Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the EXosomes in PArkiNson Disease (EXPAND) Study

 ,
,  ,
,  ,
,  ,
, 
Abstract
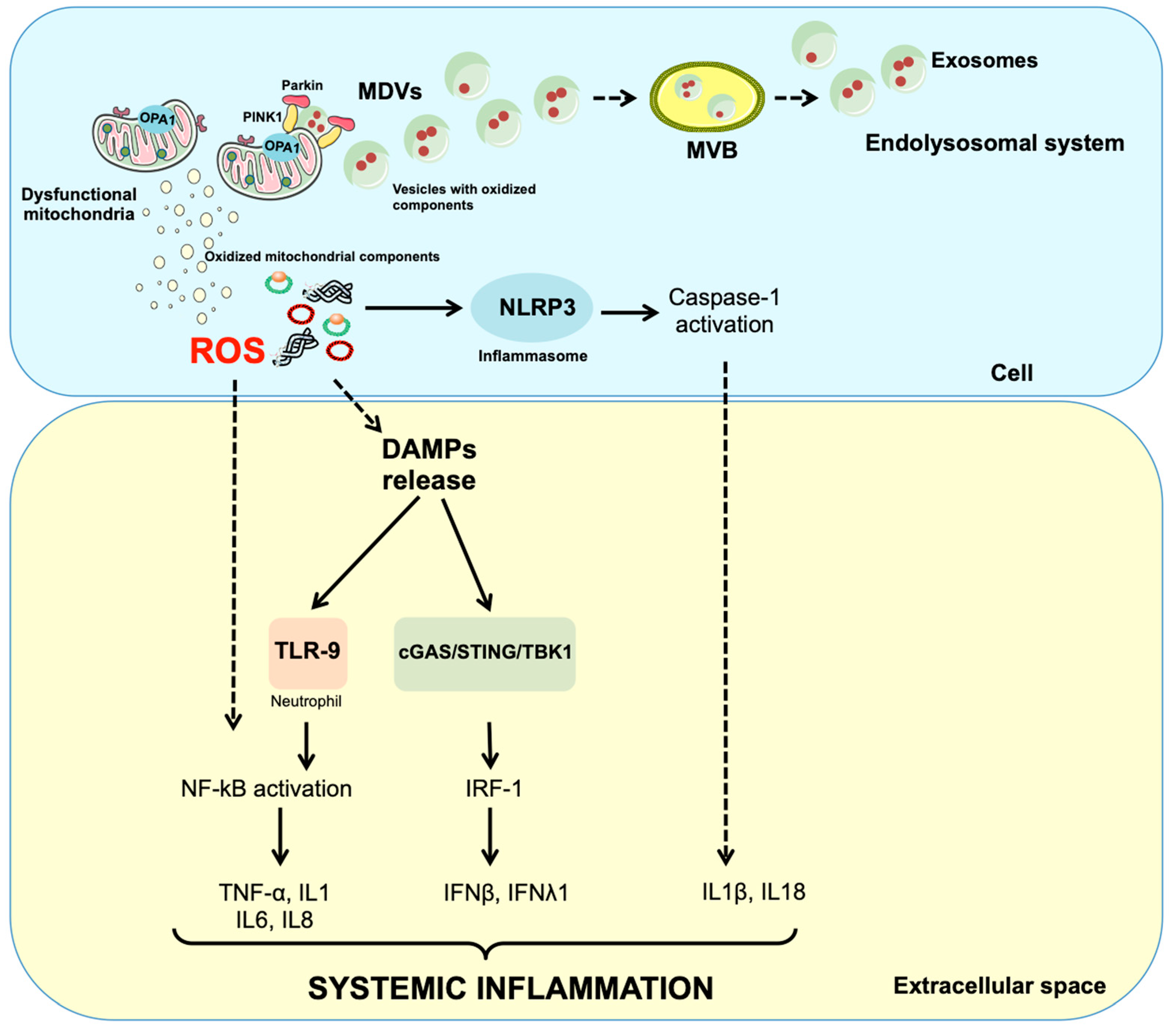
1. Introduction
2. Methods
2.1. Study Design and Population
2.2. Participant Recruitment and Assessment
2.3. Blood Sample Collection
2.4. Exosomes Isolation and Characterisation
2.4.1. Purification of Small Extracellular Vesicles/Exosomes by Differential Ultracentrifugation
2.4.2. Western Immunoblot Analysis of Small Extracellular Vesicles
2.4.3. Analysis of Mitochondrial DNA in Small Extracellular Vesicles
2.5. Determination of Inflammatory Mediators
2.6. Statistical Analysis
3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP5A | Adenosine triphosphate 5A |
| ADL | Activities of daily living |
| CCL | C-C motif chemokine ligand |
| cGAS | Cyclic GMP-AMP synthase |
| CXCL | C-X-C motif chemokine ligand |
| EV | Extracellular vesicle |
| EXPAND | EXosome PArkiNson Disease |
| FGF | Fibroblast growth factor |
| GCSF | Granulocyte colony-stimulating factor |
| GDS | Geriatric depression scale |
| GMCSF | Granulocyte macrophage colony-stimulating factor |
| IADL | Instrumental activities of daily living |
| IFN | Interferon |
| IL | Interleukin |
| IL1Ra | Interleukin 1 receptor agonist |
| LEDD | Levodopa equivalent daily dose |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MDV | Mitochondrial-derived vesicle |
| MHC | Major histocompatibility complex |
| MMSE | Mini mental state evaluation |
| mtDNA | Mitochondrial DNA |
| MTCO | Mitochondrial cytochrome C oxidase subunit |
| MVBs | Multivesicular bodies |
| MQC | Mitochondrial quality control |
| NDUFB8 | NADH:ubiquinone oxidoreductase subunit B8 |
| NDUFS3 | NADH:ubiquinone oxidoreductase core subunit |
| NLR | Nucleotide-binding oligomerisation domain-like receptor |
| NLRP3 | NLR family pyrin domain containing 3 |
| PBS | Phosphate-buffered saline |
| PD | Parkinson’s disease |
| PDGFBB | Platelet derived growth factor |
| PLS-DA | Partial least squares-discriminant analysis |
| PINK1 | Serine/threonine-protein kinase PTEN-induced putative kinase 1 |
| PRRs | Pattern recognition receptors |
| PVDF | Polyvinylidenefluoride |
| RAB | Ras-related protein in Brain |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RP | Rank product |
| SDH | Succinate dehydrogenase complex flavoprotein subunit |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| sEV | Small extracellular vesicle |
| STING | cGAS-stimulator of interferon genes |
| TFAM | Mitochondrial transcription factor A |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| UPDRS | Unified Parkinson’s disease rating scale |
| UQCRC2 | Ubiquinol-cytochrome C reductase core protein 2 |
| VIP | Variable importance in projection |
| VPS35 | Vacuolar protein sorting-associated protein 35 |
References
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259–280. [Google Scholar]
- Calvani, R.; Picca, A.; Lo Monaco, M.R.; Landi, F.; Bernabei, R.; Marzetti, E. Of microbes and minds: A narrative review on the second brain aging. Front. Med. 2018, 5, 53. [Google Scholar] [CrossRef]
