Comparative Transcriptomic and Physiological Analyses of Medicago sativa L. Indicates that Multiple Regulatory Networks Are Activated during Continuous ABA Treatment





Abstract
1. Introduction
2. Results
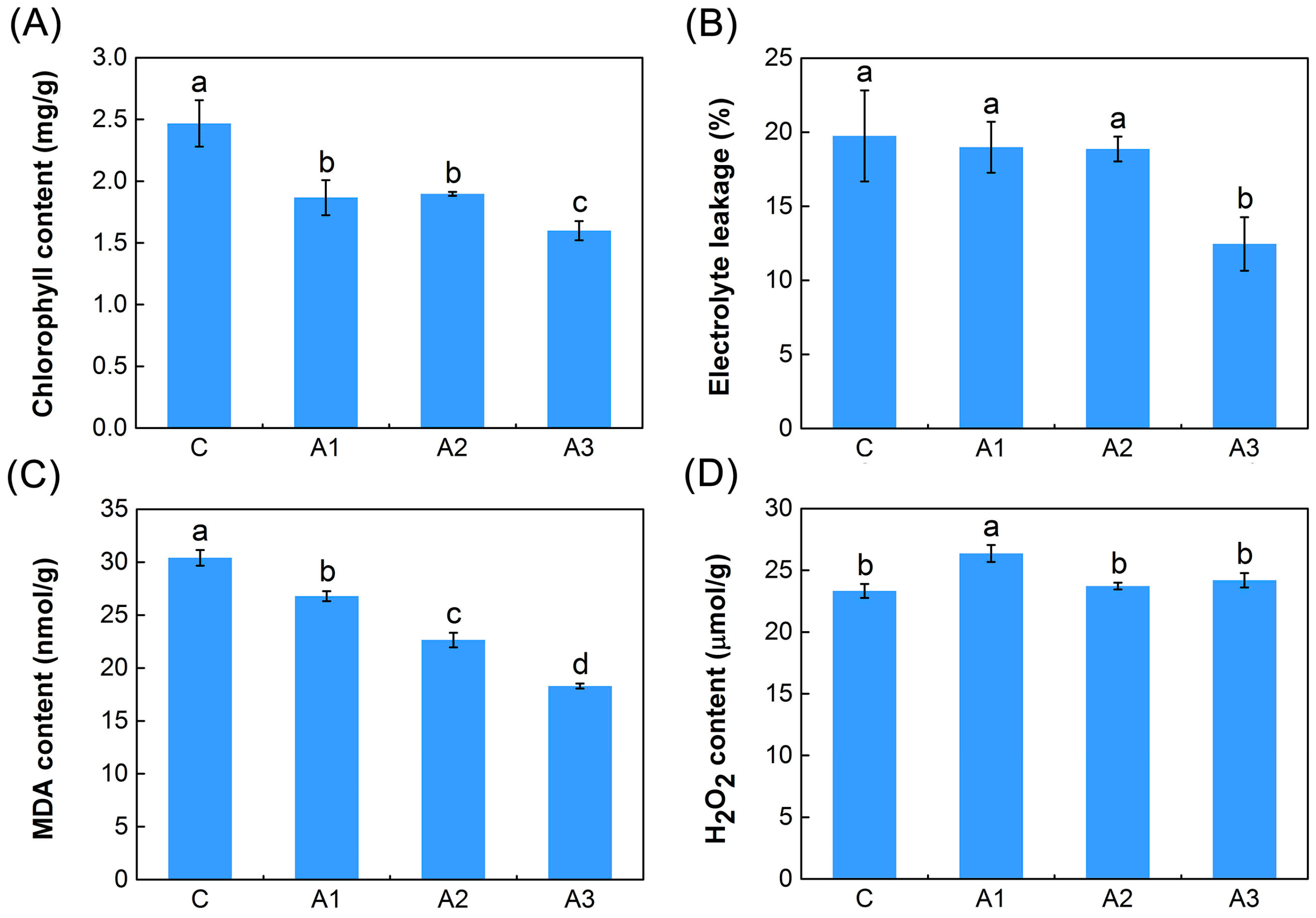
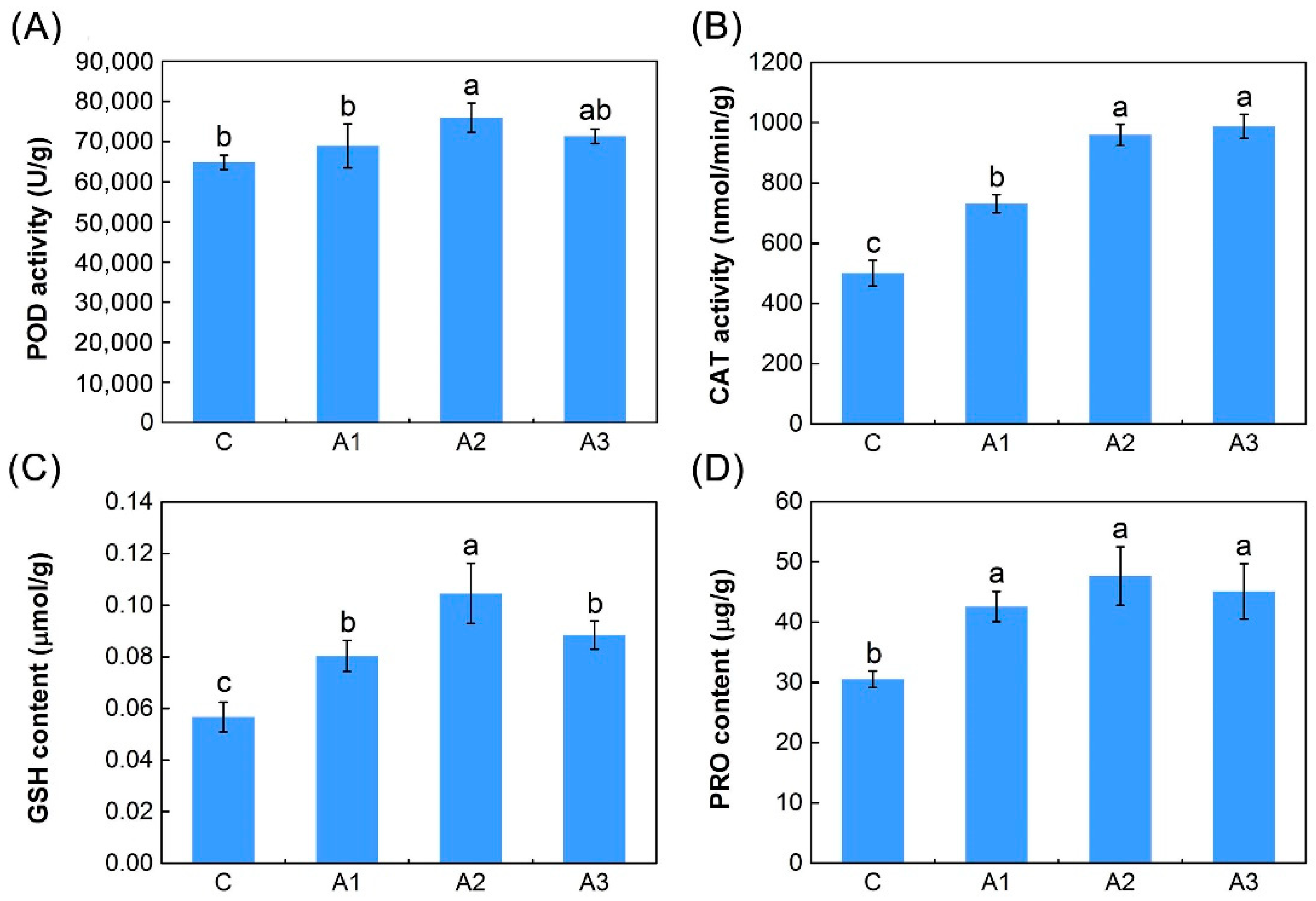
2.1. Physiological Responses to ABA
2.2. Transcriptome Sequencing, Assembly and Annotation
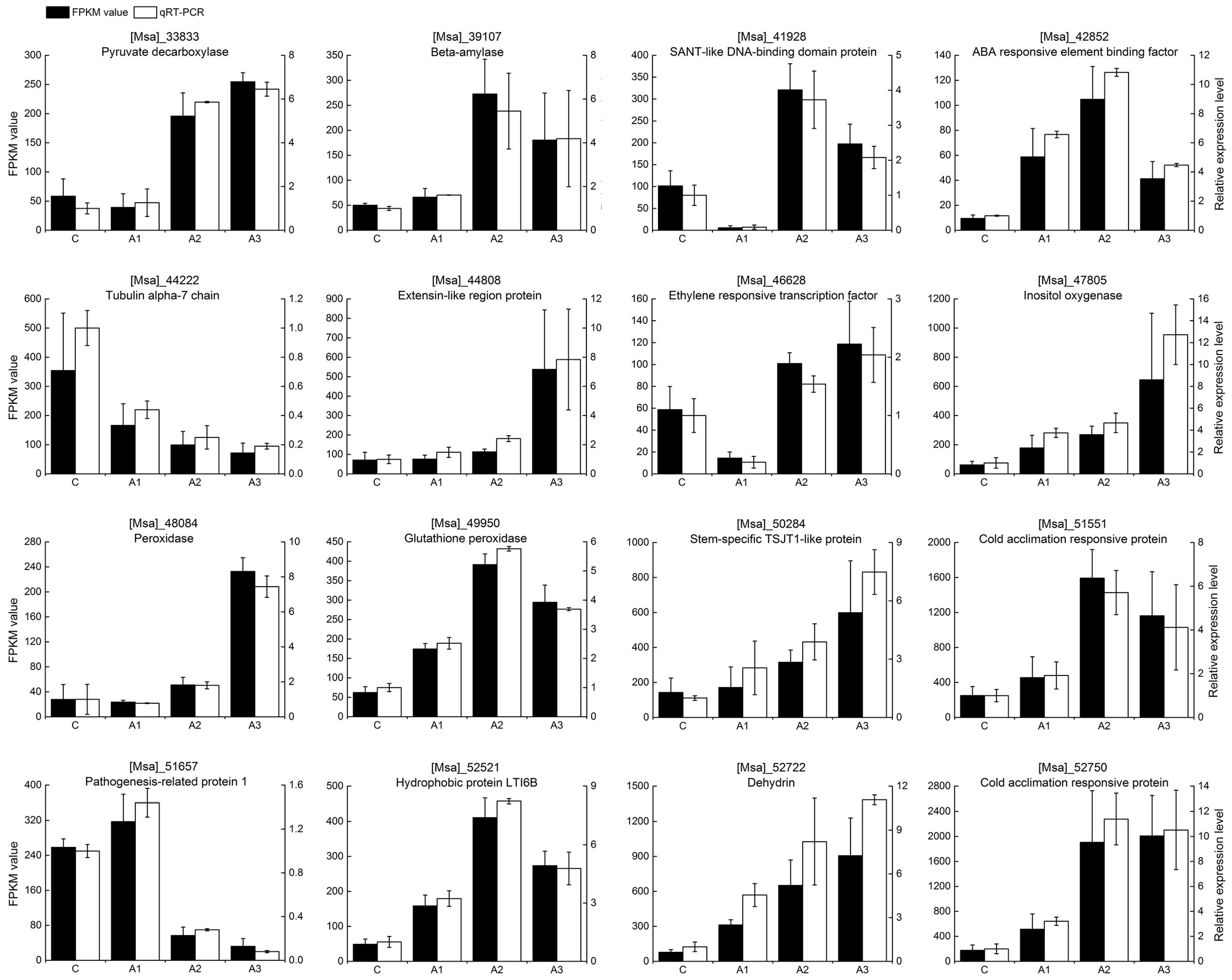
2.3. Verification of Gene Expression
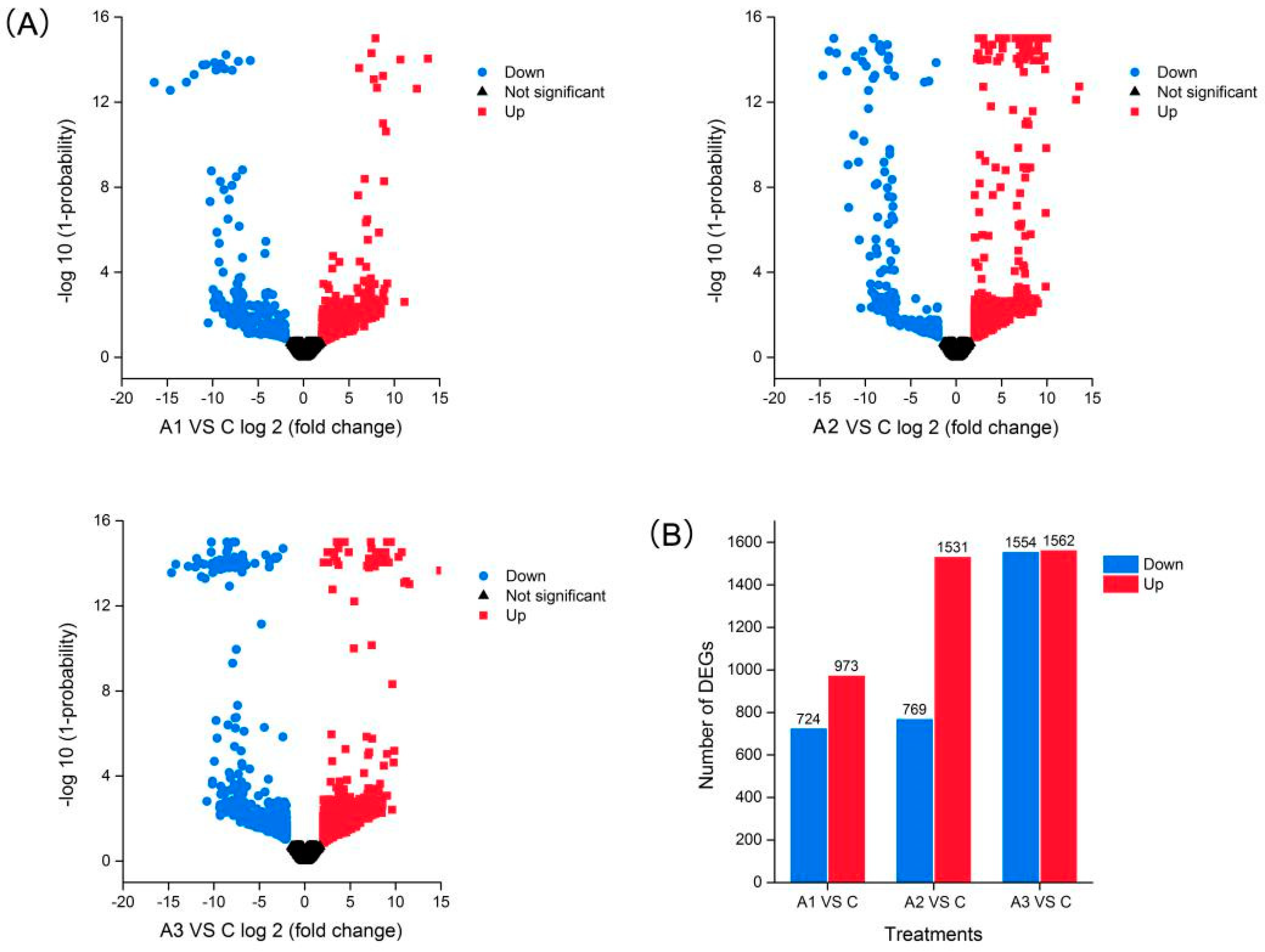
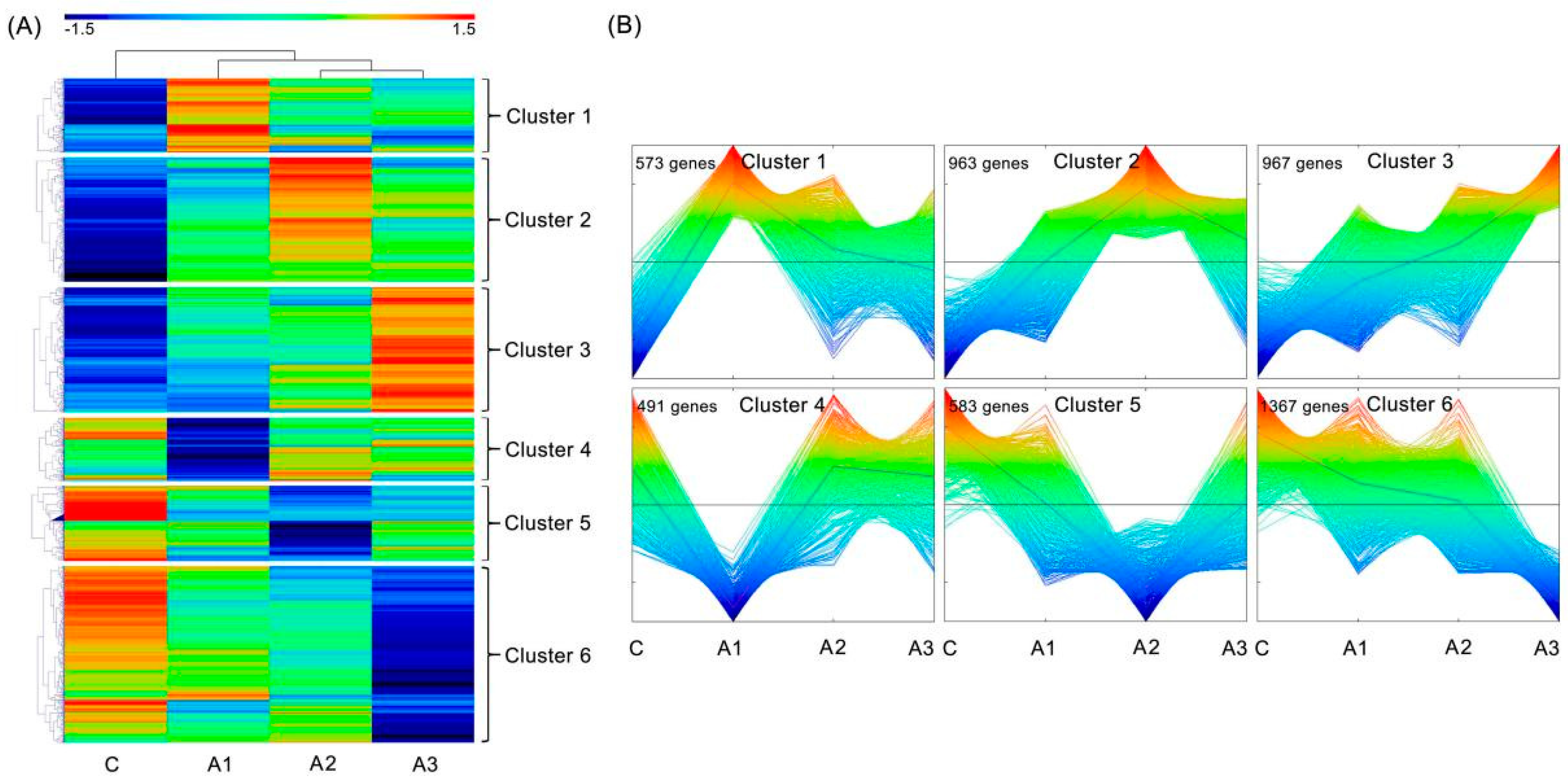
2.4. Differentially Expressed Isoforms (DEIs) Analysis
2.5. GO Enrichment Analysis of the DEIs
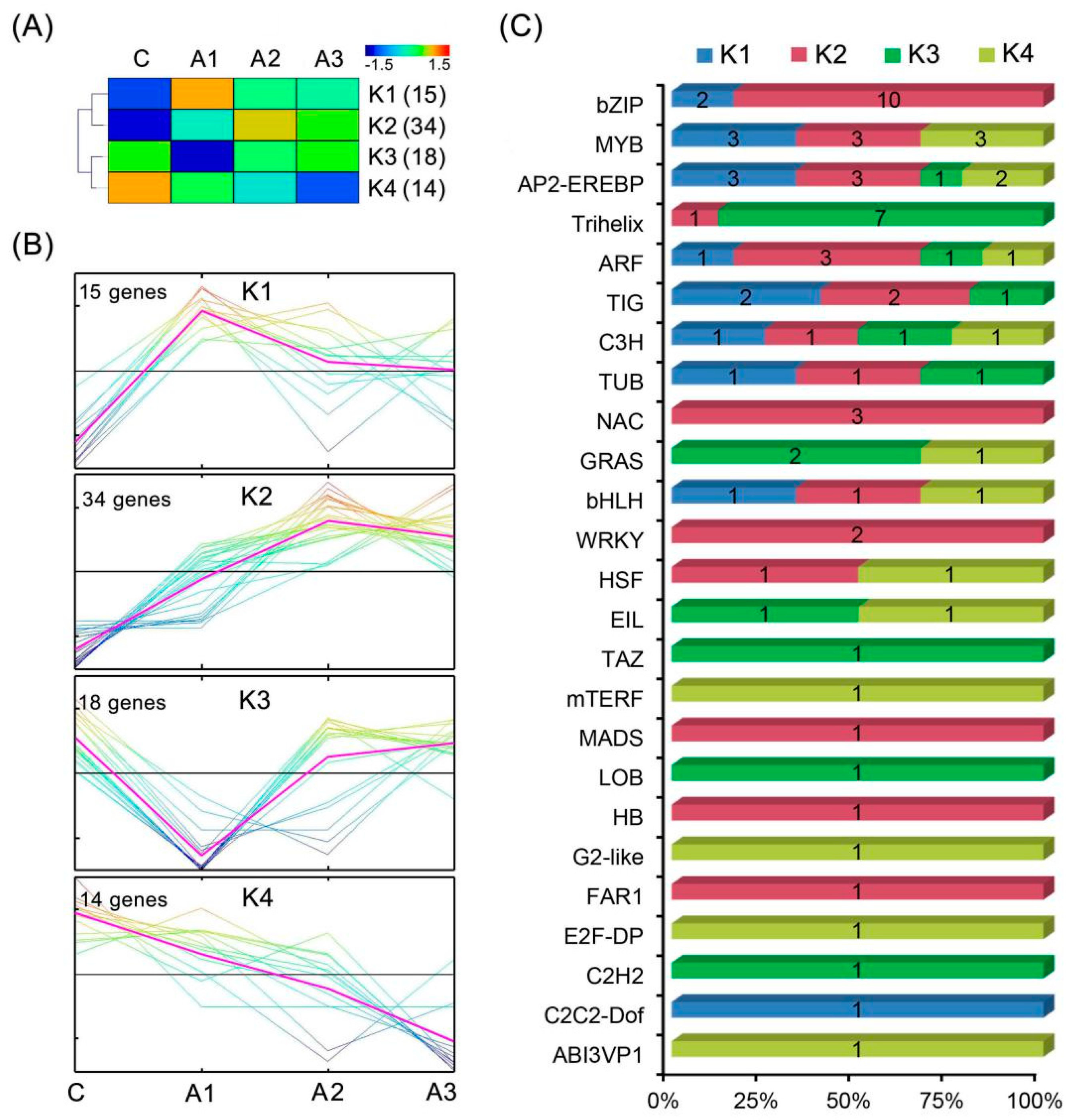
2.6. Identification of Transcription Factors (TFs) of the DEIs
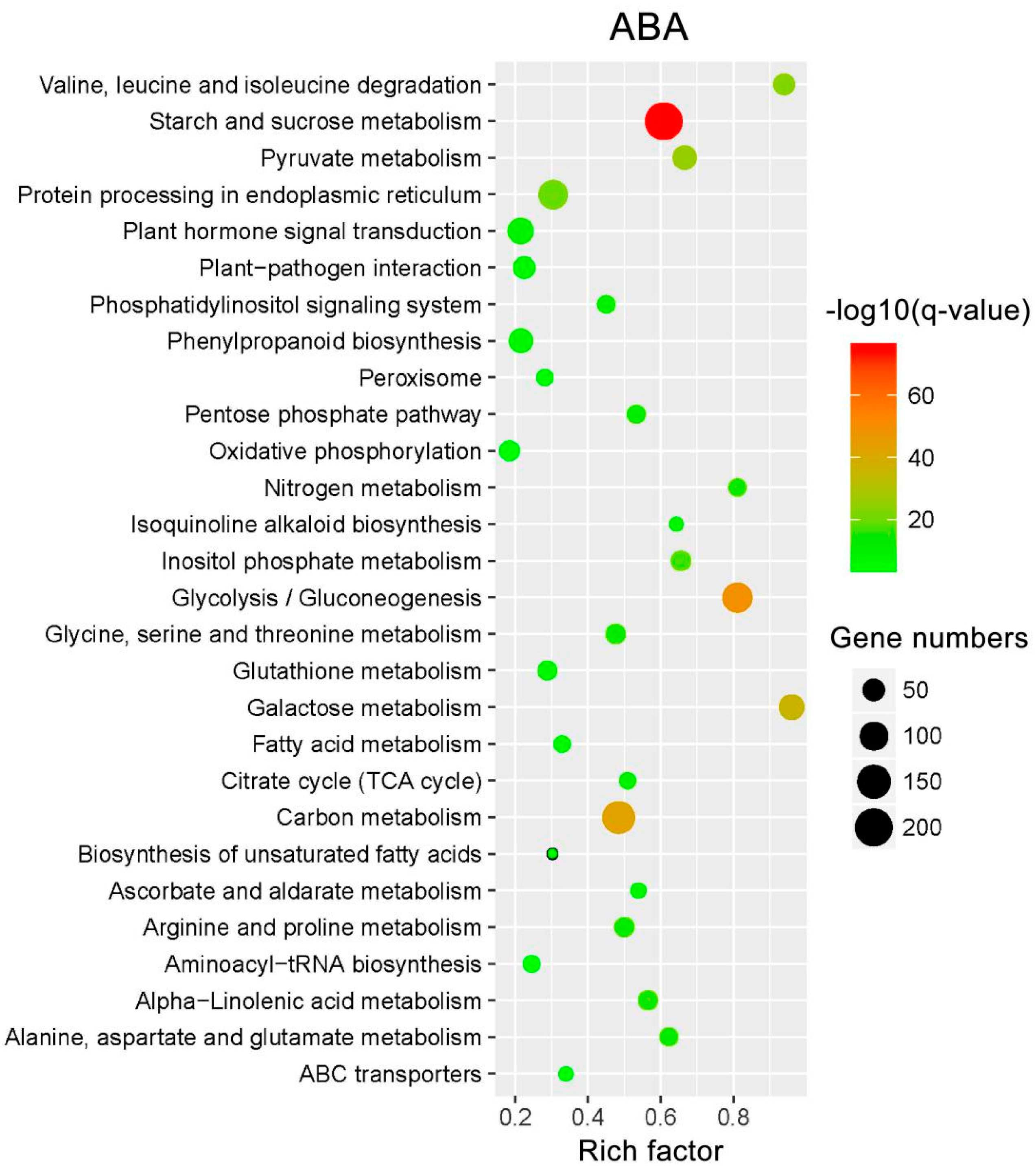
2.7. KEGG Pathway Enrichment Analysis of the DEIs
3. Discussion
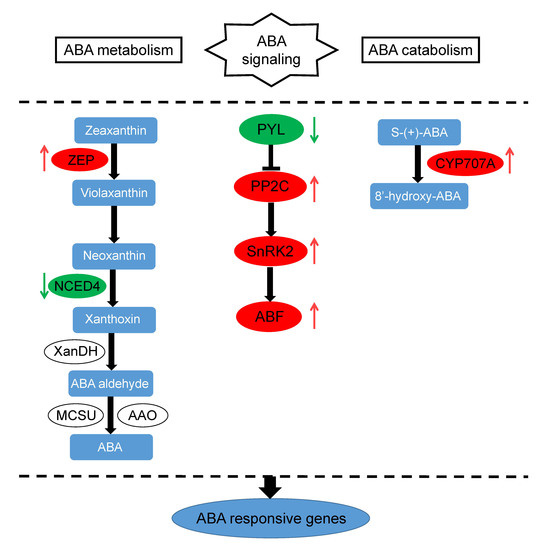
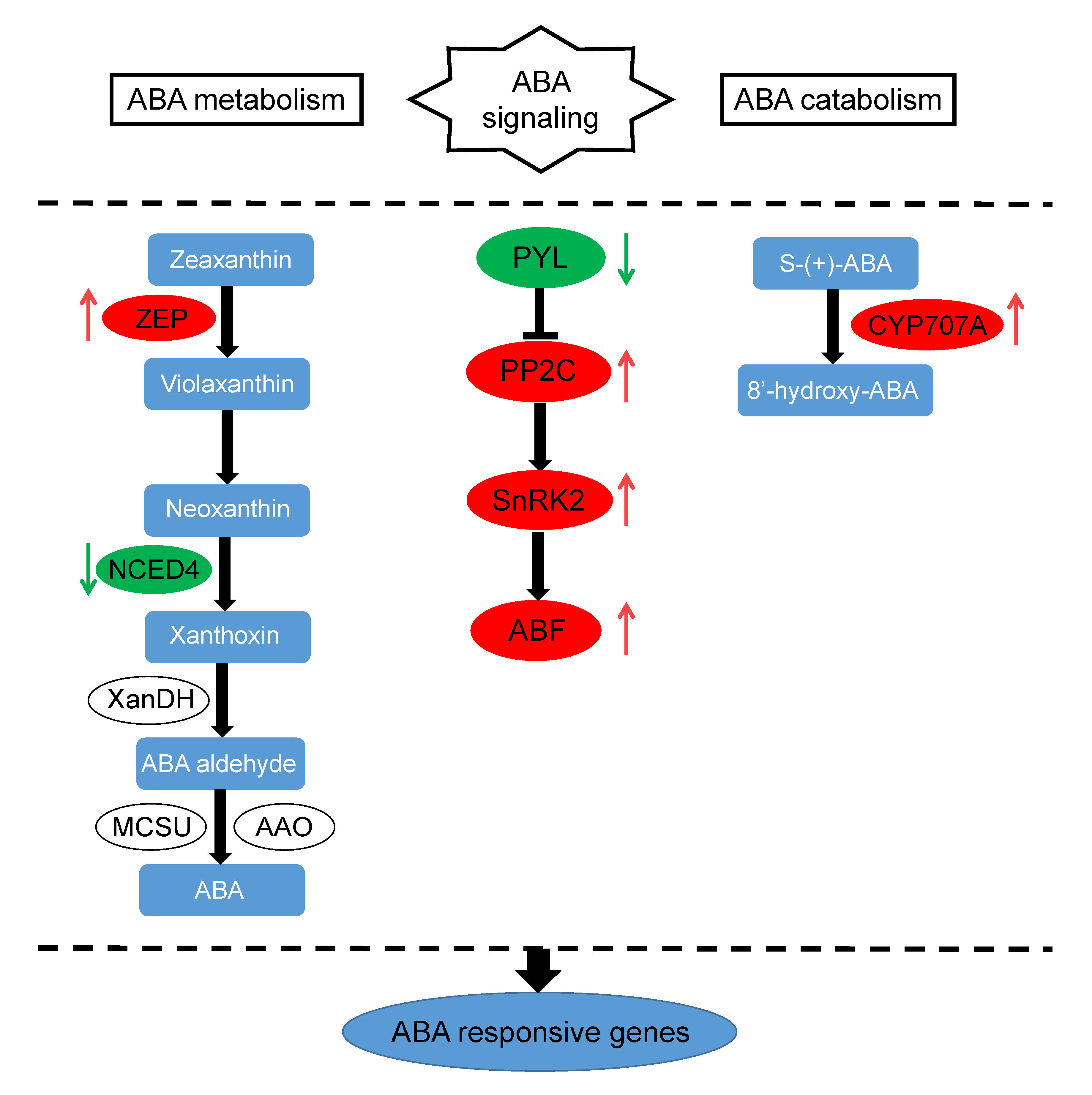
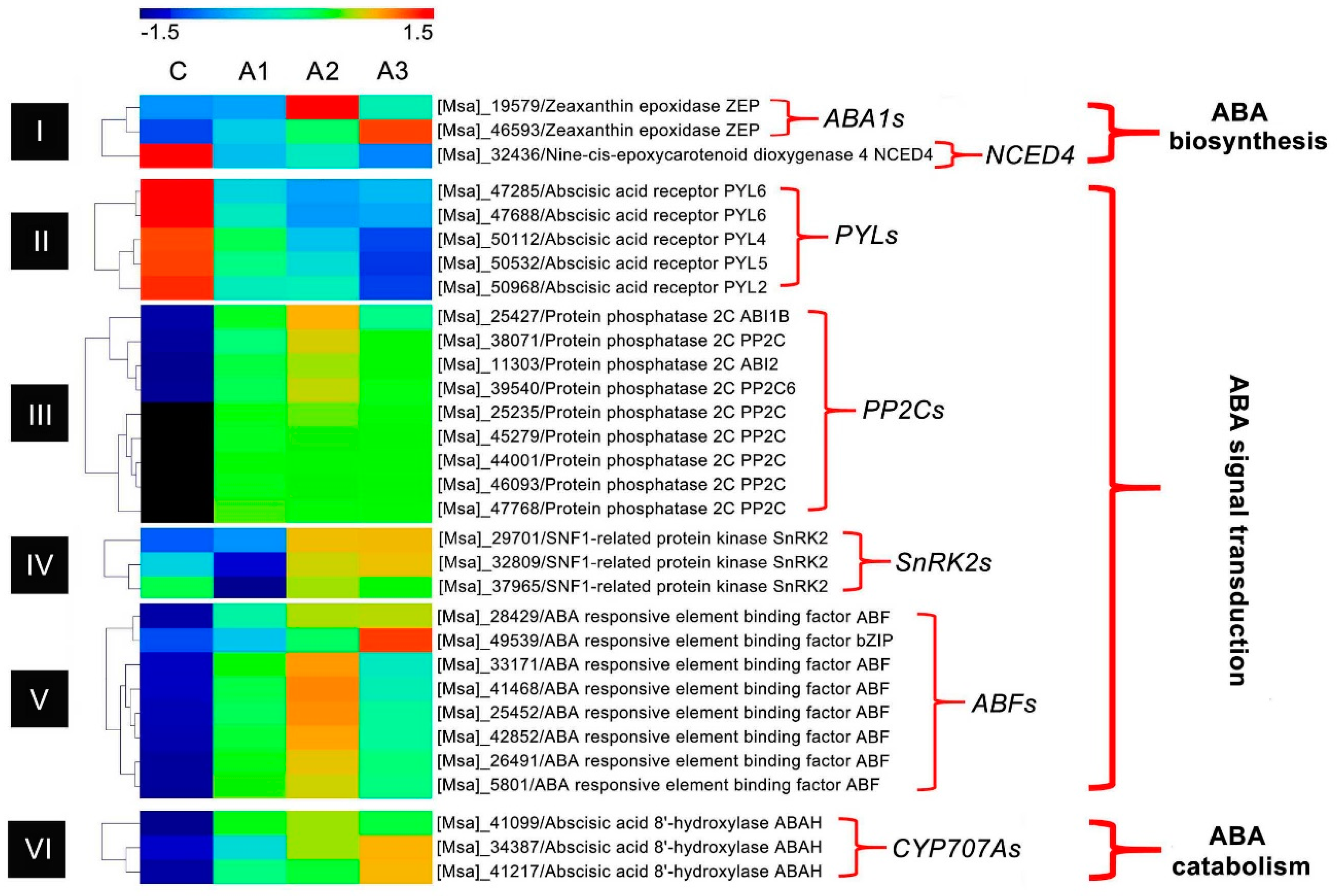
3.1. ABA Regulation Pathway-Related DEIs
3.2. Transcriptional Regulation-Related DEIs
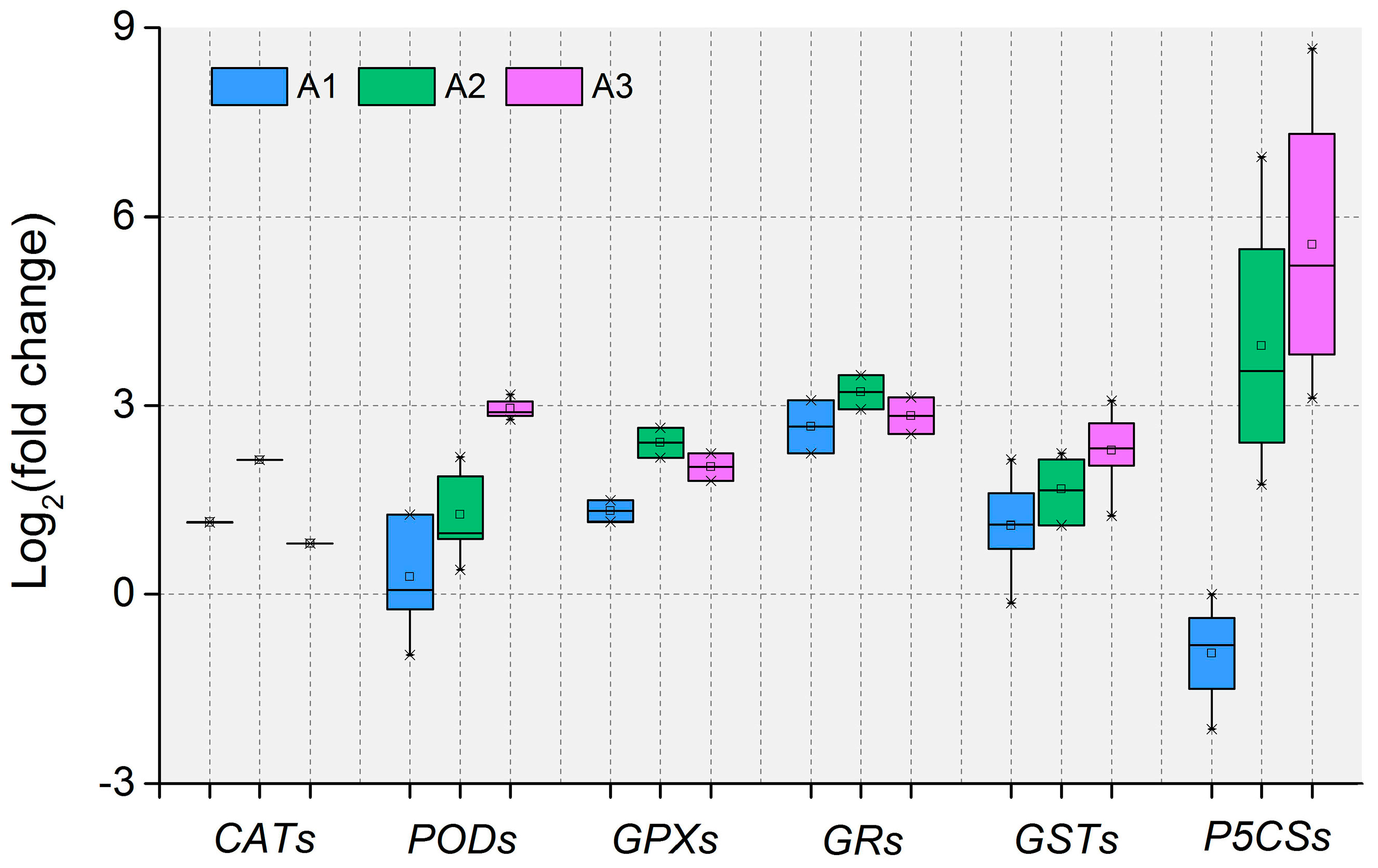
3.3. Antioxidant Defense System-Related DEIs
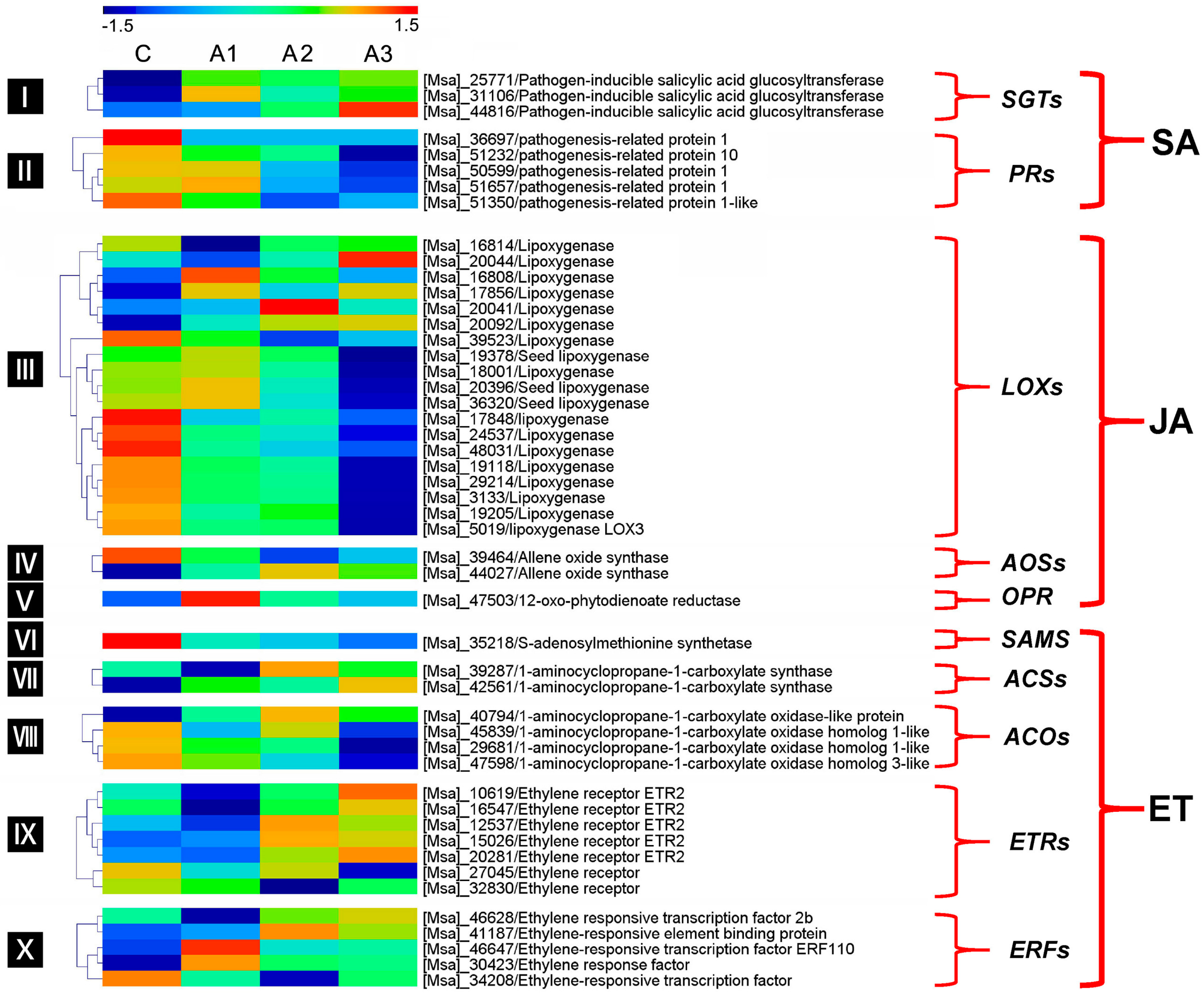
3.4. Pathogen Resistance-Related DEIs
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. ABA Treatment
4.3. Determination of Physiological Characteristics
4.4. cDNA Library Preparation, Sequencing, Assembly and Annotation
4.5. qRT-PCR Analysis
4.6. DEIs Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABA | Abscisic acid |
| ABF | ABA-responsive elements binding factor |
| CAT | Catalase |
| DEI | Differentially expressed isoform |
| ET | Ethylene |
| FPKM | Fragments per kilobase per million fragments mapped |
| GO | Gene Ontology |
| GSH | Glutathione |
| H2O2 | Hydrogen peroxide |
| JA | Jasmonate |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MDA | Malonaldehyde |
| MEV 4.9 | MultiExperiment Viewer 4.9 |
| NCBI | National Center for Biotechnology Information |
| NGS | Next-generation sequencing |
| Nr | Non-redundant protein sequences |
| POD | Peroxidase |
| PP2C | Protein phosphatase 2C |
| PRO | Proline |
| PYL | Pyrabactin resistance 1-like |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| RNA-Seq | RNA sequencing |
| ROS | Reactive oxygen species |
| SA | Salicylic acid |
| SnRK2 | Sucrose nonfermenting1-related protein kinase 2 |
| SOTA | Self-Organizing Tree Algorithm |
| TF | Transcription factor |
References
- Choi, H.K.; Mun, J.H.; Kim, D.J.; Zhu, H.; Baek, J.M.; Mudge, J.; Roe, B.; Ellis, N.; Doylr, J.; Kiss, G.B.; et al. Estimating genome conservation between crop and model legume species. Proc. Natl. Acad. Sci. USA 2004, 101, 15289–15294. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Liu, W.; Wang, Y.; Zhang, J.; Liu, Z. Development of a rapid one-step PCR protocol to distinguish between alfalfa (Medicago sativa) and sweet clover (Melilotus spp.) seeds. Seed Sci. Technol. 2014, 42, 237–246. [Google Scholar] [CrossRef]
