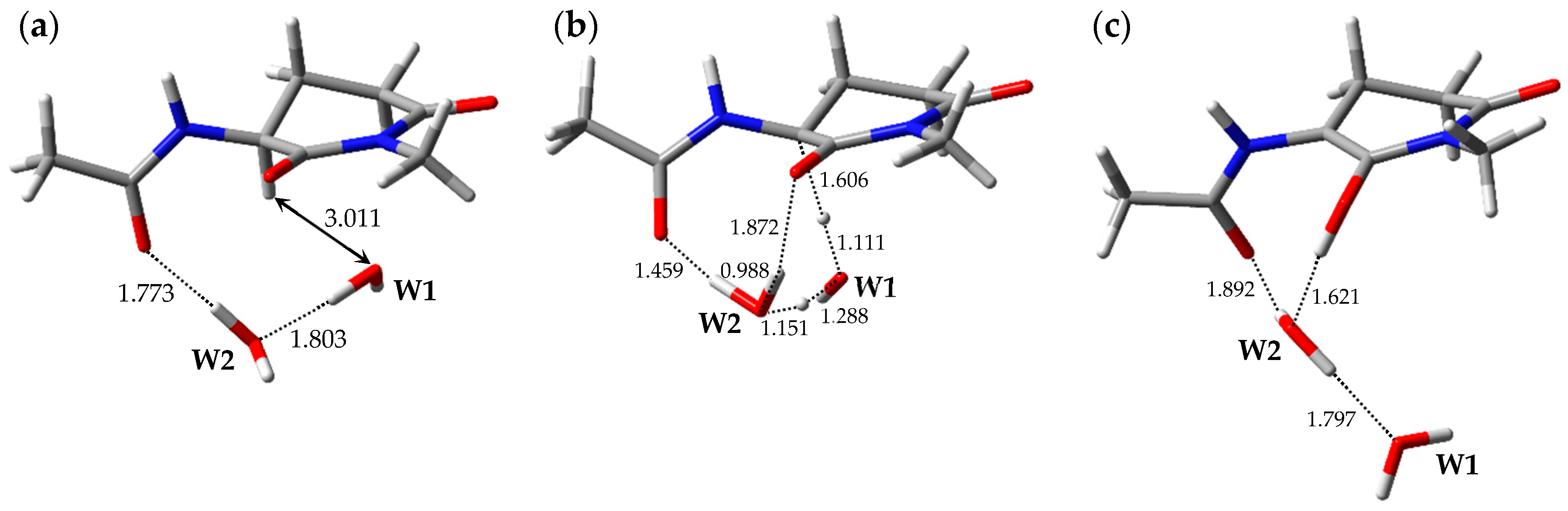
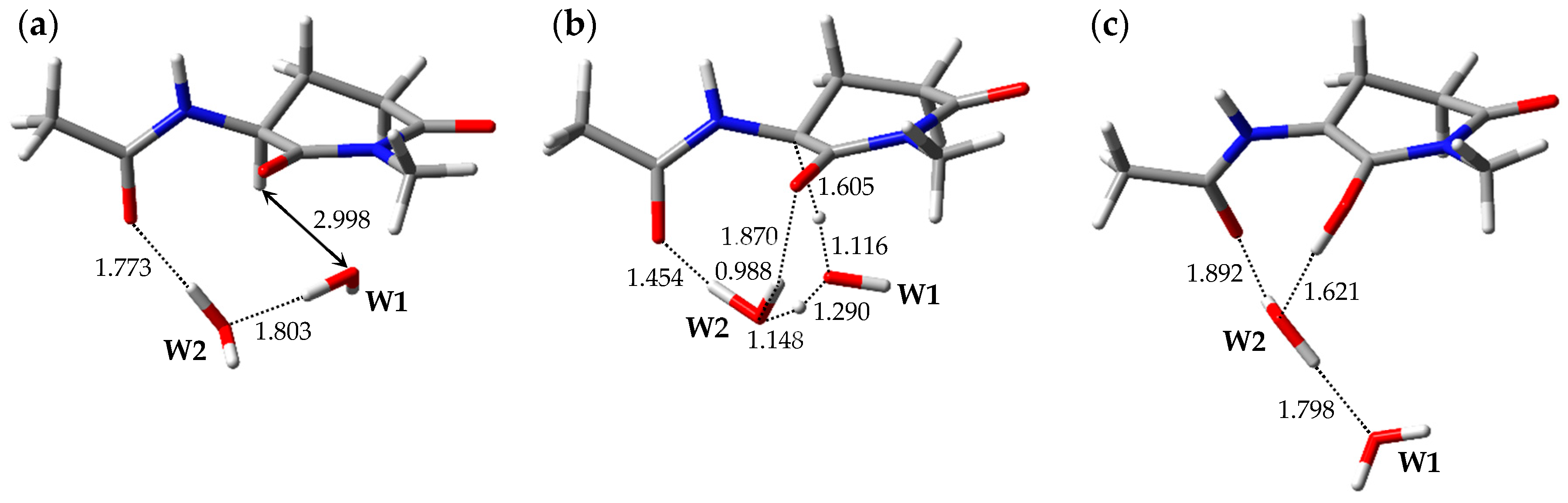
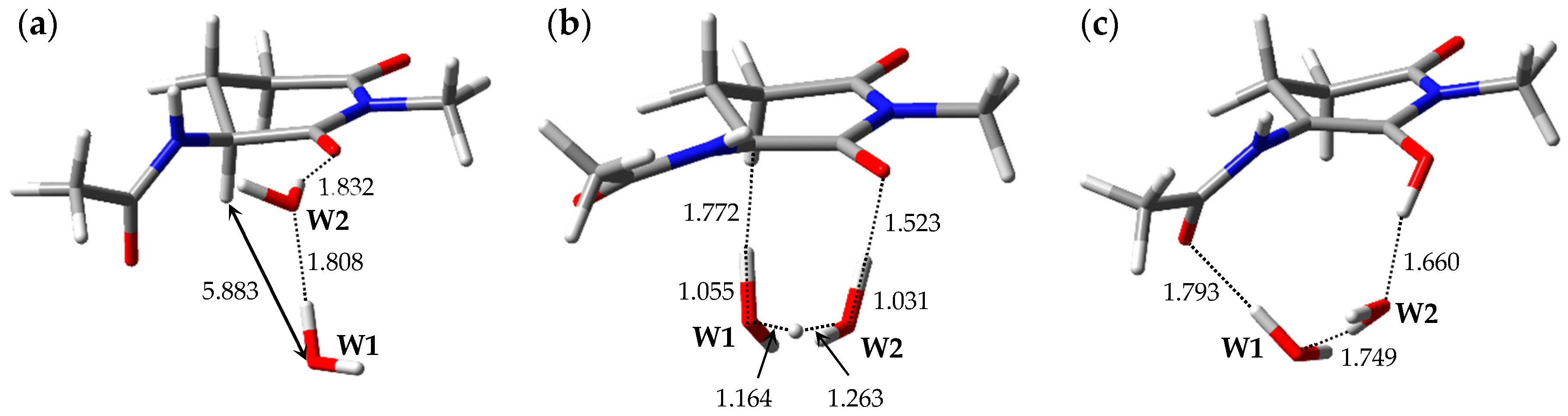
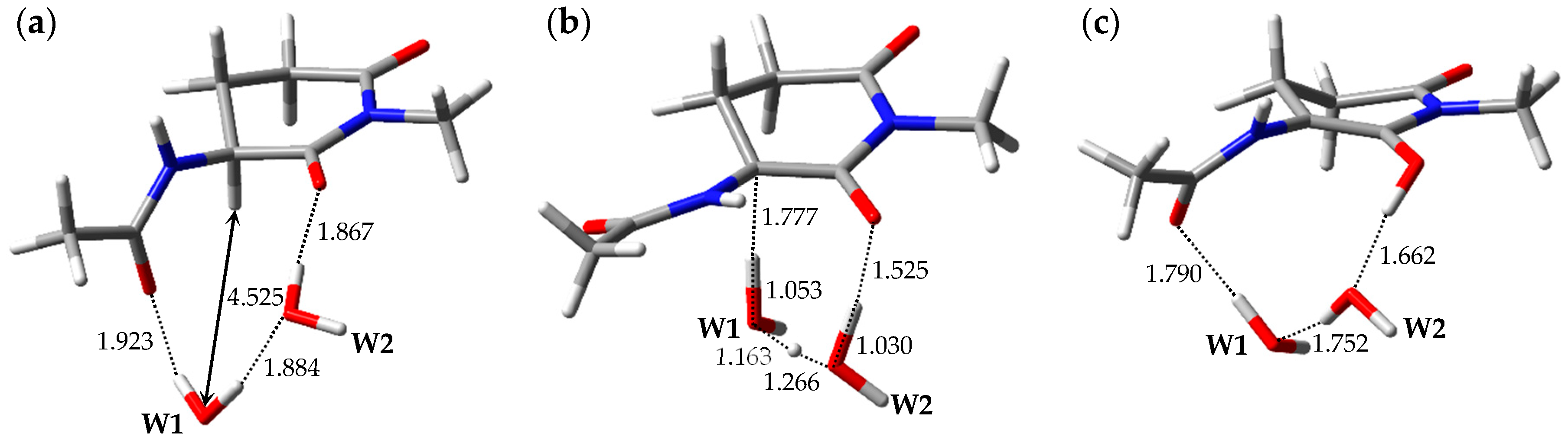
2.1. Pathway 1
2.2. Pathway 2
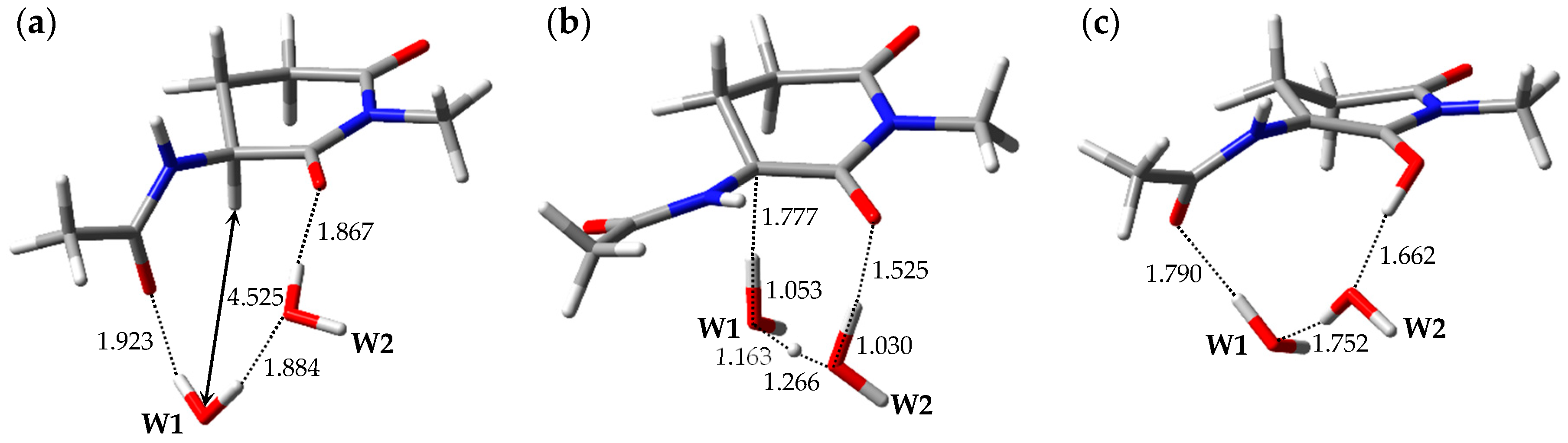
2.3. Pathway 3
2.4. Pathway 4
3. Discussion
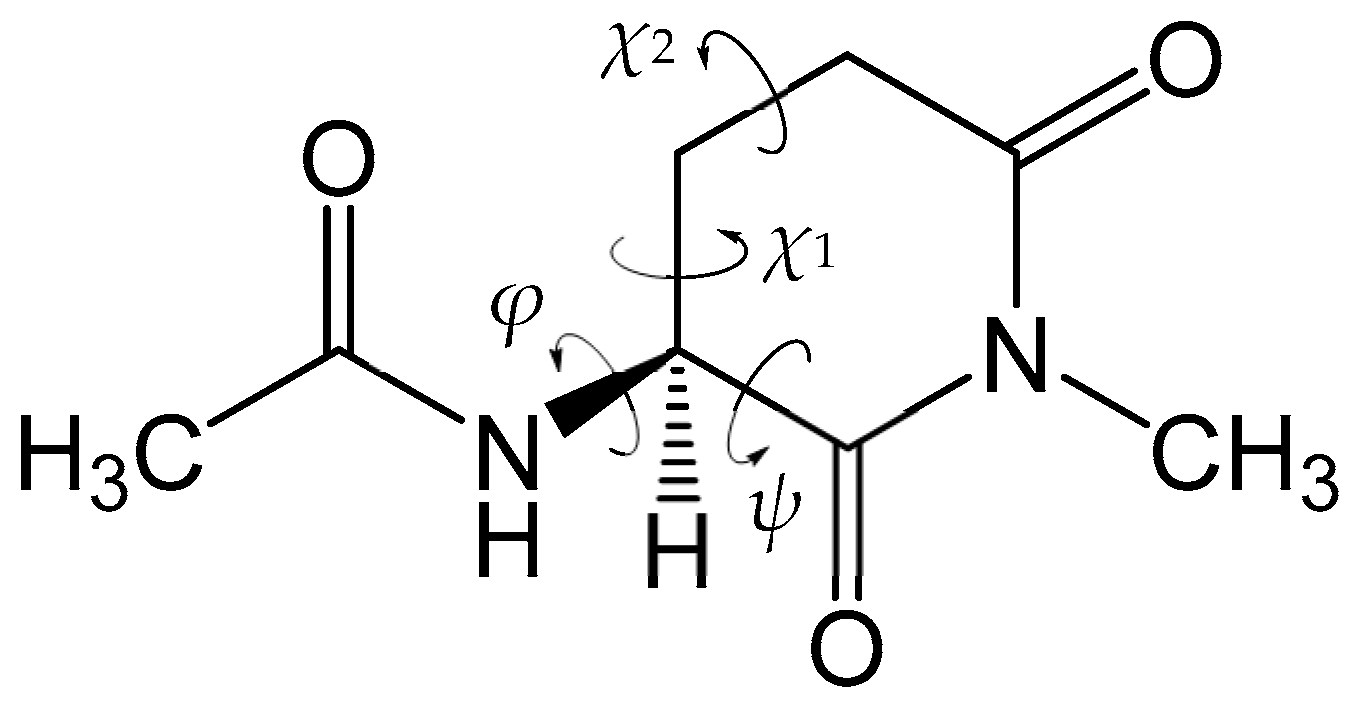
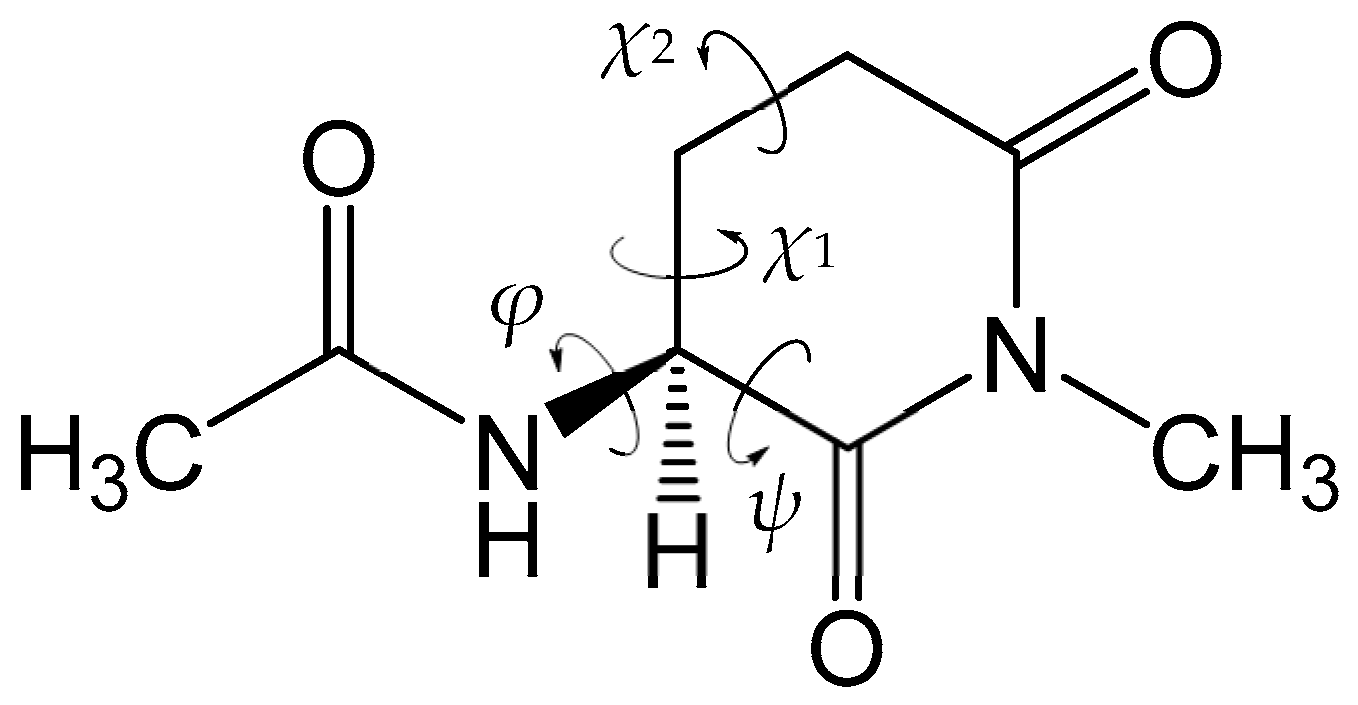
4. Material and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fujii, N.; Saito, T. Homochirality and life. Chem. Rec. 2004, 4, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N. D-Amino acids in living higher organisms. Orig. Life Evol. Biosph. 2002, 32, 103–127. [Google Scholar] [CrossRef] [PubMed]
- Ritz-Timme, S.; Collins, M.J. Racemization of aspartic acid in human proteins. Ageing Res. Rev. 2002, 1, 43–59. [Google Scholar] [CrossRef]
- Fujii, N.; Takata, T.; Fujii, N.; Aki, K.; Sakaue, H. D-Amino acids in protein: The mirror of life as a molecular index of aging. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 840–847. [Google Scholar] [CrossRef]
- Kubo, T.; Nishimura, S.; Kumagae, Y.; Kaneko, I. In vivo conversion of racemized β-amyloid ([d-Ser26]Aβ1–40) to truncated and toxic fragments ([d-Ser26]Aβ25–35/40) and fragment presence in the brains of Alzheimer’s patients. J. Neurosci. Res. 2002, 70, 474–483. [Google Scholar] [CrossRef]
- Shapira, R.; Chou, C.-H.J. Differential racemization of aspartate and serine in human myelin basic protein. Biochem. Biophys. Res. Commun. 1987, 146, 1342–1349. [Google Scholar] [CrossRef]