- Scherzer, C.R.; Eklund, A.C.; Morse, L.J.; Liao, Z.; Locascio, J.J.; Fefer, D.; Schwarzschild, M.A.; Schlossmacher, M.G.; Hauser, M.A.; Vance, J.M.; et al. Molecular markers of early Parkinson’s disease based on gene expression in blood. Proc. Natl. Acad. Sci. USA 2007, 104, 955–960. [Google Scholar] [CrossRef]
- White, A.J.; Wijeyekoon, R.S.; Scott, K.M.; Gunawardana, N.P.; Hayat, S.; Solim, I.H.; McMahon, H.T.; Barker, R.A.; Williams-Gray, C.H. The peripheral inflammatory response to alpha-synuclein and endotoxin in Parkinson’s disease. Front. Neurol. 2018, 9, 946. [Google Scholar] [CrossRef]
- Bouvier-Müller, A.; Ducongé, F. Nucleic acid aptamers for neurodegenerative diseases. Biochimie 2018, 145, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.-S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial quality control in neurodegenerative diseases: Focus on Parkinson’s disease and Huntington’s disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [PubMed]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.-B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Bossola, M.; Manes-Gravina, E.; Landi, F.; Bernabei, R.; Marzetti, E. Circulating mitochondrial dna at the crossroads of mitochondrial dysfunction and inflammation during aging and muscle wasting disorders. Rejuvenation Res. 2018, 21, 350–359. [Google Scholar] [CrossRef]
- Schifferli, J.A. Microvesicles are messengers. Semin. Immunopathol. 2011, 33, 393–394. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Kim, O.Y.; Gho, Y.S. Extracellular vesicles as emerging intercellular communicasomes. BMB Rep. 2014, 47, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Heijnen, H.F.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated platelets release two types of membrane vesicles: Microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999, 94, 3791–3799. [Google Scholar] [PubMed]
- Park, S.J.; Kim, J.M.; Kim, J.; Hur, J.; Park, S.; Kim, K.; Shin, H.-J.; Chwae, Y.-J. Molecular mechanisms of biogenesis of apoptotic exosome-like vesicles and their roles as damage-associated molecular patterns. Proc. Natl. Acad. Sci. USA 2018, 115, E11721–E11730. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial dysfunction and aging: Insights from the analysis of extracellular vesicles. Int. J. Mol. Sci. 2019, 20, 805. [Google Scholar] [CrossRef]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.-C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Anticoli, S.; Aricò, E.; Arenaccio, C.; Manfredi, F.; Chiozzini, C.; Olivetta, E.; Ferrantelli, F.; Lattanzi, L.; D’Urso, M.T.; Proietti, E.; et al. Engineered exosomes emerging from muscle cells break immune tolerance to HER2 in transgenic mice and induce antigen-specific CTLs upon challenge by human dendritic cells. J. Mol. Med. (Berl.) 2018, 96, 211–221. [Google Scholar] [CrossRef]
- Qu, M.; Lin, Q.; Huang, L.; Fu, Y.; Wang, L.; He, S.; Fu, Y.; Yang, S.; Zhang, Z.; Zhang, L.; et al. Dopamine-loaded blood exosomes targeted to brain for better treatment of Parkinson’s disease. J. Control. Release 2018, 287, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Jarmalavičiūtė, A.; Pivoriūnas, A. Exosomes as a potential novel therapeutic tools against neurodegenerative diseases. Pharmacol. Res. 2016, 113, 816–822. [Google Scholar] [CrossRef]