- Li, X.; Wei, Y.; Moore, K.J.; Michaud, R.; Viands, D.R.; Hansen, J.L.; Acharya, A.; Brummer, E.C. Association mapping of biomass yield and stem composition in a tetraploid alfalfa breeding population. Plant Genome 2011, 4, 24–35. [Google Scholar] [CrossRef]
- Samac, D.A.; Jung, H.; Lamb, J.F. Development of alfalfa (Medicago sativa L.) as a feedstock for production of ethanol and other bioproducts. In Alcoholic Fuels; Minteer, S., Ed.; CRC Press: Boca Raton, FL, USA, 2006; pp. 79–98. [Google Scholar]
- Finkelstein, R.R.; Gampala, S.S.; Rock, C.D. Abscisic acid signaling in seeds and seedlings. Plant Cell 2002, 14, 15–45. [Google Scholar] [CrossRef]
- Munemasa, S.; Hauser, F.; Park, J.; Waadt, R.; Brandt, B.; Schroeder, J.I. Mechanisms of abscisic acid-mediated control of stomatal aperture. Curr. Opin. Plant Biol. 2015, 28, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Tuberosa, R.; Sanguineti, M.C.; Landi, P. RFLP mapping of quantitative trait loci controlling abscisic acid concentration in leaves of drought-stressed maize (Zea mays L.). Theor. Appl. Genet. 1998, 97, 744–755. [Google Scholar] [CrossRef]
- McCann, S.E.; Huang, B. Drought responses of Kentucky bluegrass and creeping bentgrass as affected by abscisic acid and trinexapac-ethyl. J. Am. Soc. Hortic. Sci. 2008, 133, 20–26. [Google Scholar]
- Cheng, Z.; Jin, R.; Cao, M.; Liu, X.; Chan, Z. Exogenous application of ABA mimic 1 (AM1) improves cold stress tolerance in bermudagrass (Cynodon dactylon). Plant Cell Tissue Org. 2016, 125, 231–240. [Google Scholar] [CrossRef]
- Gurmani, A.R.; Bano, A.; Khan, S.U.; Din, J.; Zhang, J.L. Alleviation of salt stress by seed treatment with abscisic acid (ABA), 6-benzylaminopurine (BA) and chlormequat chloride (CCC) optimizes ion and organic matter accumulation and increases yield of rice (Oryza sativa L.). Aust. J. Crop Sci. 2011, 5, 1278–1285. [Google Scholar]
- Yang, Z.; Yu, J.; Merewitz, E.; Huang, B. Differential effects of abscisic acid and glycine betaine on physiological responses to drought and salinity stress for two perennial grass species. J. Am. Soc. Hortic. Sci. 2012, 137, 96–106. [Google Scholar]
- Asselbergh, B.; De Vleesschauwer, D.; Höfte, M. Global switches and fine-tuning-ABA modulates plant pathogen defense. Mol. Plant Microbe Ineract. 2008, 21, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Achuo, E.A.; Prinsen, E.; Hofte, M. Influence of drought, salt stress and abscisic acid on the resistance of tomato to Botrytis cinerea and Oidium neolycopersici. Plant Pathol. 2006, 55, 178–186. [Google Scholar] [CrossRef]
- Mohr, P.G.; Cahill, D.M. Relative roles of glyceollin, lignin and the hypersensitive response and the influence of ABA in compatible and incompatible interactions of soybeans with Phytophthora sojae. Physiol. Mol. Plant Pathol. 2001, 58, 31–41. [Google Scholar] [CrossRef]
