Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD)

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Age-Related Macular Degeneration—An Eye Disease with the Critical Role of ROS in Its Pathogenesis
3. Mitochondria—A Central Structure in AMD Pathogenesis
4. Generation and Regulation of ROS by Mitochondria
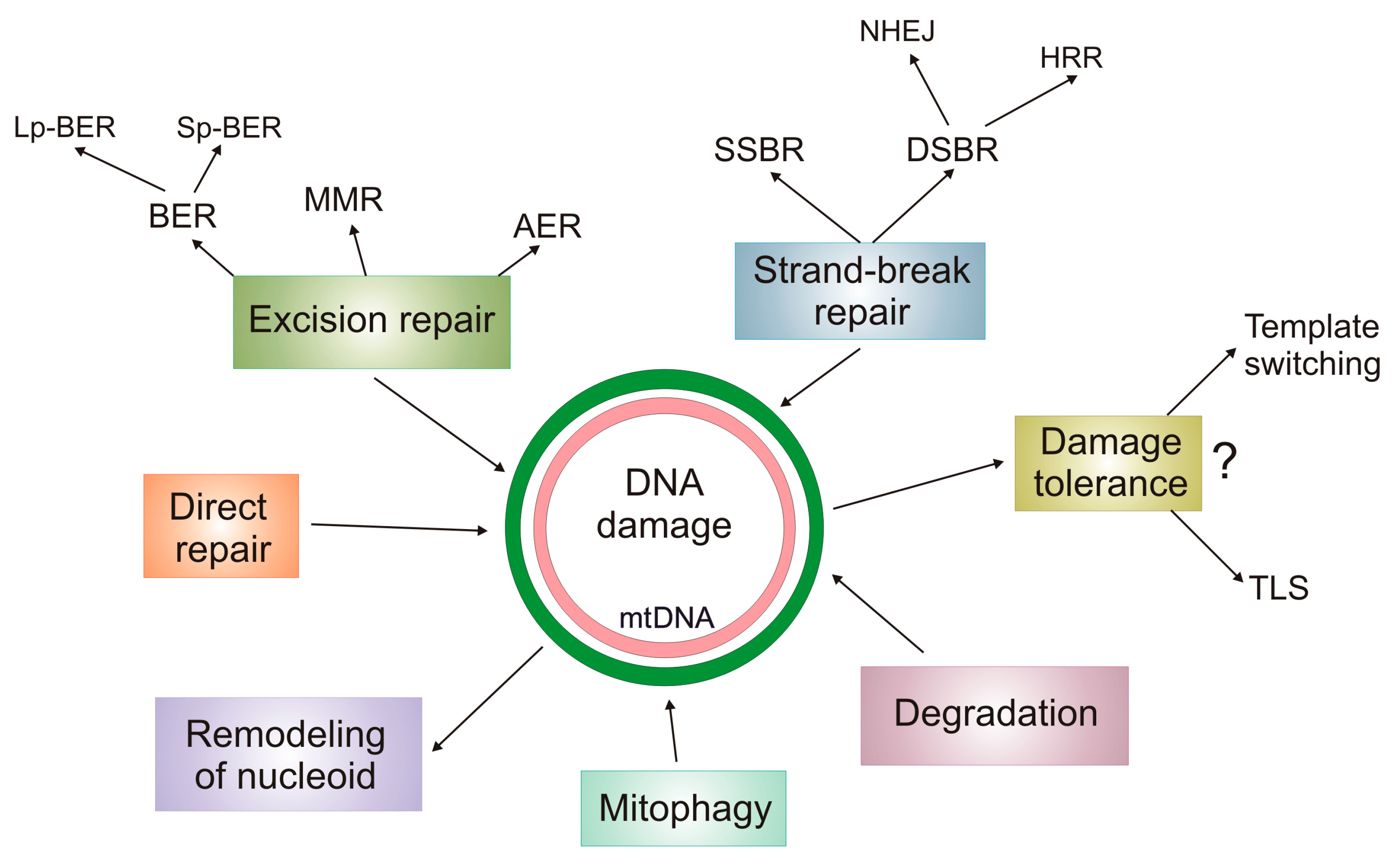
5. DNA Damage Response in mtDNA
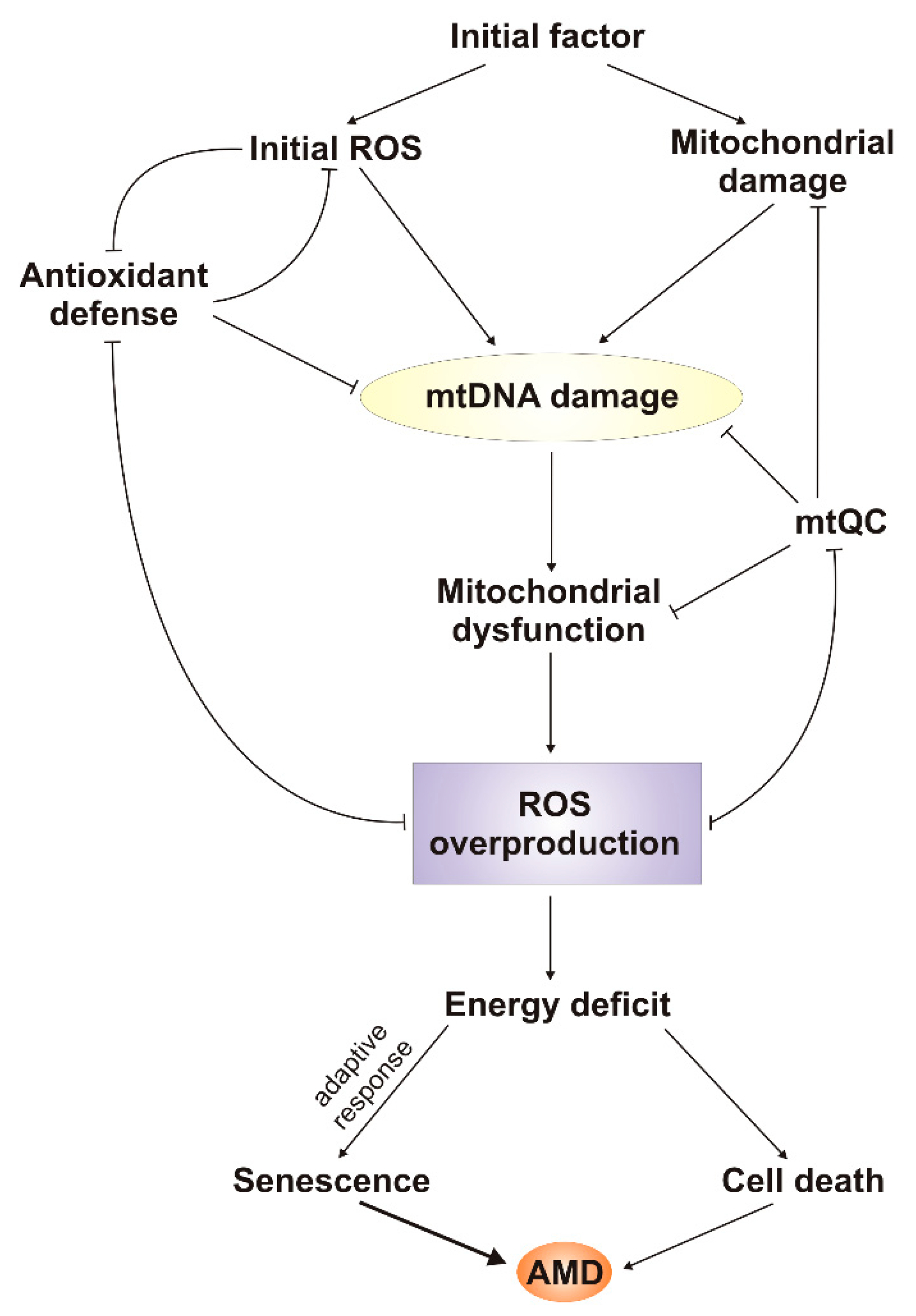
6. mtDNA Damage and Repair in AMD
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; Arca, D.; Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.; Whiteman, M. Measurement of Reactive Oxygen Species in Cells and Mitochondria. In Methods in Cell Biology; Elsevier BV: Amsterdam, The Netherlands, 2007; Volume 80, pp. 355–377. [Google Scholar]
- Hiona, A.; Leeuwenburgh, C. The role of mitochondrial DNA mutations in aging and sarcopenia: Implications for the mitochondrial vicious cycle theory of aging. Exp. Gerontol. 2008, 43, 24–33. [Google Scholar] [CrossRef]
- Kim, J.-S.; Jeong, S.-H.; Han, S.-H.; Yi, H.-K. Gomisin A modulates aging progress via mitochondrial biogenesis in human diploid fibroblast cells. Clin. Exp. Pharmacol. Physiol. 2018, 45, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free. Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef] [PubMed]
- Brand, R.M.; Wipf, P.; Durham, A.; Epperly, M.W.; Greenberger, J.S.; Falo, L.D.J. Targeting Mitochondrial Oxidative Stress to Mitigate UV-Induced Skin Damage. Front. Pharmacol. 2018, 9, 920. [Google Scholar] [CrossRef]
- Sanchez, M.C.; Lancel, S.; Boulanger, E.; Neviere, R. Targeting Oxidative Stress and Mitochondrial Dysfunction in the Treatment of Impaired Wound Healing: A Systematic Review. Antioxidants 2018, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.-H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Pottosin, I.; Dobrovinskaya, O. Mitochondria as emerging targets for therapies against T cell acute lymphoblastic leukemia. J. Leukoc. Biol. 2019, 105, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (AMD): Associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. 2016, 3, 34. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.-Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Gehrs, K.M.; Anderson, D.H.; Johnson, L.V.; Hageman, G.S. Age-related macular degeneration—emerging pathogenetic and therapeutic concepts. Ann. Med. 2006, 38, 450–471. [Google Scholar] [CrossRef]
- Klein, R.; Peto, T.; Bird, A.; VanNewkirk, M.R. The epidemiology of age-related macular degeneration. Am. J. Ophthalmol. 2004, 137, 486–495. [Google Scholar] [CrossRef]
- Bonastre, J.; Le Pen, C.; Soubrane, G.; Quentel, G. The burden of age-related macular degeneration: Results of a cohort study in two French referral centres. PharmacoEconomics 2003, 21, 181–190. [Google Scholar] [CrossRef]
- Van Leeuwen, R.; Klaver, C.C.; Vingerling, J.R.; Hofman, A.; De Jong, P.T. REVIEW: Epidemiology of age-related maculopathy: A review. Eur. J. Epidemiol. 2003, 18, 845–854. [Google Scholar] [CrossRef]
