Translatomics: The Global View of Translation


Abstract
1. Introduction
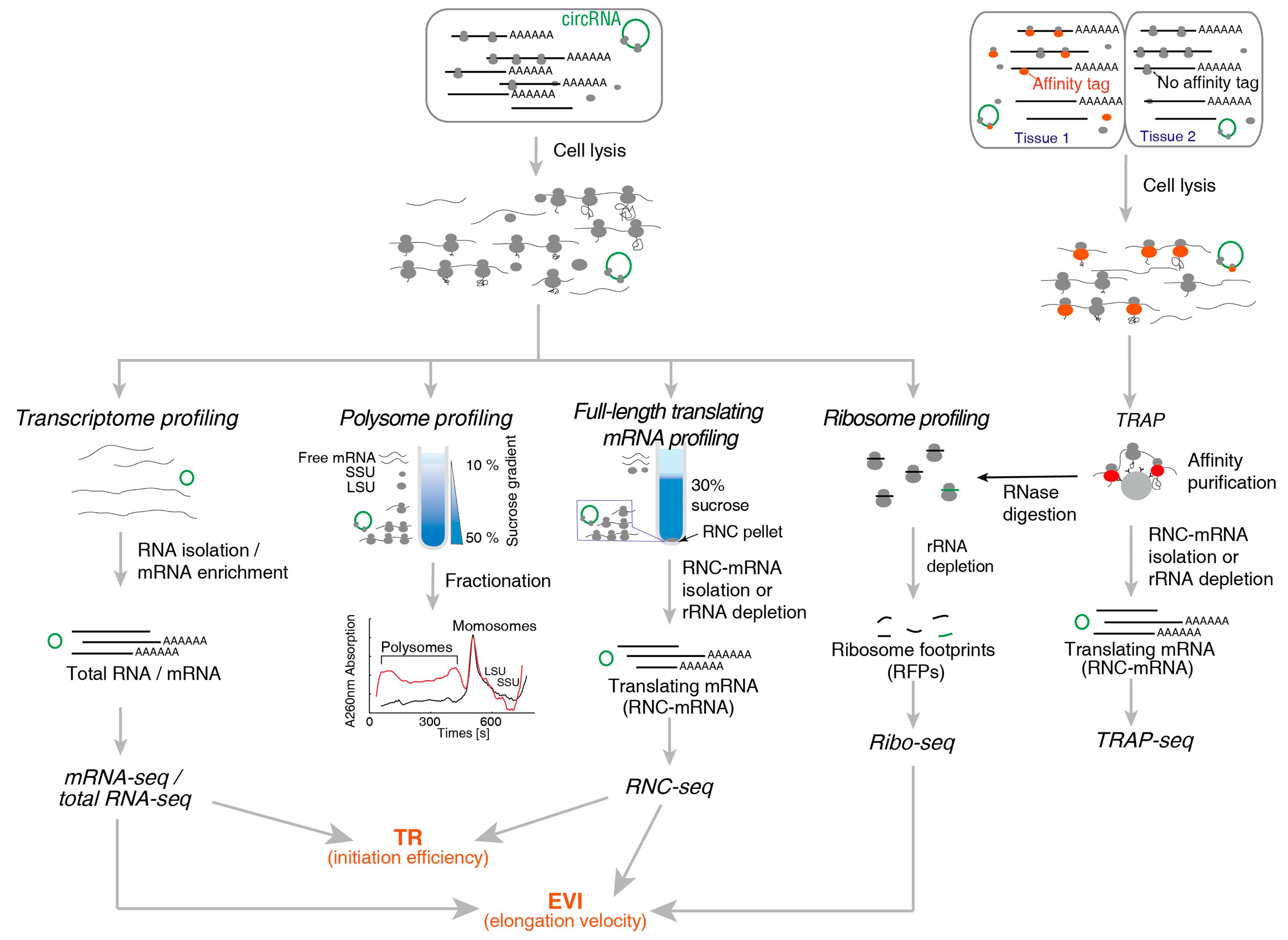
2. Methods for Translatome Research
2.1. Method for Translating mRNA
2.1.1. Polysome Profiling
2.1.2. RNC-Seq
2.1.3. Ribo-Seq
2.1.4. TRAP-Seq
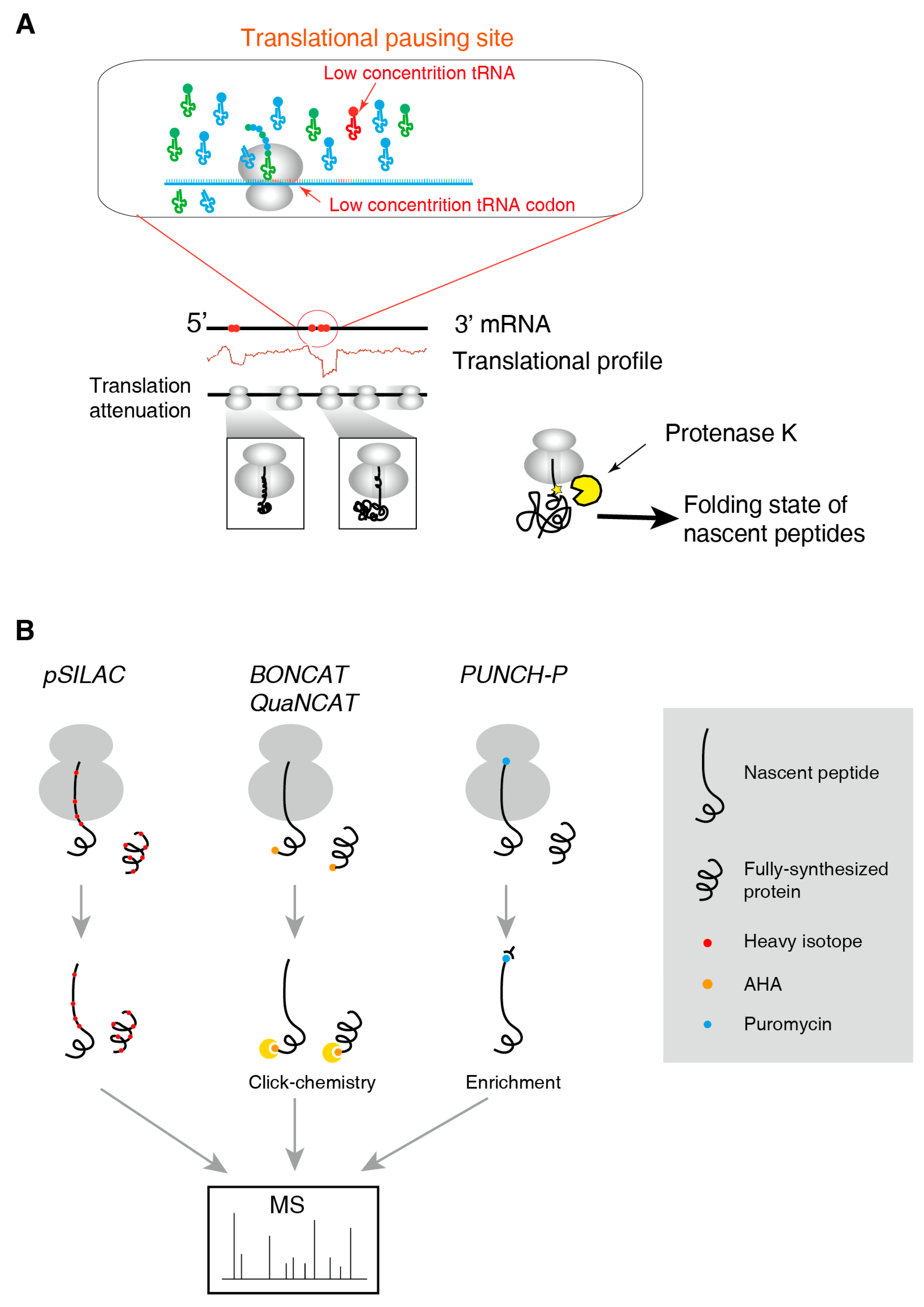
2.2. Methods for tRNAome
2.3. Methods for the Folding State of Nascent Polypeptides
2.4. Methods to Identify and to Quantify Nascent Peptides
2.5. Methods for Detecting mRNA Co-Translational Decay Intermediates
2.6. Visualization of Translation In Vivo
3. Translatomics in Fundamental Biology
3.1. The “Quantitative” Central Dogma of Molecular Biology
3.2. Translational Pausing Induces Co-Translational Folding
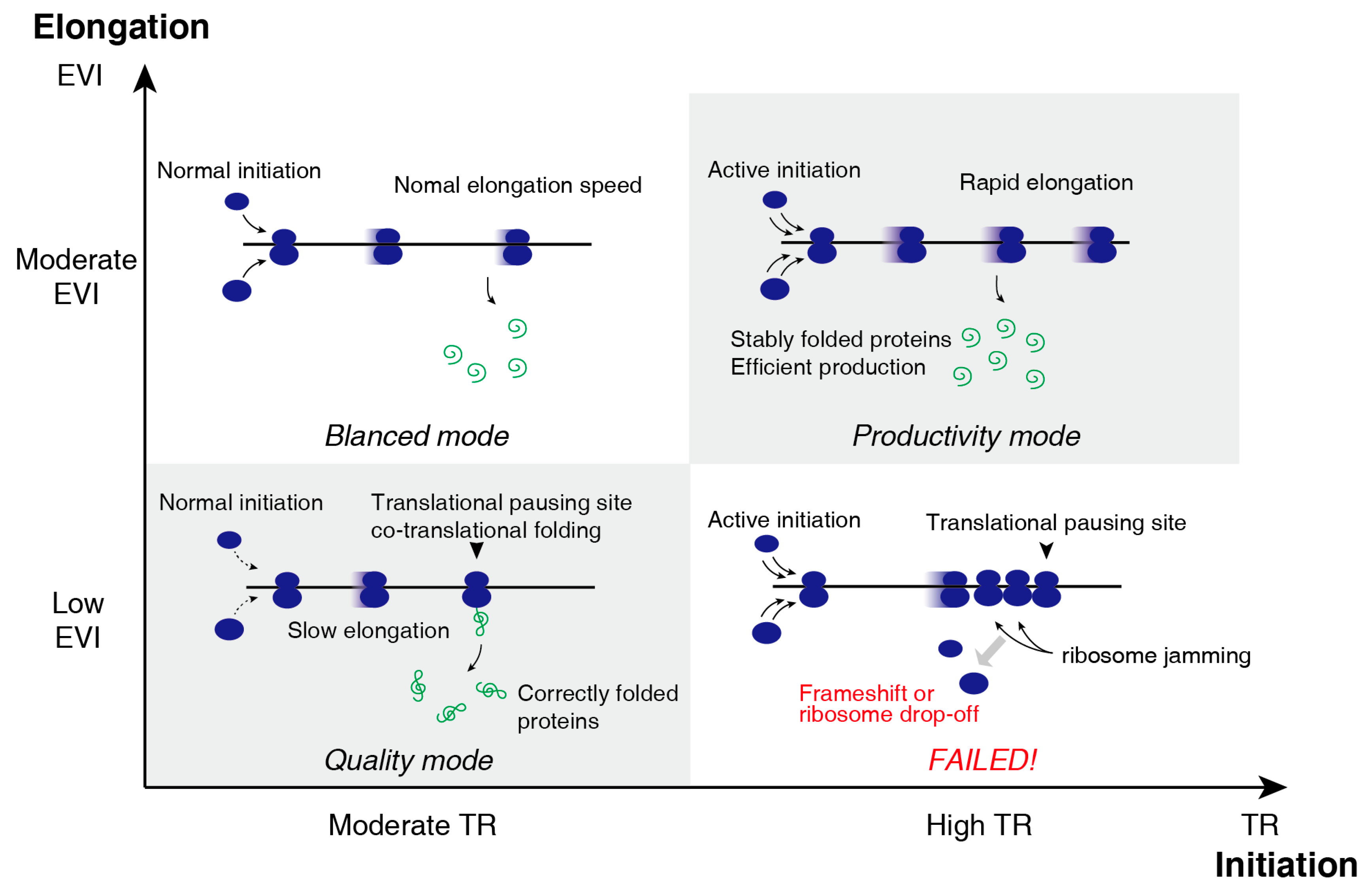
3.3. Two-Dimensional Translational Control Initiation and Elongation
3.4. Alternative Translation Start Sites and Readthrough
3.5. Ribosome Diversity
4. Translatomics in Biology/Disease-Relevant Studies
4.1. Perturbation of Global Translation in Cancer
4.2. Microbial Stress Resistance
4.3. Rhythmic Translation in Circadian Clock Regulation
4.4. Translational Control in Plants
5. Application of Translatomics
5.1. Missing Protein and New Protein Discovery
5.2. Enhancing Recombinant Protein Production
6. Internet Resources for Translatome
7. Conclusion and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2-DE | 2-dimensional electrophoresis |
| 5Pseq | 5′-monophosphorylated ends sequencing |
| AHA | Azidohomoalanine |
| BONCAT/QuaNCAT | Bio-Orthogonal/Quantitative Non-Canonical Amino acid Tagging |
| CDS | Coding sequence |
| CVN | Cyanovirin-N, an antiviral protein originated from cyanobacteria |
| FRET | Fluorescence resonance energy transfer |
| GMUCT | Genome-wide mapping of uncapped and cleaved transcripts |
| EVI | Translation elongation speed |
| FDR | False discover rate |
| HPP | The Human Proteome Project |
| miRNA | MicroRNAs |
| MS | Mass spectrometry |
| ncRNA | Non-coding RNAs |
| NCT | Nascent chain tracking |
| NGS | Next-generation sequencing |
| NGD | No-go decay |
| NMD | Nonsense-mediated mRNA decay |
| NMR | Nuclear magnetic resonance |
| NSD | Non-stop decay |
| ORF | Open reading frame |
| PARE | parallel analysis of RNA ends |
| PUNCH-P | PUromycin-associated Nascent CHain Proteomics |
| pSILAC | Pulsed-SILAC, Pulsed Stable Isotope Labelling by Amino acids |
| RFP | Ribosome footprints, equivalent to ribosome protected fragments, RPFs |
| Ribo-seq | Ribosome profiling |
| RNC | Ribosome nascent-chain complex |
| RNC-mRNA | Translating mRNAs |
| RNC-seq | Full-length translating mRNA profiling |
| ROS | Reactive oxygen species |
| SSU | Small subunit of ribosome |
| TR | Translation initiation efficiency |
| TRAP-seq | Translating ribosome affinity purification sequencing |
| tRNA-seq | Full-length tRNA sequencing |
| UTR | Untranslated regions of genes |
References
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Fedyunin, I.; Miekley, O.; Valleriani, A.; Moura, A.; Ignatova, Z. Global and local depletion of ternary complex limits translational elongation. Nucleic Acids Res. 2010, 38, 4778–4787. [Google Scholar] [CrossRef]
- Zhong, J.; Xiao, C.; Gu, W.; Du, G.; Sun, X.; He, Q.Y.; Zhang, G. Transfer RNAs Mediate the Rapid Adaptation of Escherichia coli to Oxidative Stress. PLoS Genet. 2015, 11, e1005302. [Google Scholar] [CrossRef]
- Morello, L.G.; Hesling, C.; Coltri, P.P.; Castilho, B.A.; Rimokh, R.; Zanchin, N.I. The NIP7 protein is required for accurate pre-rRNA processing in human cells. Nucleic Acids Res. 2011, 39, 648–665. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Hubalewska, M.; Ignatova, Z. Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol. 2009, 16, 274–280. [Google Scholar] [CrossRef]
- Heyer, E.E.; Moore, M.J. Redefining the Translational Status of 80S Monosomes. Cell 2016, 164, 757–769. [Google Scholar] [CrossRef] [PubMed]
- King, H.A.; Gerber, A.P. Translatome profiling: Methods for genome-scale analysis of mRNA translation. Brief. Funct. Genom. 2016, 15, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.J.D.; Wang, M.; Audas, T.E.; Kwon, D.; Carlsson, S.K.; Timpano, S.; Evagelou, S.L.; Brothers, S.; Gonzalgo, M.L.; Krieger, J.R.; et al. Systemic Reprogramming of Translation Efficiencies on Oxygen Stimulus. Cell Rep. 2016, 14, 1293–1300. [Google Scholar] [CrossRef]