- Yazawa, K.; Jiang, C.J.; Kojima, M.; Sakakibara, H.; Takatsuji, H. Reduction of abscisic acid levels or inhibition of abscisic acid signaling in rice during the early phase of Magnaporthe oryzae infection decreases its susceptibility to the fungus. Physiol. Mol. Plant Pathol. 2012, 78, 1–7. [Google Scholar] [CrossRef]
- Mauch-Mani, B.; Mauch, F. The role of abscisic acid in plant–pathogen interactions. Curr. Opin. Plant Biol. 2005, 8, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Ton, J.; Mauch-Mani, B. β-amino-butyric acid-induced resistance against necrotrophic pathogens is based on ABA-dependent priming for callose. Plant J. 2004, 38, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tao, X.; Tang, X.M.; Xiao, L.; Sun, J.L.; Yan, X.F.; Li, D.; Den, H.Y.; Ma, X.R. Comparative transcriptome analysis of tomato (Solanum lycopersicum) in response to exogenous abscisic acid. BMC Genom. 2013, 14, 841. [Google Scholar] [CrossRef]
- Matsui, A.; Ishida, J.; Morosawa, T.; Mochizuki, Y.; Kaminuma, E.; Endo, T.A.; Okamoto, M.; Nambara, E.; Nakajima, M.; Kawashima, M.; et al. Arabidopsis transcriptome analysis under drought, cold, high-salinity and ABA treatment conditions using a tiling array. Plant Cell Physiol. 2008, 49, 1135–1149. [Google Scholar] [CrossRef]
- Weiner, J.J.; Peterson, F.C.; Volkman, B.F.; Cutler, S.R. Structural and functional insights into core ABA signaling. Curr. Opin. Plant Biol. 2010, 13, 495–502. [Google Scholar] [CrossRef]
- Verhage, A.; van Wees, S.C.; Pieterse, C.M. Plant immunity: It’s the hormones talking, but what do they say? Plant Physiol. 2010, 154, 536–540. [Google Scholar] [CrossRef]
- Buchanan, C.D.; Lim, S.; Salzman, R.A.; Kagiampakis, I.; Morishige, D.T.; Weers, B.D.; Mullet, J.E.; Klein, R.R.; Pratt, L.H.; Cordonnier-Pratt, M.M.; et al. Sorghum bicolor’s transcriptome response to dehydration, high salinity and ABA. Plant Mol. Biol. 2005, 58, 699–720. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.N.; Shi, D.Q.; Ruan, M.B.; Zhang, L.L.; Meng, Z.H.; Liu, J.; Yang, W.C. Transcriptome analysis reveals crosstalk of responsive genes to multiple abiotic stresses in cotton (Gossypium hirsutum L.). PLoS ONE 2013, 8, e80218. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.A.; Maruyama, K.; Abe, H.; Khan, M.A.; Katsura, K.; Ito, Y.; Yoshiwara, K.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and abscisic acid application using cDNA microarray and RNA gel-blot analyses. Plant Physiol. 2003, 133, 1755–1767. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xiong, C.; Yan, L.; Zhang, Z.; Ma, L.; Wang, Y.; Liu, Y.; Liu, Z. Transcriptome analyses reveal candidate genes potentially involved in Al stress response in alfalfa. Front. Plant Sci. 2017, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Mao, L.; Lu, W.; Ying, T.; Luo, Z. Transcriptome profiling of postharvest strawberry fruit in response to exogenous auxin and abscisic acid. Planta 2016, 243, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, J.; Liu, S.; Liu, X.; Lin, Y.; Li, L. Identification of rapidly induced genes in the response of peanut (Arachis hypogaea) to water deficit and abscisic acid. BMC Biotechnol. 2014, 14, 58. [Google Scholar] [CrossRef]