- Malek, G.; Busik, J.; Grant, M.B.; Choudhary, M. Models of retinal diseases and their applicability in drug discovery. Expert Opin. Drug Discov. 2018, 13, 359–377. [Google Scholar] [CrossRef]
- Joachim, N.; Mitchell, P.; Rochtchina, E.; Tan, A.G.; Wang, J.J. Incidence and progression of reticular drusen in age-related macular degeneration: Findings from an older Australian cohort. Ophthalmology 2014, 121, 917–925. [Google Scholar] [CrossRef]
- Guo, M.Y.; Cheng, J.; Etminan, M.; Zafari, Z.; Maberley, D. One year effectiveness study of intravitreal aflibercept in neovascular age-related macular degeneration: A meta-analysis. Acta Ophthalmol. 2018, 97, e1–e7. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Fuhrmann, S.; Zou, C.; Levine, E.M. Retinal pigment epithelium development, plasticity, and tissue homeostasis. Exp. Eye 2014, 123, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An expanded view of complex traits: From polygenic to omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef]
- Sobrin, L.; Seddon, J.M. Nature and nurture- genes and environment- predict onset and progression of macular degeneration. Prog. Retin. Eye Res. 2014, 40, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Hoffmann, T.J.; Melles, R.B.; Sakoda, L.C.; Kvale, M.N.; Banda, Y.; Schaefer, C.; Risch, N.; Jorgenson, E. Differences in the Genetic Susceptibility to Age-Related Macular Degeneration Clinical Subtypes. Investig. Opthalmol. Sci. 2015, 56, 4290–4299. [Google Scholar] [CrossRef][Green Version]
- Ersoy, L.; Ristau, T.; Hahn, M.; Karlstetter, M.; Langmann, T.; Caramoy, A.; Hollander, A.I.D.; Fauser, S.; Dröge, K. Genetic and Environmental Risk Factors for Age-Related Macular Degeneration in Persons 90 Years and Older. Investig. Opthalmol. Sci. 2014, 55, 1842–1847. [Google Scholar] [CrossRef]
- Curcio, C.A.; Messinger, J.D.; Sloan, K.R.; McGwin, G.; Medeiros, N.E.; Spaide, R.F. Subretinal Drusenoid Deposits in Non-Neovascular Age-Related Macular Degeneration: Morphology, Prevalence, Topography, And Biogenesis Model. Retina 2013, 33, 265–276. [Google Scholar] [CrossRef]
- Spaide, R.F.; Ooto, S.; Curcio, C.A. Subretinal drusenoid deposits AKA pseudodrusen. Surv. Ophthalmol. 2018, 63, 782–815. [Google Scholar] [CrossRef]
- Zarbin, M.A. Current Concepts in the Pathogenesis of Age-Related Macular Degeneration. Arch. Ophthalmol. 2004, 122, 598. [Google Scholar] [CrossRef]
- Sachdeva, M.M.; Cano, M.; Handa, J.T. Nrf2 signaling is impaired in the aging RPE given an oxidative insult. Exp. Eye 2014, 119, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Seo, S.J.; Biswal, M.R.; Li, H.; Conners, M.; Nandyala, A.; Jones, K.; Le, Y.-Z.; Lewin, A.S. Mitochondrial Oxidative Stress in the Retinal Pigment Epithelium Leads to Localized Retinal Degeneration. Investig. Opthalmol. Sci. 2014, 55, 4613–4627. [Google Scholar] [CrossRef] [PubMed]