- Wang, T.; Cui, Y.; Jin, J.; Guo, J.; Wang, G.; Yin, X.; He, Q.Y.; Zhang, G. Translating mRNAs strongly correlate to proteins in a multivariate manner and their translation ratios are phenotype specific. Nucleic Acids Res. 2013, 41, 4743–4754. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Brar, G.A.; Rouskin, S.; McGeachy, A.M.; Weissman, J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012, 7, 1534–1550. [Google Scholar] [CrossRef] [PubMed]
- Paulet, D.; David, A.; Rivals, E. Ribo-seq enlightens codon usage bias. DNA Res. 2017, 24. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T. Ribosome Footprint Profiling of Translation throughout the Genome. Cell 2016, 165, 22–33. [Google Scholar] [CrossRef]
- Mohammad, F.; Woolstenhulme, C.J.; Green, R.; Buskirk, A.R. Clarifying the Translational Pausing Landscape in Bacteria by Ribosome Profiling. Cell Rep. 2016, 14, 686–694. [Google Scholar] [CrossRef]
- Baudin-Baillieu, A.; Hatin, I.; Legendre, R.; Namy, O. Translation Analysis at the Genome Scale by Ribosome Profiling. Methods Mol. Biol. 2016, 1361, 105–124. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T. Ribosome profiling: New views of translation, from single codons to genome scale. Nat. Rev. Genet. 2014, 15, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.Y.; Calviello, L.; Wu, H.L.; Li, F.W.; Rothfels, C.J.; Ohler, U.; Benfey, P.N. Super-resolution ribosome profiling reveals unannotated translation events in Arabidopsis. Proc. Natl. Acad. Sci. USA 2016, 113, E7126–E7135. [Google Scholar] [CrossRef] [PubMed]
- Gerashchenko, M.V.; Gladyshev, V.N. Ribonuclease selection for ribosome profiling. Nucleic Acids Res. 2017, 45, e6. [Google Scholar] [CrossRef]
- Archer, S.K.; Shirokikh, N.E.; Beilharz, T.H.; Preiss, T. Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature 2016, 535, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Shirokikh, N.E.; Archer, S.K.; Beilharz, T.H.; Powell, D.; Preiss, T. Translation complex profile sequencing to study the in vivo dynamics of mRNA-ribosome interactions during translation initiation, elongation and termination. Nat. Protoc. 2017, 12, 697–731. [Google Scholar] [CrossRef]
- Guttman, M.; Russell, P.; Ingolia, N.T.; Weissman, J.S.; Lander, E.S. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell 2013, 154, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Cui, Y.; Guo, J.; Chen, Z.; Yang, L.; He, Q.Y.; Zhang, G.; Wang, T. Resolving chromosome-centric human proteome with translating mRNA analysis: A strategic demonstration. J. Proteome Res. 2014, 13, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lu, S.; Liu, W.; Zhao, X.; Mai, Z.; Zhang, G. Optimal Settings of Mass Spectrometry Open Search Strategy for Higher Confidence. J. Proteome Res. 2018, 17, 3719–3729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhao, K.; Xu, X.; Yang, Y.; Yan, S.; Wei, P.; Liu, H.; Xu, J.; Xiao, F.; Zhou, H.; et al. A peptide encoded by circular form of LINC-PINT suppresses oncogenic transcriptional elongation in glioblastoma. Nat. Commun. 2018, 9, 4475. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Guo, J.; Gu, W.; Cui, Y.; Zhong, J.; Jin, J.; He, Q.Y.; Wang, T.; Zhang, G. Genome-Wide and Experimental Resolution of Relative Translation Elongation Speed at Individual Gene Level in Human Cells. PLoS Genet. 2016, 12, e1005901. [Google Scholar] [CrossRef] [PubMed]
- Inada, T.; Winstall, E.; Tarun, S.Z., Jr.; Yates, J.R., 3rd; Schieltz, D.; Sachs, A.B. One-step affinity purification of the yeast ribosome and its associated proteins and mRNAs. RNA 2002, 8, 948–958. [Google Scholar] [CrossRef]
- Heiman, M.; Kulicke, R.; Fenster, R.J.; Greengard, P.; Heintz, N. Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP). Nat. Protoc. 2014, 9, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Mandadi, K.K.; Scholthof, K.B. Genome-wide analysis of alternative splicing landscapes modulated during plant-virus interactions in Brachypodium distachyon. Plant Cell 2015, 27, 71–85. [Google Scholar] [CrossRef]
- Wang, Y.; Jiao, Y. Translating ribosome affinity purification (TRAP) for cell-specific translation profiling in developing flowers. Methods Mol. Biol. 2014, 1110, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, J.; Gu, W.; Wei, B.; Lei, Y.; Xiong, S.; Zhang, G. Rational design of translational pausing without altering the amino acid sequence dramatically promotes soluble protein expression. J. Biotechnol. 2014, 189, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. GtRNAdb: A database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009, 37, D93–D97. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Nilsson, L.; Kurland, C.G. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 1996, 260, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Kanaya, S.; Yamada, Y.; Kudo, Y.; Ikemura, T. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: Gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene 1999, 238, 143–155. [Google Scholar] [CrossRef]