- Shapira, R.; Austin, G.E.; Mirra, S.S. Neuritic plaque amyloid in Alzheimer’s disease is highly racemized. J. Neurochem. 1988, 50, 69–74. [Google Scholar] [CrossRef]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.J.; Reardon, I.M.; Zürcher-Neely, H.A.; Heinrikson, R.L.; Ball, M.J. Structural alterations in the peptide backbone of β-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar]
- Fujii, N.; Satoh, K.; Harada, K.; Ishibashi, Y. Simultaneous stereoinversion and isomerization at specific aspartic acid residues in αA-crystallin from human lens. J. Biochem. 1994, 116, 663–669. [Google Scholar] [CrossRef]
- Fujii, N.; Ishibashi, Y.; Satoh, K.; Fujino, M.; Harada, K. Simultaneous racemization and isomerization at specific aspartic acid residues in αB-crystallin from the aged human lens. Biochim. Biophys. Acta Struct. Mol. Enzym. 1994, 1204, 157–163. [Google Scholar] [CrossRef]
- Kaji, Y.; Oshika, T.; Takazawa, Y.; Fukayama, M.; Takata, T.; Fujii, N. Localization of D-β-aspartic acid–containing proteins in human eyes. Invest. Opthalmol. Vis. Sci. 2007, 48, 3923–3927. [Google Scholar] [CrossRef]
- Hooi, M.Y.S.; Truscott, R.J.W. Racemisation and human cataract. d-Ser, d-Asp/Asn and d-Thr are higher in the lifelong proteins of cataract lenses than in age-matched normal lenses. AGE 2011, 33, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Hosaka, D.; Mochizuki, N.; Akatsu, H.; Tsutsumiuchi, K.; Hashizume, Y.; Matsukawa, N.; Yamamoto, T.; Toyo’oka, T. Simultaneous