- Lambros, M.L.; Plafker, S.M. Oxidative Stress and the Nrf2 Anti-Oxidant Transcription Factor in Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2016, 854, 67–72. [Google Scholar] [PubMed]
- Jarrett, S.G.; Boulton, M.E. Consequences of oxidative stress in age-related macular degeneration. Mol. Asp. Med. 2012, 33, 399–417. [Google Scholar] [CrossRef]
- Blasiak, J.; Piechota, M.; Pawlowska, E.; Szatkowska, M.; Sikora, E.; Kaarniranta, K. Cellular Senescence in Age-Related Macular Degeneration: Can Autophagy and DNA Damage Response Play a Role? Oxid. Med. Cell. Longev. 2017, 2017, 5293258. [Google Scholar] [CrossRef]
- Dieguez, H.H.; Romeo, H.E.; Alaimo, A.; Fleitas, M.F.G.; Aranda, M.L.; Rosenstein, R.E.; Dorfman, D. Oxidative stress damage circumscribed to the central temporal retinal pigment epithelium in early experimental non-exudative age-related macular degeneration. Radic. Biol. Med. 2018, 131, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yasumura, D.; Li, X.; Matthes, M.; Lloyd, M.; Nielsen, G.; Ahern, K.; Snyder, M.; Bok, D.; Dunaief, J.L.; et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J. Clin. Investig. 2011, 121, 369–383. [Google Scholar] [CrossRef]
- Fehér, J.; Kovács, I.; Artico, M.; Cavallotti, C.; Papale, A.; Gabrieli, C.B. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 2006, 27, 983–993. [Google Scholar] [CrossRef]
- Nordgaard, C.L.; Berg, K.M.; Kapphahn, R.J.; Reilly, C.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Proteomics of the Retinal Pigment Epithelium Reveals Altered Protein Expression at Progressive Stages of Age-Related Macular Degeneration. Investig. Opthalmol. Sci. 2006, 47, 815–822. [Google Scholar] [CrossRef]
- Nordgaard, C.L.; Karunadharma, P.P.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Investig. Opthalmol. Sci. 2008, 49, 2848–2855. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef]
- Iacovelli, J.; Rowe, G.C.; Khadka, A.; Diaz-Aguilar, D.; Spencer, C.; Arany, Z.; Saint-Geniez, M. PGC-1alpha Induces Human RPE Oxidative Metabolism and Antioxidant Capacity. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1038–1051. [Google Scholar] [CrossRef]
- Golestaneh, N.; Chu, Y.; Xiao, Y.-Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death 2017, 8, e2537. [Google Scholar] [CrossRef]
- Starkov, A.A. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef]
- Dunn, J.D.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Nanayakkara, G.K.; Wang, H.; Yang, X. Proton leak regulates mitochondrial reactive oxygen species generation in endothelial cell activation and inflammation—A novel concept. Arch. Biochem. Biophys. 2018, 662, 68–74. [Google Scholar] [CrossRef]
- Cline, S.D. Mitochondrial DNA Damage and its Consequences for Mitochondrial Gene Expression. Biochim. Biophys. Acta 2012, 1819, 979–991. [Google Scholar] [CrossRef]
- Pinto, M.; Moraes, C.T. Mechanisms linking mtDNA damage and aging. Radic. Biol. Med. 2015, 85, 250–258. [Google Scholar] [CrossRef]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Persistent damage induces mitochondrial DNA degradation. DNA Repair 2013, 12, 488–499. [Google Scholar] [CrossRef]