- Kanduc, D. Changes of tRNA population during compensatory cell proliferation: Differential expression of methionine-tRNA species. Arch. Biochem. Biophys. 1997, 342, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Czech, A.; Fedyunin, I.; Zhang, G.; Ignatova, Z. Silent mutations in sight: Co-variations in tRNA abundance as a key to unravel consequences of silent mutations. Mol. Biosyst. 2010, 6, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.-L.; Mai, Z.-B.; Lian, X.-L.; Zhong, J.-Y.; Jin, J.-J.; He, Q.-Y.; Zhang, G. FANSe2: A robust and cost-efficient alignment tool for quantitative next-generation sequencing applications. PLoS ONE 2014, 9, e94250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Fedyunin, I.; Kirchner, S.; Xiao, C.; Valleriani, A.; Ignatova, Z. FANSe: An accurate algorithm for quantitative mapping of large scale sequencing reads. Nucleic Acids Res. 2012, 40, e83. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Qin, Y.; Clark, W.C.; Dai, Q.; Yi, C.; He, C.; Lambowitz, A.M.; Pan, T. Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 2015, 12, 835–837. [Google Scholar] [CrossRef] [PubMed]
- Cozen, A.E.; Quartley, E.; Holmes, A.D.; Hrabeta-Robinson, E.; Phizicky, E.M.; Lowe, T.M. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods 2015, 12, 879–884. [Google Scholar] [CrossRef]
- Gogakos, T.; Brown, M.; Garzia, A.; Meyer, C.; Hafner, M.; Tuschl, T. Characterizing Expression and Processing of Precursor and Mature Human tRNAs by Hydro-tRNAseq and PAR-CLIP. Cell Rep. 2017, 20, 1463–1475. [Google Scholar] [CrossRef]
- Chen, C.W.; Tanaka, M. Genome-wide Translation Profiling by Ribosome-Bound tRNA Capture. Cell Rep. 2018, 23, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.T.; Fucini, P.; Cabrita, L.D.; Launay, H.; Dobson, C.M.; Christodoulou, J. Structure and dynamics of a ribosome-bound nascent chain by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2007, 104, 16516–16521. [Google Scholar] [CrossRef] [PubMed]
- Deckert, A.; Waudby, C.A.; Wlodarski, T.; Wentink, A.S.; Wang, X.; Kirkpatrick, J.P.; Paton, J.F.; Camilloni, C.; Kukic, P.; Dobson, C.M.; et al. Structural characterization of the interaction of alpha-synuclein nascent chains with the ribosomal surface and trigger factor. Proc. Natl. Acad. Sci. USA 2016, 113, 5012–5017. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Wei, S.S.; Ji, Y.L.; Guo, X.J.; Yang, F.Q. Quantitative proteomics using SILAC: Principles, applications and developments. Proteomics 2015, 15, 3175–3192. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.K.; Hammond, D.E.; Clagule, M.J.; Gaskell, S.J.; Beynon, R.J. Turnover of the Human Proteome: Determination of Protein Intracellular Stability by Dynamic SILAC. J. Proteome Res. 2009, 8, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Aviner, R.; Geiger, T.; Elroy-Stein, O. Novel proteomic approach (PUNCH-P) reveals cell cycle-specific fluctuations in mRNA translation. Gene Dev. 2013, 27, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, D.C.; Lee, J.J.; Link, A.J.; Graumann, J.; Tirrell, D.A.; Schuman, E.M. Labelling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat. Protoc. 2007, 2, 532–540. [Google Scholar] [CrossRef]
- Howden, A.J.M.; Geoghegan, V.; Katsch, K.; Efstathiou, G.; Bhushan, B.; Boutureira, O.; Thomas, B.; Trudgian, D.C.; Kessler, B.M.; Dieterich, D.C.; et al. QuaNCAT: Quantitating proteome dynamics in primary cells. Nat. Methods 2013, 10, 343–346. [Google Scholar] [CrossRef]
- Kramer, G.; Sprenger, R.R.; Back, J.; Dekker, H.L.; Nessen, M.A.; van Maarseveen, J.H.; de Koning, L.J.; Hellingwerf, K.J.; de Jong, L.; de Koster, C.G. Identification and Quantitation of Newly Synthesized Proteins in Escherichia coli by Enrichment of Azidohomoalanine-labelled Peptides with Diagonal Chromatography. Mol. Cell. Proteom. 2009, 8, 1599–1611. [Google Scholar] [CrossRef]
- Zhang, G.A.; Bowling, H.; Hom, N.; Kirshenbaum, K.; Klann, E.; Chao, M.V.; Neubert, T.A. In-Depth Quantitative Proteomic Analysis of de Novo Protein Synthesis Induced by Brain-Derived Neurotrophic Factor. J. Proteome Res. 2014, 13, 5707–5714. [Google Scholar] [CrossRef]