- Halušková, L.U.; Valentovičová, K.; Huttová, J.; Mistrík, I.; Tamás, L. Effect of abiotic stresses on glutathione peroxidase and glutathione S-transferase activity in barley root tips. Plant Physiol. Biochem. 2009, 47, 1069–1074. [Google Scholar] [CrossRef]
- Latz, E.; Eisenhauer, N.; Rall, B.C.; Allan, E.; Roscher, C.; Scheu, S.; Jousset, A. Plant diversity improves protection against soil-borne pathogens by fostering antagonistic bacterial communities. J. Ecol. 2012, 100, 597–604. [Google Scholar] [CrossRef]
- Parent, B.; Hachez, C.; Redondo, E.; Simonneau, T.; Chaumont, F.; Tardieu, F. Drought and abscisic acid effects on aquaporin content translate into changes in hydraulic conductivity and leaf growth rate: A trans-scale approach. Plant Physiol. 2009, 149, 2000–2012. [Google Scholar] [CrossRef]
- Chan, Z. Expression profiling of ABA pathway transcripts indicates crosstalk between abiotic and biotic stress responses in Arabidopsis. Genomics 2012, 100, 110–115. [Google Scholar] [CrossRef]
- Chen, C.W.; Yang, Y.W.; Lur, H.S.; Tsai, Y.G.; Chang, M.C. A novel function of abscisic acid in the regulation of rice (Oryza sativa L.) root growth and development. Plant Cell Physiol. 2006, 47, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Umezawa, T.; Urano, K.; Shinozaki, K. Regulatory metabolic networks in drought stress responses. Curr. Opin. Plant Biol. 2007, 10, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Ma, S.; Bai, L.; Zhang, L.; Ma, H.; Jia, P.; Liu, J.; Zhong, M.; Guo, Z. Signal transduction during cold, salt, and drought stresses in plants. Mol. Biol. Rep. 2012, 39, 969–987. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.K.; Wang, C.E.; Fei, Y.Y.; Gai, J.Y.; Zhao, T.J. Genome-wide identification and transcription analysis of soybean carotenoid oxygenase genes during abiotic stress treatments. Mol. Biol. Rep. 2013, 40, 4737–4745. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Kushiro, T.; Jikumaru, Y.; Abrams, S.R.; Kamiya, Y.; Seki, M.; Nambara, E. ABA 9′-hydroxylation is catalyzed by CYP707A in Arabidopsis. Phytochemistry 2011, 72, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Szostkiewicz, I.; Korte, A.; Moes, D.; Yang, Y.; Christmann, A.; Grill, E. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science 2009, 324, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Chen, S.; Yang, W.; Wang, T.; Zheng, K.; Wang, Y.; Cheng, Y.; Zhang, N.; Liu, S.; Li, D.; et al. A novel family of transcription factors conserved in angiosperms is required for ABA signalling. Plant Cell Environ. 