- Murphy, M.P. Modulating mitochondrial intracellular location as a redox signal. Sci. Signal. 2012, 5, pe39. [Google Scholar] [CrossRef]
- Caston, R.A.; Demple, B. Risky repair: DNA-protein crosslinks formed by mitochondrial base excision DNA repair enzymes acting on free radical lesions. Radic. Biol. Med. 2017, 107, 146–150. [Google Scholar] [CrossRef]
- Sigurðardóttir, S.; Helgason, A.; Gulcher, J.R.; Stefansson, K.; Donnelly, P. The Mutation Rate in the Human mtDNA Control Region. Am. J. Hum. Genet. 2000, 66, 1599–1609. [Google Scholar] [CrossRef]
- Phillips, A.F.; Millet, A.R.; Tigano, M.; Dubois, S.M.; Crimmins, H.; Babin, L.; Charpentier, M.; Piganeau, M.; Brunet, E.; Sfeir, A. Single-Molecule Analysis of mtDNA Replication Uncovers the Basis of the Common Deletion. Mol. Cell 2017, 65, 527–538. [Google Scholar] [CrossRef]
- Russell, O.; Turnbull, D. Mitochondrial DNA disease—Molecular insights and potential routes to a cure. Exp. Cell 2014, 325, 38–43. [Google Scholar] [CrossRef]
- Stefano, G.B.; Bjenning, C.; Wang, F.; Wang, N.; Kream, R.M. Mitochondrial Heteroplasmy. Adv. Exp. Med. Biol. 2017, 982, 577–594. [Google Scholar]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 226, 274–286. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. Endogenous oxidative damage of mtDNA. Res. Mol. Mech. Mutagen. 1999, 424, 51–58. [Google Scholar] [CrossRef]
- Nakamura, J.; Swenberg, J.A. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 1999, 59, 2522–2526. [Google Scholar]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids 2009, 37, 2539–2548. [Google Scholar] [CrossRef]
- Vaishnav, R.A.; Singh, I.N.; Miller, D.M.; Hall, E.D. Lipid Peroxidation-Derived Reactive Aldehydes Directly and Differentially Impair Spinal Cord and Brain Mitochondrial Function. J. Neurotrauma 2010, 27, 1311–1320. [Google Scholar] [CrossRef]
- Henderson, P.T.; Delaney, J.C.; Gu, F.; Tannenbaum, S.R.; Essigmann, J.M. Oxidation of 7,8-Dihydro-8-oxoguanine Affords Lesions That Are Potent Sources of Replication Errors in Vivo†. Biochemistry 2002, 41, 914–921. [Google Scholar] [CrossRef]
- Henderson, P.T.; Delaney, J.C.; Muller, J.G.; Neeley, W.L.; Tannenbaum, S.R.; Burrows, C.J.; Essigmann, J.M. The Hydantoin Lesions Formed from Oxidation of 7,8-Dihydro-8-oxoguanine Are Potent Sources of Replication Errors in Vivo†. Biochemistry 2003, 42, 9257–9262. [Google Scholar] [CrossRef]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The Maintenance of Mitochondrial DNA Integrity—Critical Analysis and Update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef]
- Vasileiou, P.V.S.; Mourouzis, I.; Pantos, C. Principal Aspects Regarding the Maintenance of Mammalian Mitochondrial Genome Integrity. Int. J. Mol. Sci. 2017, 18, 1821. [Google Scholar] [CrossRef]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2016, 68, 20–48. [Google Scholar] [CrossRef]