- Hughes, A.J.; Daniel, S.E.; Kilford, L.; Lees, A.J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 1992, 55, 181–184. [Google Scholar] [CrossRef]
- Katz, S.; Ford, A.B.; Moskowitz, R.W.; Jackson, B.A.; Jaffe, M.W. Studies of illness in the aged. The index of ADL: A standardized measure of biological and psychosocial function. JAMA 1963, 185, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Lawton, M.P.; Brody, E.M. Assessment of older people: Self-maintaining and instrumental activities of daily living. Gerontologist 1969, 9, 179–186. [Google Scholar] [CrossRef]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Yesavage, J.A.; Sheikh, J.I. 9/Geriatric Depression Scale (GDS). Clin. Gerontol. 1986, 5, 165–173. [Google Scholar] [CrossRef]
- Antonini, A.; Abbruzzese, G.; Ferini-Strambi, L.; Tilley, B.; Huang, J.; Stebbins, G.T.; Goetz, C.G.; Barone, P.; MDS-UPDRS Italian Validation Study Group, M.; Bandettini di Poggio, M.; et al. Validation of the Italian version of the Movement Disorder Society—Unified Parkinson’s Disease Rating Scale. Neurol. Sci. 2013, 34, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martin, P. Hoehn and Yahr staging scale. Encycl. Mov. Disord. 2010, 23–25. [Google Scholar] [CrossRef]
- Tomlinson, C.L.; Stowe, R.; Patel, S.; Rick, C.; Gray, R.; Clarke, C.E. Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov. Disord. 2010, 25, 2649–2653. [Google Scholar] [CrossRef]
- Guerra, F.; Paiano, A.; Migoni, D.; Girolimetti, G.; Perrone, A.M.; De Iaco, P.; Fanizzi, F.P.; Gasparre, G.; Bucci, C. Modulation of RAB7A protein expression determines resistance to cisplatin through late endocytic pathway impairment and extracellular vesicular secretion. Cancers (Basel) 2019, 11, 52. [Google Scholar] [CrossRef]
- Théry, C.; Regnault, A.; Garin, J.; Wolfers, J.; Zitvogel, L.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J. Cell Biol. 1999, 147, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3.22.1–3.22.29. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef]
- Picca, A.; Pesce, V.; Lezza, A.M.S. Does eating less make you live longer and better? An update on calorie restriction. Clin. Interv. Aging 2017, 12, 1887. [Google Scholar] [CrossRef]
- Girolimetti, G.; Guerra, F.; Iommarini, L.; Kurelac, I.; Vergara, D.; Maffia, M.; Vidone, M.; Amato, L.B.; Leone, G.; Dusi, S.; et al. Platinum-induced mitochondrial DNA mutations confer lower sensitivity to paclitaxel by impairing tubulin cytoskeletal organization. Hum. Mol. Genet. 2017, 26, 2961–2974. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.; Karlsson, O.; Baus, J.A.; Wellner, E.; Tacke, R. Si- and C-functional organosilicon building blocks for synthesis based on 4-silacyclohexan-1-ones containing the silicon protecting groups MOP (4-methoxyphenyl), DMOP (2,6-dimethoxyphenyl), or TMOP (2,4,6-trimethoxyphenyl). J. Org. Chem. 2015, 80, 5804–5811. [Google Scholar] [CrossRef]