- Chang, C.; Li, L.; Zhang, C.; Wu, S.; Guo, K.; Zi, J.; Chen, Z.; Jiang, J.; Ma, J.; Yu, Q. Systematic analyses of the transcriptome, translatome and proteome provide a global view and potential strategy for the C-HPP. J. Proteome Res. 2013, 13, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Aviner, R.; Geiger, T.; Elroy-Stein, O. Genome-wide identification and quantification of protein synthesis in cultured cells and whole tissues by puromycin-associated nascent chain proteomics (PUNCH-P). Nat. Protoc. 2014, 9, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Overall, C.M.; Van Eyk, J.E.; Baker, M.S.; Palk, Y.K.; Weintraub, S.T.; Lane, L.; Martens, L.; Vandenbrouck, Y.; Kusebauch, U.; et al. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 2.1. J. Proteome Res. 2016, 15, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Puighermanal, E.; Biever, A.; Pascoli, V.; Melser, S.; Pratlong, M.; Cutando, L.; Rialle, S.; Severac, D.; Boubaker-Vitre, J.; Meyuhas, O.; et al. Ribosomal Protein S6 Phosphorylation Is Involved in Novelty-Induced Locomotion, Synaptic Plasticity and mRNA Translation. Front. Mol. Neurosci. 2017, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef] [PubMed]
- Zur, H.; Aviner, R.; Tuller, T. Complementary Post Transcriptional Regulatory Information is Detected by PUNCH-P and Ribosome Profiling. Sci. Rep. 2016, 6, 21635. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhong, J.; Liu, W.; Zhao, J.; Zhang, G. Protein-Level Integration Strategy of Multiengine MS Spectra Search Results for Higher Confidence and Sequence Coverage. J. Proteome Res. 2017, 16, 4446–4454. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, D.; Zhao, J.; He, Q.-Y.; Zhang, G. Deep coverage of nascent polypeptides using mass spectrometry and translatome sequencing. Manuscript in preparation.
- Heck, A.M.; Wilusz, J. The Interplay between the RNA Decay and Translation Machinery in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2018, 10, 2398–2416. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Sweet, T.J.; Chamnongpol, S.; Baker, K.E.; Coller, J. Co-translational mRNA decay in Saccharomyces cerevisiae. Nature 2009, 461, 225–229. [Google Scholar] [CrossRef]
- Mazzoni-Putman, S.M.; Stepanova, A.N. A Plant Biologist’s Toolbox to Study Translation. Front. Plant Sci. 2018, 9, 873. [Google Scholar] [CrossRef]
- Gregory, B.D.; O’Malley, R.C.; Lister, R.; Urich, M.A.; Tonti-Filippini, J.; Chen, H.; Millar, A.H.; Ecker, J.R. A link between RNA metabolism and silencing affecting Arabidopsis development. Dev. Cell 2008, 14, 854–866. [Google Scholar] [CrossRef] [PubMed]
- German, M.A.; Pillay, M.; Jeong, D.H.; Hetawal, A.; Luo, S.; Janardhanan, P.; Kannan, V.; Rymarquis, L.A.; Nobuta, K.; German, R.; et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 2008, 26, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Pelechano, V.; Wei, W.; Steinmetz, L.M. Widespread Co-translational RNA Decay Reveals Ribosome Dynamics. Cell 2015, 161, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Willmann, M.R.; Berkowitz, N.D.; Gregory, B.D. Improved genome-wide mapping of uncapped and cleaved transcripts in eukaryotes—GMUCT 2.0. Methods 2014, 67, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Willmann, M.R.; Anderson, S.J.; Gregory, B.D. Genome-Wide Mapping of Uncapped and Cleaved Transcripts Reveals a Role for the Nuclear mRNA Cap-Binding Complex in Cotranslational RNA Decay in Arabidopsis. Plant Cell 2016, 28, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.Y.; Lee, W.C.; Chou, H.C.; Chen, A.P.; Chou, S.J.; Chen, H.M. Global Analysis of Truncated RNA Ends Reveals New Insights into Ribosome Stalling in Plants. Plant Cell 2016, 28, 2398–2416. [Google Scholar] [CrossRef]
- Pelechano, V.; Alepuz, P. eIF5A facilitates translation termination globally and promotes the elongation of many non polyproline-specific tripeptide sequences. Nucleic Acids Res. 2017, 45, 7326–7338. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, H.; Broitman, S.L.; Reiche, M.; Farrell, I.; Cooperman, B.S.; Goldman, Y.E. Dynamics of translation by single ribosomes through mRNA secondary structures. Nat. Struct. Mol. Biol. 2013, 20, 582–588. [Google Scholar] [CrossRef]
- Stevens, B.; Chen, C.; Farrell, I.; Zhang, H.; Kaur, J.; Broitman, S.L.; Smilansky, Z.; Cooperman, B.S.; Goldman, Y.E. FRET-based identification of mRNAs undergoing translation. PLoS ONE 2012, 7, e38344. [Google Scholar] [CrossRef]
- Morisaki, T.; Lyon, K.; DeLuca, K.F.; DeLuca, J.G.; English, B.P.; Zhang, Z.; Lavis, L.D.; Grimm, J.B.; Viswanathan, S.; Looger, L.L.; et al. Real-time quantification of single RNA translation dynamics in living cells. Science 2016, 352, 1425–1429. [Google Scholar] [CrossRef]