2017, 40, 2958–2971. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Schumaker, K.S.; Zhu, J.K. Cell signaling during cold, drought, and salt stress. Plant Cell 2002, 14, S165–S183. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Uribe, L.; O’Connell, M.A. A root-specific bZIP transcription factor is responsive to water deficit stress in tepary bean (Phaseolus acutifolius) and common bean (P. vulgaris). J. Exp. Bot. 2006, 57, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.H.; Li, C.W.; Su, R.C.; Cheng, C.P.; Tsai, Y.C.; Chan, M.T. A tomato bZIP transcription factor, SlAREB, is involved in water deficit and salt stress response. Planta 2010, 231, 1459–1473. [Google Scholar] [CrossRef] [PubMed]
- Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AP2/ERF family transcription factors in plant abiotic stress responses. BBA-Gene Regul. Mech. 2012, 1819, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.S.; Chen, M.; Li, L.C.; Ma, Y.Z. Functions and application of the AP2/ERF transcription factor family in crop improvement. J. Integr. Plant Biol. 2011, 53, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Khurana, J.P. Transcript profiling reveals diverse roles of auxin-responsive genes during reproductive development and abiotic stress in rice. FEBS J. 2009, 276, 3148–3162. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Kaplan-Levy, R.N.; Brewer, P.B.; Quon, T.; Smyth, D.R. The trihelix family of transcription factors—light, stress and development. Trends Plant Sci. 2012, 17, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Zhu, G.; Liu, Y.; Li, Y.; Zhang, J. ABA controls H2O2 accumulation through the induction of OsCATB in rice leaves under water stress. Plant Cell Physiol. 2011, 52, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhang, J. Effect of abscisic acid on active oxygen species, antioxidative defence system and oxidative damage in leaves of maize seedlings. Plant Cell Physiol. 2001, 42, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Deis, L.; Cavagnaro, B.; Bottini, R.; Wuilloud, R.; Silva, M.F. Water deficit and exogenous ABA significantly affect grape and wine phenolic composition under in field and in-vitro conditions. Plant Growth Regul. 2011, 65, 11–21. [Google Scholar] [CrossRef]
- Ghassemian, M.; Nambara, E.; Cutler, S.; Kawaide, H.; Kamiya, Y.; McCourt, P. Regulation of abscisic acid signaling by the ethylene response pathway in Arabidopsis. Plant Cell 2000, 12, 1117–1126. [Google Scholar] [CrossRef]
- Xie, Z.; Jiang, D.; Dai, T.; Jing, Q.; Cao, W. Effects of exogenous ABA and cytokinin on leaf photosynthesis and grain protein accumulation in wheat ears cultured in vitro. Plant Growth Regul. 2004, 44, 25–32. [Google Scholar] [CrossRef]