determination of post-translational racemization and isomerization of N-terminal amyloid-β in Alzheimer’s brain tissues by covalent chiral derivatized ultraperformance liquid chromatography tandem mass spectrometry. Anal. Chem. 2014, 86, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.T.; Vine, N.; Crossman, M. On the accumulation of d-aspartate in elastin and other proteins of the ageing aorta. Atherosclerosis 1992, 97, 201–208. [Google Scholar] [CrossRef]
- Ritz-Timme, S.; Laumeier, I.; Collins, M. Age estimation based on aspartic acid racemization in elastin from the yellow ligaments. Int. J. Legal Med. 2003, 117, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Ritz-Timme, S.; Laumeier, I.; Collins, M.J. Aspartic acid racemization: Evidence for marked longevity of elastin in human skin. Brit. J. Derm. 2003, 149, 951–959. [Google Scholar] [CrossRef]
- Fujii, N.; Fujii, N.; Kida, M.; Kinouchi, T. Influence of Lβ-, Dα- and Dβ-Asp isomers of the Asp-76 residue on the properties of αA-crystallin 70–88 peptide. Amino Acids 2010, 39, 1393–1399. [Google Scholar] [CrossRef] [PubMed]
- Oda, A.; Kobayashi, K.; Takahashi, O. Molecular-dynamics simulations for amyloid β1–42 Monomer with D-aspartic acid residues using continuous solvent. Chem. Biodivers. 2010, 7, 1357–1363. [Google Scholar] [CrossRef]