- Plesca, D.; Mazumder, S.; Almasan, A. DNA damage response and apoptosis. Methods Enzymol. 2008, 446, 107–122. [Google Scholar]
- Chocron, E.S.; Munkacsy, E.; Pickering, A.M. Cause or casualty: The role of mitochondrial DNA in aging and age-associated disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1865, 285–297. [Google Scholar] [CrossRef]
- Jezek, P.; Spacek, T.; Tauber, J.; Pavluch, V. Mitochondrial Nucleoids: Superresolution microscopy analysis. Int. J. Biochem. Cell Biol. 2018, 106, 21–25. [Google Scholar] [CrossRef]
- Pohjoismäki, J.L.O.; Forslund, J.M.E.; Goffart, S.; Torregrosa-Muñumer, R.; Wanrooij, S. Known Unknowns of Mammalian Mitochondrial DNA Maintenance. BioEssays 2018, 40, 1800102. [Google Scholar] [CrossRef]
- Torregrosa-Muñumer, R.; Forslund, J.M.E.; Goffart, S.; Pfeiffer, A.; Stojkovič, G.; Carvalho, G.; Al-Furoukh, N.; Blanco, L.; Wanrooij, S.; Pohjoismäki, J.L.O. PrimPol is required for replication reinitiation after mtDNA damage. Proc. Natl. Acad. Sci. USA 2017, 114, 11398–11403. [Google Scholar] [CrossRef]
- Medeiros, T.C.; Thomas, R.L.; Ghillebert, R.; Graef, M. Autophagy balances mtDNA synthesis and degradation by DNA polymerase POLG during starvation. J. Cell Biol. 2018, 217, 1601–1611. [Google Scholar] [CrossRef]
- Nogueira, C.; Almeida, L.S.; Nesti, C.; Pezzini, I.; Videira, A.; Vilarinho, L.; Santorelli, F.M. Syndromes associated with mitochondrial DNA depletion. Ital. J. Pediatr. 2014, 40, 34. [Google Scholar] [CrossRef]
- Aamann, M.D.; Sorensen, M.M.; Hvitby, C.; Berquist, B.R.; Muftuoglu, M.; Tian, J.; de Souza-Pinto, N.C.; Scheibye-Knudsen, M.; Wilson, D.M., 3rd; Stevnsner, T.; et al. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J. 2010, 24, 2334–2346. [Google Scholar] [CrossRef]
- Kamenisch, Y.; Fousteri, M.; Knoch, J.; Von Thaler, A.-K.; Fehrenbacher, B.; Kato, H.; Becker, T.; Dollé, M.E.; Kuiper, R.; Majora, M.; et al. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J. Exp. Med. 2010, 207, 379–390. [Google Scholar] [CrossRef]
- Prakash, A.; Doublié, S. Base Excision Repair in the Mitochondria. J. Cell. Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef]
- Yasuhira, S.; Yasui, A. Alternative Excision Repair Pathway of UV-damaged DNA in Schizosaccharomyces pombe Operates Both in Nucleus and in Mitochondria. J. Biol. Chem. 2000, 275, 11824–11828. [Google Scholar] [CrossRef]
- Dahal, S.; Dubey, S.; Raghavan, S.C. Homologous recombination-mediated repair of DNA double-strand breaks operates in mammalian mitochondria. Cell. Mol. Life Sci. 2018, 75, 1641–1655. [Google Scholar] [CrossRef]
- Tadi, S.K.; Sebastian, R.; Dahal, S.; Babu, R.K.; Choudhary, B.; Raghavan, S.C.; Cohen-Fix, O. Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions. Mol. Biol. Cell 2016, 27, 223–235. [Google Scholar] [CrossRef]