- Maier, T.; Guell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ignatova, Z. Generic algorithm to predict the speed of translational elongation: Implications for protein biogenesis. PLoS ONE 2009, 4, e5036. [Google Scholar] [CrossRef] [PubMed]
- Zwanzig, R.; Szabo, A.; Bagchi, B. Levinthal’s paradox. Proc. Natl. Acad. Sci. USA 1992, 89, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Sauna, Z.E.; Kimchi-Sarfaty, C. Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 2011, 12, 683–691. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Lareau, L.F.; Weissman, J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 2011, 147, 789–802. [Google Scholar] [CrossRef]
- Yamamoto, H.; Wittek, D.; Gupta, R.; Qin, B.; Ueda, T.; Krause, R.; Yamamoto, K.; Albrecht, R.; Pech, M.; Nierhaus, K.H. 70S-scanning initiation is a novel and frequent initiation mode of ribosomal translation in bacteria. Proc. Natl. Acad. Sci. USA 2016, 113, E1180–E1189. [Google Scholar] [CrossRef]
- Milon, P.; Rodnina, M.V. Kinetic control of translation initiation in bacteria. Crit. Rev. Biochem. Mol. 2012, 47, 334–348. [Google Scholar] [CrossRef]
- Tsai, A.; Petrov, A.; Marshall, R.A.; Korlach, J.; Uemura, S.; Puglisi, J.D. Heterogeneous pathways and timing of factor departure during translation initiation. Nature 2012, 487, 390–394. [Google Scholar] [CrossRef]
- McCutcheon, J.P.; Agrawal, R.K.; Philips, S.M.; Grassucci, R.A.; Gerchman, S.E.; Clemons, W.M., Jr.; Ramakrishnan, V.; Frank, J. Location of translational initiation factor IF3 on the small ribosomal subunit. Proc. Natl. Acad. Sci. USA 1999, 96, 4301–4306. [Google Scholar] [CrossRef] [PubMed]
- Kaempfer, R. Initiation factor IF-3: A specific inhibitor of ribosomal subunit association. J. Mol. Biol. 1972, 71, 583–598. [Google Scholar] [CrossRef]
- Grunberg-Manago, M.; Dessen, P.; Pantaloni, D.; Godefroy-Colburn, T.; Wolfe, A.D.; Dondon, J. Light-scattering studies showing the effect of initiation factors on the reversible dissociation of Escherichia coli ribosomes. J. Mol. Biol. 1975, 94, 461–478. [Google Scholar] [PubMed]
- Zavialov, A.V.; Hauryliuk, V.V.; Ehrenberg, M. Splitting of the posttermination ribosome into subunits by the concerted action of RRF and EF-G. Mol. Cell 2005, 18, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Risuleo, G.; Gualerzi, C.; Pon, C. Specificity and properties of the destabilization, induced by initiation factor IF-3, of ternary complexes of the 30-S ribosomal subunit, aminoacyl-tRNA and polynucleotides. Eur. J. Biochem. 1976, 67, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, G.C.; Kuzmenko, A.; Kamenski, P.; Vysokikh, M.Y.; Lakunina, V.; Tankov, S.; Smirnova, E.; Soosaar, A.; Tenson, T.; Hauryliuk, V. Evolutionary and genetic analyses of mitochondrial translation initiation factors identify the missing mitochondrial IF3 in S. cerevisiae. Nucleic Acids Res. 2012, 40, 6122–6134. [Google Scholar] [CrossRef] [PubMed]
- Cummings, H.S.; Hershey, J.W. Translation initiation factor IF1 is essential for cell viability in Escherichia coli. J. Bacteriol. 1994, 176, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.P.; Clemons, W.M., Jr.; Brodersen, D.E.; Morgan-Warren, R.J.; Hartsch, T.; Wimberly, B.T.; Ramakrishnan, V. Crystal structure of an initiation factor bound to the 30S ribosomal subunit. Science 2001, 291, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Pon, C.L.; Gualerzi, C.O. Mechanism of protein biosynthesis in prokaryotic cells. Effect of initiation factor IF1 on the initial rate of 30 S initiation complex formation. FEBS Lett. 1984, 175, 203–207. [Google Scholar] [CrossRef]
- Gao, X.; Wan, J.; Qian, S.B. Genome-Wide Profiling of Alternative Translation Initiation Sites. Methods Mol. Biol. 2016, 1358, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hou, J.; Quedenau, C.; Chen, W. Pervasive isoform-specific translational regulation via alternative transcription start sites in mammals. Mol. Syst. Biol. 2016, 12, 875. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Huber, W. Alternative start and termination sites of transcription drive most transcript isoform differences across human tissues. Nucleic Acids Res. 2018, 46, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Harrell, L.; Melcher, U.; Atkins, J.F. Predominance of six different hexanucleotide recoding signals 3′ of read-through stop codons. Nucleic Acids Res. 2002, 30, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Loughran, G.; Chou, M.Y.; Ivanov, I.P.; Jungreis, I.; Kellis, M.; Kiran, A.M.; Baranov, P.V.; Atkins, J.F. Evidence of efficient stop codon readthrough in four mammalian genes. Nucleic Acids Res. 2014, 42, 8928–8938. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.G.; Foo, C.K.; Belletier, N.G.; Gavis, E.R.; Weissman, J.S. Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. eLife 2013, 2, e01179. [Google Scholar] [CrossRef]