- Sakaue, H.; Kinouchi, T.; Fujii, N.; Takata, T.; Fujii, N. Isomeric replacement of a single aspartic acid induces a marked change in protein function: The example of ribonuclease A. ACS Omega 2017, 2, 260–267. [Google Scholar] [CrossRef]
- Radkiewicz, J.L.; Zipse, H.; Clarke, S.; Houk, K.N. Accelerated racemization of aspartic acid and asparagine residues via succinimide intermediates: an ab initio theoretical exploration of mechanism. J. Am. Chem. Soc. 1996, 118, 9148–9155. [Google Scholar] [CrossRef]
- Kuge, K.; Fujii, N.; Miura, Y.; Tajima, S.; Saito, T. Kinetic study of racemization of aspartyl residues in synthetic elastin peptides. Amino Acids 2004, 27, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Catak, S.; Monard, G.; Aviyente, V.; Ruiz-López, M.F. Deamidation of asparagine residues: Direct hydrolysis versus succinimide-mediated deamidation mechanisms. J. Phys. Chem. A 2009, 113, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, O.; Kobayashi, K.; Oda, A. Modeling the enolization of succinimide derivatives, a key step of racemization of aspartic acid residues: Importance of a two-H2O mechanism. Chem. Biodivers. 2010, 7, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, O. Two-water-assisted racemization of the succinimide intermediate formed in proteins. A computational model study. Health 2013, 5, 2018–2021. [Google Scholar] [CrossRef]