- Mansouri, H.; Asrar, Z.; Szopa, J. Effects of ABA on primary terpenoids and Δ9-tetrahydrocannabinol in Cannabis sativa L. at flowering stage. Plant Growth Regul. 2009, 58, 269–277. [Google Scholar] [CrossRef]
- Ortiz, D.; Hu, J.Y.; Salas Fernandez, M.G. Genetic architecture of photosynthesis in Sorghum bicolor under non-stress and cold stress conditions. J. Exp. Bot. 2017, 68, 4545–4557. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Hartung, W. Long-distance signalling of abscisic acid (ABA): The factors regulating the intensity of the ABA signal. J. Exp. Bot. 2007, 59, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, F.; Zhang, J.; Davies, W.J. What information is conveyed by an ABA signal from maize roots in drying field soil? Plant Cell Environ. 1992, 15, 185–191. [Google Scholar] [CrossRef]
- Tardieu, F.; Zhang, J.; Katerji, N. Xylem ABA controls the stomatal conductance of field-grown maize subjected to soil compaction or soil drying. Plant Cell Environ. 1992, 15, 193–197. [Google Scholar] [CrossRef]
- Zhang, J.; Davies, W.J. Changes in the concentration of ABA in xylem sap as a function of changing soil water status can account for changes in leaf conductance and growth. Plant Cell Environ. 1990, 13, 277–285. [Google Scholar] [CrossRef]
- Sauter, A.; Davies, W.J.; Hartung, W. The long-distance abscisic acid signal in the droughted plant: the fate of the hormone on its way from root to shoot. J. Exp. Bot. 2001, 52, 1991–1997. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Luo, D.; Chai, X.; Wu, Y.; Wang, Y.; Nan, Z.; Yang, Q.; Liu, W.; Liu, Z. Multiple regulatory networks are activated during cold stress in Medicago sativa L. Int. J. Mol. Sci. 2018, 19, 3169. [Google Scholar] [CrossRef]
- Bari, R.; Jones, J.D. Role of plant hormones in plant defence responses. Plant Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef]
- Spoel, S.H.; Dong, X. How do plants achieve immunity? Defence without specialized immune cells. Nat. Rev. Immunol. 2012, 12, 89–100. [Google Scholar] [CrossRef]
- De Vleesschauwer, D.; Gheysen, G.; Höfte, M. Hormone defense networking in rice: tales from a different world. Trends Plant Sci. 2013, 18, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Song, J.T.; Koo, Y.J.; Seo, H.S.; Kim, M.C.; Do Choi, Y.; Kim, J.H. Overexpression of AtSGT1, an Arabidopsis salicylic acid glucosyltransferase, leads to increased susceptibility to Pseudomonas syringae. Phytochemistry 2008, 69, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Sarowar, S.; Kim, Y.J.; Kim, E.N.; Kim, K.D.; Hwang, B.K.; Islam, R.; Shin, J.S. Overexpression of a pepper basic pathogenesis-related protein 1 gene in tobacco plants enhances resistance to heavy metal and pathogen stresses. Plant Cell Rep. 2005, 24, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Stumpe, M.; Göbel, C.; Faltin, B.; Beike, A.K.; Hause, B.; Himmelsbach, K.; Bode, J.; Kramell, R.; Wasternack, C.; Frank, W.; et al. The moss Physcomitrella patens contains cyclopentenones but no jasmonates: mutations in allene oxide cyclase lead to reduced fertility and altered sporophyte morphology. New Phytol. 