- Schueren, F.; Thoms, S. Functional Translational Readthrough: A Systems Biology Perspective. PLoS Genet. 2016, 12, e1006196. [Google Scholar] [CrossRef]
- Faller, W.J.; Jackson, T.J.; Knight, J.R.; Ridgway, R.A.; Jamieson, T.; Karim, S.A.; Jones, C.; Radulescu, S.; Huels, D.J.; Myant, K.B.; et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 2015, 517, 497–500. [Google Scholar] [CrossRef]
- Beznoskova, P.; Wagner, S.; Jansen, M.E.; von der Haar, T.; Valasek, L.S. Translation initiation factor eIF3 promotes programmed stop codon readthrough. Nucleic Acids Res. 2015, 43, 5099–5111. [Google Scholar] [CrossRef]
- Filipovska, A.; Rackham, O. Specialization from synthesis: How ribosome diversity can customize protein function. FEBS Lett. 2013, 587, 1189–1197. [Google Scholar] [CrossRef]
- Xue, S.; Barna, M. Specialized ribosomes: A new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol. 2012, 13, 355–369. [Google Scholar] [CrossRef]
- Gunderson, J.H.; Sogin, M.L.; Wollett, G.; Hollingdale, M.; de la Cruz, V.F.; Waters, A.P.; McCutchan, T.F. Structurally distinct, stage-specific ribosomes occur in Plasmodium. Science 1987, 238, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Velichutina, I.V.; Rogers, M.J.; McCutchan, T.F.; Liebman, S.W. Chimeric rRNAs containing the GTPase centers of the developmentally regulated ribosomal rRNAs of Plasmodium falciparum are functionally distinct. RNA 1998, 4, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Parenteau, J.; Durand, M.; Morin, G.; Gagnon, J.; Lucier, J.F.; Wellinger, R.J.; Chabot, B.; Abou Elela, S. Introns within Ribosomal Protein Genes Regulate the Production and Function of Yeast Ribosomes. Cell 2011, 147, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Marygold, S.J.; Roote, J.; Reuter, G.; Lambertsson, A.; Ashburner, M.; Millburn, G.H.; Harrison, P.M.; Yu, Z.; Kenmochi, N.; Kaufman, T.C.; et al. The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol. 2007, 8, R216. [Google Scholar] [CrossRef]
- Ramagopal, S.; Ennis, H.L. Regulation of synthesis of cell-specific ribosomal proteins during differentiation of Dictyostelium discoideum. Proc. Natl. Acad. Sci. USA 1981, 78, 3083–3087. [Google Scholar] [CrossRef]
- Ramagopal, S. Induction of cell-specific ribosomal proteins in aggregation-competent nonmorphogenetic Dictyostelium discoideum. Biochem. Cell Biol. 1990, 68, 1281–1287. [Google Scholar] [CrossRef]
- Lopes, A.M.; Miguel, R.N.; Sargent, C.A.; Ellis, P.J.; Amorim, A.; Affara, N.A. The human RPS4 paralogue on Yq11.223 encodes a structurally conserved ribosomal protein and is preferentially expressed during spermatogenesis. BMC Mol. Biol. 2010, 11, 33. [Google Scholar] [CrossRef]
- Bortoluzzi, S.; d’Alessi, F.; Romualdi, C.; Danieli, G.A. Differential expression of genes coding for ribosomal proteins in different human tissues. Bioinformatics 2001, 17, 1152–1157. [Google Scholar] [CrossRef]
- Colon-Ramos, D.A.; Shenvi, C.L.; Weitzel, D.H.; Gan, E.C.; Matts, R.; Cate, J.; Kornbluth, S. Direct ribosomal binding by a cellular inhibitor of translation. Nat. Struct. Mol. Biol. 2006, 13, 103–111. [Google Scholar] [CrossRef]
- Guo, J.; Lian, X.; Zhong, J.; Wang, T.; Zhang, G. Length-dependent translation initiation benefits the functional proteome of human cells. Mol. Biosyst. 2015, 11, 370–378. [Google Scholar] [CrossRef]
- Yang, X.Y.; He, K.; Du, G.; Wu, X.; Yu, G.; Pan, Y.; Zhang, G.; Sun, X.; He, Q.Y. Integrated Translatomics with Proteomics to Identify Novel Iron-Transporting Proteins in Streptococcus pneumoniae. Front. Microbiol. 2016, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.B.; Karp, N.A.; Maywood, E.S.; Sage, E.A.; Deery, M.; O’Neill, J.S.; Wong, G.K.; Chesham, J.; Odell, M.; Lilley, K.S.; et al. Circadian orchestration of the hepatic proteome. Curr. Biol. 2006, 16, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Janich, P.; Arpat, A.B.; Castelo-Szekely, V.; Lopes, M.; Gatfield, D. Ribosome profiling reveals the rhythmic liver translatome and circadian clock regulation by upstream open reading frames. Genome Res. 2015, 25, 1848–1859. [Google Scholar] [CrossRef]