- Takahashi, O.; Oda, A. Amide-iminol tautomerization of the C-terminal peptide groups of aspartic acid residues: Two-water-assisted mechanism, cyclization from the iminol tautomer leading to the tetrahedral intermediate of succinimide formation, and implication to peptide group hydrogen exchange. In Tyrosine and Aspartic Acid: Properties, Source and Health Benefits; Jones, J.E., Morano, D.M., Eds.; Nova Science Publishers: New York, NY, USA, 2012; pp. 131–147. [Google Scholar]
- Takahashi, O.; Kirikoshi, R. Intramolecular cyclization of aspartic acid residues assisted by three water molecules: A density functional theory study. Comput. Sci. Disc. 2014, 7. Number 1. [Google Scholar] [CrossRef]
- Takahashi, O.; Kirikoshi, R.; Manabe, N. Roles of intramolecular and intermolecular hydrogen bonding in a three-water-assisted mechanism of succinimide formation from aspartic acid residues. Molecules 2014, 19, 11440–11452. [Google Scholar] [CrossRef] [PubMed]
- Fukuyoshi, S.; Nakayoshi, T.; Takahashi, O.; Oda, A. Theoretical study on keto-enol tautomerisation of glutarimide for exploration of the isomerization reaction pathway of glutamic acid in proteins using density functional theory. Mol. Phys. 2017, 115, 560–565. [Google Scholar] [CrossRef]
- Nakayoshi, T.; Kato, K.; Fukuyoshi, S.; Takahashi, O.; Kurimoto, E.; Oda, A. Computational studies on cyclic imide formation mechanism of glutamic acid residues catalyzed by two water molecules. J. Phys.: Conf. Ser. 2018, 1136, 012020. [Google Scholar] [CrossRef]
- Nakayoshi, T.; Kato, K.; Fukuyoshi, S.; Takahashi, O.; Kurimoto, E.; Oda, A. Computational studies on the water-catalyzed stereoinversion mechanism of glutamic acid residues in peptides and proteins. Chirality 2018, 30, 527–535. [Google Scholar] [CrossRef]