- Ngo, J.K.; Pomatto, L.C.D.; Davies, K.J.A. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease and aging. Redox Biol. 2013, 1, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Preste, R.; Vitale, O.; Clima, R.; Gasparre, G.; Attimonelli, M. HmtVar: A new resource for human mitochondrial variations and pathogenicity data. Nucleic Acids Res. 2019, 47, D1202–D1210. [Google Scholar] [CrossRef]
- Norambuena, A.; Wallrabe, H.; Cao, R.; Wang, D.B.; Silva, A.; Svindrych, Z.; Periasamy, A.; Hu, S.; Tanzi, R.E.; Kim, D.Y.; et al. A novel lysosome-to-mitochondria signaling pathway disrupted by amyloid-β oligomers. EMBO J. 2018, 37, e100241. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Landi, F.; Marini, F.; Cesari, M.; Buford, T.W.; Manini, T.M.; Onder, G.; Pahor, M.; Bernabei, R.; Leeuwenburgh, C.; et al. Patterns of circulating inflammatory biomarkers in older persons with varying levels of physical performance: A partial least squares-discriminant analysis approach. Front. Med. 2014, 1, 27. [Google Scholar] [CrossRef]
- Calvani, R.; Picca, A.; Marini, F.; Biancolillo, A.; Gervasoni, J.; Persichilli, S.; Primiano, A.; Coelho-Junior, H.J.; Bossola, M.; Urbani, A.; et al. A distinct pattern of circulating amino acids characterizes older persons with physical frailty and sarcopenia: Results from the BIOSPHERE study. Nutrients 2018, 10, 1691. [Google Scholar] [CrossRef] [PubMed]
- Westerhuis, J.A.; Hoefsloot, H.C.J.; Smit, S.; Vis, D.J.; Smilde, A.K.; van Velzen, E.J.J.; van Duijnhoven, J.P.M.; van Dorsten, F.A. Assessment of PLSDA cross validation. Metabolomics 2008, 4, 81–89. [Google Scholar] [CrossRef]
- Wold, S.; Martens, H.; Wold, H. The Multivariate Calibration Problem in Chemistry Solved by the PLS Method; Springer: Berlin/Heidelberg, Germany, 1983; pp. 286–293. [Google Scholar]
- Smit, S.; van Breemen, M.J.; Hoefsloot, H.C.J.; Smilde, A.K.; Aerts, J.M.F.G.; de Koster, C.G. Assessing the statistical validity of proteomics based biomarkers. Anal. Chim. Acta 2007, 592, 210–217. [Google Scholar] [CrossRef]
- Larsen, S.B.; Hanss, Z.; Krüger, R. The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef]
- Amo, T.; Saiki, S.; Sawayama, T.; Sato, S.; Hattori, N. Detailed analysis of mitochondrial respiratory chain defects caused by loss of PINK1. Neurosci. Lett. 2014, 580, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Tzoulis, C.; Schwarzlmüller, T.; Biermann, M.; Haugarvoll, K.; Bindoff, L.A. Mitochondrial DNA homeostasis is essential for nigrostriatal integrity. Mitochondrion 2016, 28, 33–37. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.-L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.-E.; Burelle, Y.; et al. Parkinson’s Disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Lázaro, S.; Gamarra, D.; Del Val, M. Proteolytic enzymes involved in MHC class I antigen processing: A guerrilla army that partners with the proteasome. Mol. Immunol. 2015, 68, 72–76. [Google Scholar] [CrossRef]
- Cebrián, C.; Zucca, F.A.; Mauri, P.; Steinbeck, J.A.; Studer, L.; Scherzer, C.R.; Kanter, E.; Budhu, S.; Mandelbaum, J.; Vonsattel, J.P.; et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat. Commun. 2014, 5, 3633. [Google Scholar] [CrossRef]