2010, 188, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.S.; Hwang, B.K. The pepper 9-lipoxygenase gene CaLOX1 functions in defense and cell death responses to microbial pathogens. Plant Physiol. 2010, 152, 948–967. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Xu, W.; Li, D.; Hu, X.; Liu, J.; Zhang, R.; Tong, Z.; Dong, J.; Su, Z.; Zhang, L.; et al. De novo transcriptome analysis of Medicago falcata reveals novel insights about the mechanisms underlying abiotic stress-responsive pathway. BMC Genom. 2015, 16, 818. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Tan, J.; Zhuo, C.; Wang, C.; Xiang, B.; Wang, Z. Abscisic acid, H2O2 and nitric oxide interactions mediated cold-induced S-adenosylmethionine synthetase in Medicago sativa subsp. falcata that confers cold tolerance through up-regulating polyamine oxidation. Plant Biotechnol. J. 2014, 12, 601–612. [Google Scholar] [CrossRef]
- Prathuangwong, S.; Buensanteai, N. Bacillus amyloliquefaciens induced systemic resistance against bacterial pustule pathogen with increased phenols, phenylalanine ammonia lyase, peroxidases and 1, 3-β-glucanases in soybean plants. Acta Phytopathologica et Entomologica Hungarica 2007, 42, 321–330. [Google Scholar] [CrossRef]
- Singh, S.D.; Robbins, N.; Zaas, A.K.; Schell, W.A.; Perfect, J.R.; Cowen, L.E. Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog. 2009, 5, e1000532. [Google Scholar] [CrossRef]
- Gitelson, A.A.; Gritz, Y.; Merzlyak, M.N. Relationships between leaf chlorophyll content and spectral reflectance and algorithms for non-destructive chlorophyll assessment in higher plant leaves. J. Plant Physiol. 2003, 160, 271–282. [Google Scholar] [CrossRef]
- Su, L.Y.; Dai, Z.W.; Li, S.H.; Xin, H.P. A novel system for evaluating drought-cold tolerance of grapevines using chlorophyll fluorescence. BMC Plant Biol. 2015, 15, 82. [Google Scholar] [CrossRef] [PubMed]
- Castrejón, S.E.; Yatsimirsky, A.K. Cyclodextrin enhanced fluorimetric determination of malonaldehyde by the thiobarbituric acid method. Talanta 1997, 44, 951–957. [Google Scholar] [CrossRef]
- Men, Y.; Wang, D.; Li, B.; Su, Y.; Chen, G. Effects of drought stress on the antioxidant system, osmolytes and secondary metabolites of Saposhnikovia divaricata seedlings. Acta Physiol. Plant. 2018, 40, 191. [Google Scholar] [CrossRef]
- Toivonen, P.M.A.; Sweeney, M. Differences in chlorophyll loss at 13 C for two broccoli (Brassica oleracea L.) cultivars associated with antioxidant enzyme activities. J. Agric. Food Chem. 1998, 46, 20–24. [Google Scholar] [CrossRef]