- Foresman, J.B.; Keith, T.A.; Wiberg, K.B.; Snoonian, J.; Frisch, M.J. Solvent effects. 5. Influence of cavity shape, truncation of electrostatics, and electron correlation on ab initio reaction field calculations. J. Phys. Chem. 1996, 100, 16098–16104. [Google Scholar] [CrossRef]
- Takenaka, N.; Koyano, Y.; Nakagawa, Y.; Nagaoka, M. An optimum strategy for solution chemistry using semiempirical molecular orbital method: Importance of description of charge distribution. J. Comput. Chem. 2009, 31, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03. Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Fukuyoshi, S.; Nakayoshi, T.; Takahashi, O.; Oda, A. Evaluations of density functionals and solvent effect for racemization reaction of amino acid residues. (manuscript in preparation).
- Kato, K.; Nakayoshi, T.; Kurimoto, E.; Oda, A. Computational studies on the nonenzymatic deamidation mechanisms of glutamine residues. ACS Omega 2019, 4, 3508–3513. [Google Scholar] [CrossRef]
- Nakayoshi, T.; Kato, K.; Kurimoto, E.; Oda, A. Possible mechanisms of nonenzymatic formation of dehydroalanine residue catalyzed by dihydrogen phosphate ion. J. Phys. Chem. B 2019, 123, 3147–3155. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Geometry | Relative Energy |
|---|---|---|
| 1 | 1-RC | 0.641 |
| 1-TS | 35.7 | |
| 1-PC | 21.0 | |
| 2 | 2-RC | 0.655 |