- Castelo-Szekely, V.; Arpat, A.B.; Janich, P.; Gatfield, D. Translational contributions to tissue specificity in rhythmic and constitutive gene expression. Genome Biol. 2017, 18, 116. [Google Scholar] [CrossRef] [PubMed]
- Yanguez, E.; Castro-Sanz, A.B.; Fernandez-Bautista, N.; Oliveros, J.C.; Castellano, M.M. Analysis of genome-wide changes in the translatome of Arabidopsis seedlings subjected to heat stress. PLoS ONE 2013, 8, e71425. [Google Scholar] [CrossRef] [PubMed]
- Branco-Price, C.; Kawaguchi, R.; Ferreira, R.B.; Bailey-Serres, J. Genome-wide analysis of transcript abundance and translation in Arabidopsis seedlings subjected to oxygen deprivation. Ann. Bot. 2005, 96, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Peviani, A.; van der Horst, S.; Gamm, M.; Snel, B.; Bentsink, L.; Hanson, J. Extensive translational regulation during seed germination revealed by polysomal profiling. New Phytol. 2017, 214, 233–244. [Google Scholar] [CrossRef]
- Meteignier, L.V.; El Oirdi, M.; Cohen, M.; Barff, T.; Matteau, D.; Lucier, J.F.; Rodrigue, S.; Jacques, P.E.; Yoshioka, K.; Moffett, P. Translatome analysis of an NB-LRR immune response identifies important contributors to plant immunity in Arabidopsis. J. Exp. Bot. 2017, 68, 2333–2344. [Google Scholar] [CrossRef]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef]
- Hu, Z.; Scott, H.S.; Qin, G.; Zheng, G.; Chu, X.; Xie, L.; Adelson, D.L.; Oftedal, B.E.; Venugopal, P.; Babic, M.; et al. Revealing Missing Human Protein Isoforms Based on Ab Initio Prediction, RNA-seq and Proteomics. Sci. Rep. 2015, 5, 10940. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.S.; Ahn, S.B.; Mohamedali, A.; Islam, M.T.; Cantor, D.; Verhaert, P.D.; Fanayan, S.; Sharma, S.; Nice, E.C.; Connor, M.; et al. Accelerating the search for the missing proteins in the human proteome. Nat. Commun. 2017, 8, 14271. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Y.; Zhong, J.; Zhang, J.; Chen, Z.; Yang, L.; Cao, X.; He, Q.-Y.; Zhang, G.; Wang, T. Identification of missing proteins defined by chromosome-centric proteome project in the cytoplasmic detergent-insoluble proteins. J. Proteome Res. 2015, 14, 3693–3709. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lian, X.; Zhang, W.; Guo, J.; Wang, Q.; Li, Y.; Chen, Y.; Yin, X.; Yang, P.; Lan, F. Finding missing proteins from the epigenetically manipulated human cell with stringent quality criteria. J. Proteome Res. 2015, 14, 3645–3657. [Google Scholar] [CrossRef] [PubMed]
- Hussmann, J.A.; Patchett, S.; Johnson, A.; Sawyer, S.; Press, W.H. Understanding Biases in Ribosome Profiling Experiments Reveals Signatures of Translation Dynamics in Yeast. PLoS Genet. 2015, 11, e1005732. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Brar, G.A.; Stern-Ginossar, N.; Harris, M.S.; Talhouarne, G.J.; Jackson, S.E.; Wills, M.R.; Weissman, J.S. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014, 8, 1365–1379. [Google Scholar] [CrossRef]
- Pauli, A.; Valen, E.; Schier, A.F. Identifying (non-)coding RNAs and small peptides: Challenges and opportunities. BioEssays News Rev. Mol. Cell. Dev. Biol. 2015, 37, 103–112. [Google Scholar] [CrossRef]
- Calviello, L.; Mukherjee, N.; Wyler, E.; Zauber, H.; Hirsekorn, A.; Selbach, M.; Landthaler, M.; Obermayer, B.; Ohler, U. Detecting actively translated open reading frames in ribosome profiling data. Nat. Methods 2016, 13, 165–170. [Google Scholar] [CrossRef]
- Li, Q.; Ahsan, M.A.; Chen, H.; Xue, J.; Chen, M. Discovering putative peptides encoded from non-coding RNAs in ribosome profiling data of Arabidopsis thaliana. ACS Synth. Biol. 2018. [Google Scholar] [CrossRef]
- Sterne-Weiler, T.; Martinez-Nunez, R.T.; Howard, J.M.; Cvitovik, I.; Katzman, S.; Tariq, M.A.; Pourmand, N.; Sanford, J.R. Frac-seq reveals isoform-specific recruitment to polyribosomes. Genome Res. 2013, 23, 1615–1623. [Google Scholar] [CrossRef]
- Liu, Y.; Ying, W.; Ren, Z.; Gu, W.; Zhang, Y.; Yan, G.; Yang, P.; Liu, Y.; Yin, X.; Chang, C.; et al. Chromosome-8-coded proteome of Chinese Chromosome Proteome Data set (CCPD) 2.0 with partial immunohistochemical verifications. J. Proteome Res. 2014, 13, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wen, B.; Wang, T.; Xu, Z.; Yin, X.; Xu, S.; Ren, Z.; Hou, G.; Zhou, R.; Zhao, H.; et al. Omics evidence: Single nucleotide variants transmissions on chromosome 20 in liver cancer cell lines. J. Proteome Res. 2014, 13, 200–211. [Google Scholar] [CrossRef]