- Benner, E.J.; Banerjee, R.; Reynolds, A.D.; Sherman, S.; Pisarev, V.M.; Tsiperson, V.; Nemachek, C.; Ciborowski, P.; Przedborski, S.; Mosley, R.L.; et al. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE 2008, 3, e1376. [Google Scholar] [CrossRef] [PubMed]
- Theodore, S.; Cao, S.; McLean, P.J.; Standaert, D.G. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.S.; Cao, S.; Rowse, A.L.; Thome, A.D.; Li, X.; Mangieri, L.R.; Cron, R.Q.; Shacka, J.J.; Raman, C.; Standaert, D.G. MHCII is required for α-synuclein-induced activation of microglia, CD4 T cell proliferation and dopaminergic neurodegeneration. J. Neurosci. 2013, 33, 9592–9600. [Google Scholar] [CrossRef]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling inflamm-aging through mitochondrial dysfunction: Mechanisms and molecular targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles 2013, 2, 20389. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Antibody | Manufacturer and Catalogue Number | Type | Species | Detected Band MW (kDa) |
|---|---|---|---|---|
| ATP5A (complex V) | Abcam (ab1104413) | Monoclonal | Mouse | 55 |
| UQCRC2 (complex III) | 48 | |||
| MTCOI (complex IV) | 40 | |||
| SDHB (complex II) | 30 | |||
| NDUFB8 (complex I) | 20 | |||
| CD63 | Santa Cruz Biotechnology (sc-5275) | Monoclonal | Mouse | 26 |
| CD81 | Santa Cruz Biotechnology (sc-166020) | Monoclonal | Mouse | 25 |
| CD9 | Santa Cruz Biotechnology (sc-13118) | Monoclonal | Mouse | 25 |
| MTCOII (complex IV) | Santa Cruz Biotechnology (sc-514489) | Monoclonal | Mouse | 25 |
| NDUFS3 (complex I) | Santa Cruz Biotechnology (sc-374283) | Monoclonal | Mouse | 25 |
| Parkin | R&D Systems (MAB14381) | Monoclonal | Mouse | 52 |
| PINK1 | AbD Serotec (HCA150) | Monoclonal | Mouse | 70 |
| SDHA (complex II) | Santa Cruz Biotechnology (sc-390381) | Monoclonal | Mouse | 70 |
| Type | Biomarkers |
|---|---|
| Cytokines | IFNγ, IL1β, IL1Ra, IL2, IL4, IL5, IL6, IL7, IL8, IL9, IL10, IL12, IL13, IL15, IL17, TNF-α |
| Chemokines | CCL2, CCL3, CCL4, CCL5, CCL11, CXCL |
| Growth factors | FGF-β, GCSF, GMCSF, PDGF BB |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the EXosomes in PArkiNson Disease (EXPAND) Study. Int. J. Mol. Sci. 2019, 20, 2373. https://doi.org/10.3390/ijms20102373
Picca A, Guerra F, Calvani R, Bucci C, Lo Monaco MR, Bentivoglio AR, Landi F, Bernabei R, Marzetti E. Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the EXosomes in PArkiNson Disease (EXPAND) Study. International Journal of Molecular Sciences. 2019; 20(10):2373. https://doi.org/10.3390/ijms20102373
Chicago/Turabian StylePicca, Anna, Flora Guerra, Riccardo Calvani, Cecilia Bucci, Maria Rita Lo Monaco, Anna Rita Bentivoglio, Francesco Landi, Roberto Bernabei, and Emanuele Marzetti. 2019. "Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the EXosomes in PArkiNson Disease (EXPAND) Study" International Journal of Molecular Sciences 20, no. 10: 2373. https://doi.org/10.3390/ijms20102373
APA StylePicca, A., Guerra, F., Calvani, R., Bucci, C., Lo Monaco, M. R., Bentivoglio, A. R., Landi, F., Bernabei, R., & Marzetti, E. (2019). Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the EXosomes in PArkiNson Disease (EXPAND) Study. International Journal of Molecular Sciences, 20(10), 2373. https://doi.org/10.3390/ijms20102373