| 2-TS | 36.1 | |
| 2-PC | 20.9 | |
| 3 | 3-RC | 1.53 |
| 3-TS | 35.6 | |
| 3-PC | 18.8 | |
| 4 | 4-RC | 0 |
| 4-TS | 35.8 | |
| 4-PC | 18.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakayoshi, T.; Fukuyoshi, S.; Kato, K.; Kurimoto, E.; Oda, A. Computational Studies on Water-Catalyzed Mechanisms for Stereoinversion of Glutarimide Intermediates Formed from Glutamic Acid Residues in Aqueous Phase. Int. J. Mol. Sci. 2019, 20, 2410. https://doi.org/10.3390/ijms20102410
Nakayoshi T, Fukuyoshi S, Kato K, Kurimoto E, Oda A. Computational Studies on Water-Catalyzed Mechanisms for Stereoinversion of Glutarimide Intermediates Formed from Glutamic Acid Residues in Aqueous Phase. International Journal of Molecular Sciences. 2019; 20(10):2410. https://doi.org/10.3390/ijms20102410
Chicago/Turabian StyleNakayoshi, Tomoki, Shuichi Fukuyoshi, Koichi Kato, Eiji Kurimoto, and Akifumi Oda. 2019. "Computational Studies on Water-Catalyzed Mechanisms for Stereoinversion of Glutarimide Intermediates Formed from Glutamic Acid Residues in Aqueous Phase" International Journal of Molecular Sciences 20, no. 10: 2410. https://doi.org/10.3390/ijms20102410
APA StyleNakayoshi, T., Fukuyoshi, S., Kato, K., Kurimoto, E., & Oda, A. (2019). Computational Studies on Water-Catalyzed Mechanisms for Stereoinversion of Glutarimide Intermediates Formed from Glutamic Acid Residues in Aqueous Phase. International Journal of Molecular Sciences, 20(10), 2410. https://doi.org/10.3390/ijms20102410