- Liu, P.; Qian, L.; Sung, J.-S.; De Souza-Pinto, N.C.; Zheng, L.; Bogenhagen, D.F.; Bohr, V.A.; Wilson, D.M.; Shen, B.; Demple, B. Removal of Oxidative DNA Damage via FEN1-Dependent Long-Patch Base Excision Repair in Human Cell Mitochondria. Mol. Cell. Biol. 2008, 28, 4975–4987. [Google Scholar] [CrossRef]
- Leung, W.; Baxley, R.M.; Moldovan, G.-L.; Bielinsky, A.-K. Mechanisms of DNA Damage Tolerance: Post-Translational Regulation of PCNA. Genes 2018, 10, 10. [Google Scholar] [CrossRef]
- Graziewicz, M.A.; Longley, M.J.; Copeland, W.C. DNA polymerase gamma in mitochondrial DNA replication and repair. Chem. Rev. 2006, 106, 383–405. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Samuels, D.C.; Elson, J.; Turnbull, D.M. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: Is there a common mechanism? Lancet 2002, 360, 1323–1325. [Google Scholar] [CrossRef]
- Barreau, E.; Brossas, J.Y.; Courtois, Y.; Tréton, J.A. Accumulation of mitochondrial DNA deletions in human retina during aging. Investig. Ophthalmol. Sci. 1996, 37, 384–391. [Google Scholar]
- Jones, M.M.; Manwaring, N.; Wang, J.J.; Rochtchina, E.; Mitchell, P.; Sue, C.M. Mitochondrial DNA Haplogroups and Age-Related Maculopathy. Arch. Ophthalmol. 2007, 125, 1235–1240. [Google Scholar] [CrossRef]
- Joachim, N.; Mitchell, P.; Burlutsky, G.; Kifley, A.; Wang, J.J. The Incidence and Progression of Age-Related Macular Degeneration over 15 Years: The Blue Mountains Eye Study. Ophthalmology 2015, 122, 2482–2489. [Google Scholar] [CrossRef]
- Udar, N.; Atilano, S.R.; Memarzadeh, M.; Boyer, D.S.; Chwa, M.; Lu, S.; Maguen, B.; Langberg, J.; Coskun, P.; Wallace, D.C.; et al. Mitochondrial DNA Haplogroups Associated with Age-Related Macular Degeneration. Investig. Opthalmol. Sci. 2009, 50, 2966–2974. [Google Scholar] [CrossRef]
- Canter, J.A.; Olson, L.M.; Spencer, K.; Schnetz-Boutaud, N.; Anderson, B.; Hauser, M.A.; Schmidt, S.; Postel, E.A.; Agarwal, A.; Pericak-Vance, M.A.; et al. Mitochondrial DNA polymorphism A4917G is independently associated with age-related macular degeneration. PLoS ONE 2008, 3, e2091. [Google Scholar] [CrossRef]
- SanGiovanni, J.P.; Arking, D.E.; Iyengar, S.K.; Elashoff, M.; Clemons, T.E.; Reed, G.F.; Henning, A.K.; Sivakumaran, T.A.; Xu, X.; DeWan, A.; et al. Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PLoS ONE 2009, 4, e5508. [Google Scholar] [CrossRef]
- Kenney, M.C.; Chwa, M.; Falatoonzadeh, P.; Ramirez, C.; Malik, D.; Tarek, M.; Cáceres-Del-Carpio, J.; Vawter, M.; Jazwinski, S.M.; Miceli, M.; et al. Inherited mitochondrial DNA variants can affect complement, inflammation and apoptosis pathways: Insights into mitochondrial-nuclear interactions. Hum. Mol. Genet. 2014, 23, 3537–3551. [Google Scholar] [CrossRef]
- Nashine, S.; Cohen, P.; Chwa, M.; Lu, S.; Nesburn, A.B.; Kuppermann, B.D.; Kenney, M.C. Humanin G (HNG) protects age-related macular degeneration (AMD) transmitochondrial ARPE-19 cybrids from mitochondrial and cellular damage. Cell Death 2017, 8, e2951. [Google Scholar] [CrossRef]