- Vieira, S.M.; Silva, T.M.; Glória, M.B.A. Influence of processing on the levels of amines and proline and on the physico-chemical characteristics of concentrated orange juice. Food Chem. 2010, 119, 7–11. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Z.; Chen, S.; Ma, L.; Wang, H.; Dong, R.; Wang, Y.; Liu, Z. Global transcriptome profiling analysis reveals insight into saliva-responsive genes in alfalfa. Plant Cell Rep. 2016, 35, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011, 12, 323. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.I.N.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodríguez, P.; Riano-Pachon, D.M.; Corrêa, L.G.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2009, 38, D822–D827. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TF Family | A1 | A2 | A3 | Total |
|---|---|---|---|---|
| ABI3VP1 | 0 | 1 | 0 | 1 |
| AP2-EREBP | 3 | 6 | 3 | 9 |
| ARF | 2 | 4 | 3 | 6 |
| bHLH | 1 | 1 | 1 | 3 |
| bZIP | 11 | 9 | 7 | 12 |
| C2C2-Dof | 1 | 1 | 0 | 1 |
| C2H2 | 1 | 1 | 0 | 1 |
| C3H | 2 | 1 | 1 | 4 |
| E2F-DP | 0 | 0 | 1 | 1 |
| EIL | 2 | 1 | 1 | 2 |
| FAR1 | 0 | 1 | 0 | 1 |
| G2-like | 0 | 0 | 1 | 1 |
| GRAS | 1 | 3 | 1 | 3 |
| HB | 0 | 1 | 0 | 1 |
| HSF | 0 | 1 | 1 | 2 |
| LOB | 1 | 0 | 0 | 1 |
| MADS | 0 | 0 | 1 | 1 |
| mTERF | 0 | 0 | 1 | 1 |
| MYB | 4 | 6 | 6 | 9 |
| NAC | 3 | 3 | 2 | 3 |
| TAZ | 0 | 1 | 0 | 1 |
| TIG | 3 | 3 | 1 | 5 |
| Trihelix | 7 | 1 | 0 | 8 |
| TUB | 2 | 1 | 1 | 3 |
| WRKY | 0 | 0 | 2 | 2 |
| Total | 44 | 46 | 34 | 82 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, D.; Wu, Y.; Liu, J.; Zhou, Q.; Liu, W.; Wang, Y.; Yang, Q.; Wang, Z.; Liu, Z. Comparative Transcriptomic and Physiological Analyses of Medicago sativa L. Indicates that Multiple Regulatory Networks Are Activated during Continuous ABA Treatment. Int. J. Mol. Sci. 2019, 20, 47. https://doi.org/10.3390/ijms20010047
Luo D, Wu Y, Liu J, Zhou Q, Liu W, Wang Y, Yang Q, Wang Z, Liu Z. Comparative Transcriptomic and Physiological Analyses of Medicago sativa L. Indicates that Multiple Regulatory Networks Are Activated during Continuous ABA Treatment. International Journal of Molecular Sciences. 2019; 20(1):47. https://doi.org/10.3390/ijms20010047
Chicago/Turabian StyleLuo, Dong, Yuguo Wu, Jie Liu, Qiang Zhou, Wenxian Liu, Yanrong Wang, Qingchuan Yang, Zengyu Wang, and Zhipeng Liu. 2019. "Comparative Transcriptomic and Physiological Analyses of Medicago sativa L. Indicates that Multiple Regulatory Networks Are Activated during Continuous ABA Treatment" International Journal of Molecular Sciences 20, no. 1: 47. https://doi.org/10.3390/ijms20010047
APA StyleLuo, D., Wu, Y., Liu, J., Zhou, Q., Liu, W., Wang, Y., Yang, Q., Wang, Z., & Liu, Z. (2019). Comparative Transcriptomic and Physiological Analyses of Medicago sativa L. Indicates that Multiple Regulatory Networks Are Activated during Continuous ABA Treatment. International Journal of Molecular Sciences, 20(1), 47. https://doi.org/10.3390/ijms20010047