- Mueller, E.E.; Schaier, E.; Brunner, S.M.; Eder, W.; Mayr, J.A.; Egger, S.F.; Nischler, C.; Oberkofler, H.; Reitsamer, H.A.; Patsch, W.; et al. Mitochondrial haplogroups and control region polymorphisms in age-related macular degeneration: A case-control study. PLoS ONE 2012, 7, e30874. [Google Scholar] [CrossRef]
- Tilleul, J.; Richard, F.; Puche, N.; Zerbib, J.; Leveziel, N.; Sahel, J.A.; Cohen, S.Y.; Korobelnik, J.-F.; Feingold, J.; Munnich, A.; et al. Genetic association study of mitochondrial polymorphisms in neovascular age-related macular degeneration. Mol. Vis. 2013, 19, 1132–1140. [Google Scholar]
- Ballinger, S.W.; Van Houten, B.; Conklin, C.A.; Jin, G.-F.; Godley, B.F. Hydrogen Peroxide Causes Significant Mitochondrial DNA Damage in Human RPE Cells. Exp. Eye 1999, 68, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Glowacki, S.; Kauppinen, A.; Kaarniranta, K. Mitochondrial and Nuclear DNA Damage and Repair in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2013, 14, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Karunadharma, P.P.; Nordgaard, C.L.; Olsen, T.W.; Ferrington, D.A. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Investig. Opthalmol. Sci. 2010, 51, 5470–5479. [Google Scholar] [CrossRef]
- Godley, B.F.; Shamsi, F.A.; Liang, F.-Q.; Jarrett, S.G.; Davies, S.; Boulton, M. Blue Light Induces Mitochondrial DNA Damage and Free Radical Production in Epithelial Cells. J. Biol. Chem. 2005, 280, 21061–21066. [Google Scholar] [CrossRef]
- Atilano, S.R.; Boyer, D.; Chwa, M.; Chak, G.; Chinichian, S.; Coskun, P.; Nesburn, A.B.; Udar, N.S.; Kenney, M.C.; Wallace, D.C. Characterization of Retinal and Blood Mitochondrial DNA from Age-Related Macular Degeneration Patients. Investig. Opthalmol. Sci. 2010, 51, 4289–4297. [Google Scholar]
- Rothfuss, O.; Gasser, T.; Patenge, N. Analysis of differential DNA damage in the mitochondrial genome employing a semi-long run real-time PCR approach. Nucleic Acids Res. 2010, 38, e24. [Google Scholar] [CrossRef]
- Lin, H.; Xu, H.; Liang, F.-Q.; Liang, H.; Gupta, P.; Havey, A.N.; Boulton, M.E.; Godley, B.F. Mitochondrial DNA Damage and Repair in RPE Associated with Aging and Age-Related Macular Degeneration. Investig. Opthalmol. Sci. 2011, 52, 3521–3529. [Google Scholar] [CrossRef]
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating Mitochondria as a Target for Treating Age-Related Macular Degeneration. J. Neurosci. 2015, 35, 7304–7311. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Neufeld, A.H. Increased mitochondrial DNA damage and down-regulation of DNA repair enzymes in aged rodent retinal pigment epithelium and choroid. Mol. Vis. 2008, 14, 644–651. [Google Scholar]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Neufeld, A.H. Age-related increase in mitochondrial DNA damage and loss of DNA repair capacity in the neural retina. Neurobiol. Aging 2010, 31, 2002–2010. [Google Scholar] [CrossRef] [PubMed]
- Miceli, M.V.; Jazwinski, S.M. Nuclear Gene Expression Changes Due to Mitochondrial Dysfunction in ARPE-19 Cells: Implications for Age-Related Macular Degeneration. Investig. Opthalmol. Sci. 2005, 46, 1765–1773. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Kajdanek, J.; Morawiec, J.; Pawlowska, E.; Blasiak, J. PGC-1alpha Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis. Int. J. Mol. Sci. 2018, 19, 2317. [Google Scholar] [CrossRef]
- Satish, S.; Philipose, H.; Rosales, M.A.B.; Saint-Geniez, M. Pharmaceutical Induction of PGC-1alpha Promotes Retinal Pigment Epithelial Cell Metabolism and Protects against Oxidative Damage. Oxid. Med. Cell. Longev. 2018, 2018, 9248640. [Google Scholar] [CrossRef]
- Salero, E.; Blenkinsop, T.A.; Corneo, B.; Harris, A.; Rabin, D.; Stern, J.H.; Temple, S. Adult Human RPE Can Be Activated into a Multipotent Stem Cell that Produces Mesenchymal Derivatives. Cell Stem Cell 2012, 10, 88–95. [Google Scholar] [CrossRef]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.T.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1alpha genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Zhang, M.; Chu, Y.; Mowery, J.; Konkel, B.; Galli, S.; Theos, A.C.; Golestaneh, N. Pgc-1alpha repression and high-fat diet induce age-related macular degeneration-like phenotypes in mice. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Saint-Geniez, M.; Jiang, A.; Abend, S.; Liu, L.; Sweigard, H.; Connor, K.M.; Arany, Z. PGC-1alpha regulates normal and pathological angiogenesis in the retina. Am. J. Pathol. 2013, 182, 255–265. [Google Scholar] [CrossRef]
- Golestaneh, N.; Chu, Y.; Cheng, S.K.; Cao, H.; Poliakov, E.; Berinstein, D.M. Repressed SIRT1/PGC-1alpha pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J. Transl. Med. 2016, 14, 344. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.-F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef]
- Lawrenson, J.G.; Evans, J.R. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst. Rev. 2017, 2017, CD000254. [Google Scholar]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaarniranta, K.; Pawlowska, E.; Szczepanska, J.; Jablkowska, A.; Blasiak, J. Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD). Int. J. Mol. Sci. 2019, 20, 2374. https://doi.org/10.3390/ijms20102374
Kaarniranta K, Pawlowska E, Szczepanska J, Jablkowska A, Blasiak J. Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD). International Journal of Molecular Sciences. 2019; 20(10):2374. https://doi.org/10.3390/ijms20102374
Chicago/Turabian StyleKaarniranta, Kai, Elzbieta Pawlowska, Joanna Szczepanska, Aleksandra Jablkowska, and Janusz Blasiak. 2019. "Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD)" International Journal of Molecular Sciences 20, no. 10: 2374. https://doi.org/10.3390/ijms20102374
APA StyleKaarniranta, K., Pawlowska, E., Szczepanska, J., Jablkowska, A., & Blasiak, J. (2019). Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD). International Journal of Molecular Sciences, 20(10), 2374. https://doi.org/10.3390/ijms20102374