- Zhang, C.; Li, N.; Zhai, L.; Xu, S.; Liu, X.; Cui, Y.; Ma, J.; Han, M.; Jiang, J.; Yang, C.; et al. Systematic analysis of missing proteins provides clues to help define all of the protein-coding genes on human chromosome 1. J. Proteome Res. 2014, 13, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Khatun, J.; Yu, Y.; Wrobel, J.A.; Risk, B.A.; Gunawardena, H.P.; Secrest, A.; Spitzer, W.J.; Xie, L.; Wang, L.; Chen, X.; et al. Whole human genome proteogenomic mapping for ENCODE cell line data: Identifying protein-coding regions. BMC Genom. 2013, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Z.; Chen, M.; Chen; Gao, X.C.; Zhu, S.; Huang, H.; Hu, M.; Zhu, H.; Yan, G.R. A Peptide Encoded by a Putative lncRNA HOXB-AS3 Suppresses Colon Cancer Growth. Mol. Cell 2017, 68, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Baneyx, F.; Mujacic, M. Recombinant protein folding and misfolding in Escherichia coli. Nat. Biotechnol. 2004, 22, 1399–1408. [Google Scholar] [CrossRef]
- Hess, A.K.; Saffert, P.; Liebeton, K.; Ignatova, Z. Optimization of translation profiles enhances protein expression and solubility. PLoS ONE 2015, 10, e0127039. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Liu, W.; Jin, J.; Xiao, Q.; Lu, R.; Chen, W.; Xiong, S.; Zhang, G. Steady-state structural fluctuation is a predictor of the necessity of pausing-mediated co-translational folding for small proteins. Biochem. Biophys. Res. Commun. 2018, 498, 186–192. [Google Scholar] [CrossRef]
- Wang, H.; Yang, L.; Wang, Y.; Chen, L.; Li, H.; Xie, Z. RPFdb v2.0: An updated database for genome-wide information of translated mRNA generated from ribosome profiling. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Michel, A.M.; Kiniry, S.J.; O’Connor, P.B.F.; Mullan, J.P.; Baranov, P.V. GWIPS-viz: 2018 update. Nucleic Acids Res. 2018, 46, D823–D830. [Google Scholar] [CrossRef]
- Sharipov, R.N.; Yevshin, I.S.; Kondrakhin, Y.V.; Volkova, O.A. RiboSeqDB–a repository of selected human and mouse ribosome footprint and RNA-seq data. Virtual Biol. 2014, 1, 37–46. [Google Scholar] [CrossRef]
- Liu, W.; Xiang, L.; Zheng, T.; Jin, J.; Zhang, G. TranslatomeDB: A comprehensive database and cloud-based analysis platform for translatome sequencing data. Nucleic Acids Res. 2018, 46, D206–D212. [Google Scholar] [CrossRef] [PubMed]
- Olexiouk, V.; Van Criekinge, W.; Menschaert, G. An update on sORFs.org: A repository of small ORFs identified by ribosome profiling. Nucleic Acids Res. 2018, 46, D497–D502. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Qian, S.B. TISdb: A database for alternative translation initiation in mammalian cells. Nucleic Acids Res. 2014, 42, D845–D850. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Y.; Xie, Z. Computational resources for ribosome profiling: From database to Web server and software. Brief. Bioinform. 2017. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Technical Aspects | Polysome Profiling | RNC-Seq | Ribo-Seq | TRAP-Seq |
|---|---|---|---|---|
| Recovery of RNC-mRNA | Full-length | Full-length | Ribosome protected fragments | Full-length |
| Difficulty in Recovering translating mRNA | Demanding | Simple | Demanding | Simple |
| High-throughput methods can be used | Microarray, NGS | Microarray, NGS | NGS | Microarray, NGS |
| Throughput requirement | Low | Low | High | Low |
| Read length | Any | Any | 22–35 nt | Any |
| Detecting sequence variations | Simple | Simple | Demanding | Simple |
| UTR | Yes | Yes | No | Yes |
| Obtain the position of ribosome, densities, ORF, uORFs | No | No | Yes | No |
| Obtaining the amount of ribosomes in single mRNA | Yes | No | No | No |
| Tissue specific | No | No | No | Yes |
| Under physiological conditions | Yes | Yes | Yes | No |
| Required experimental steps | Simple | Simple | Complex | Complex |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Qin, B.; Nikolay, R.; Spahn, C.M.T.; Zhang, G. Translatomics: The Global View of Translation. Int. J. Mol. Sci. 2019, 20, 212. https://doi.org/10.3390/ijms20010212
Zhao J, Qin B, Nikolay R, Spahn CMT, Zhang G. Translatomics: The Global View of Translation. International Journal of Molecular Sciences. 2019; 20(1):212. https://doi.org/10.3390/ijms20010212
Chicago/Turabian StyleZhao, Jing, Bo Qin, Rainer Nikolay, Christian M. T. Spahn, and Gong Zhang. 2019. "Translatomics: The Global View of Translation" International Journal of Molecular Sciences 20, no. 1: 212. https://doi.org/10.3390/ijms20010212
APA StyleZhao, J., Qin, B., Nikolay, R., Spahn, C. M. T., & Zhang, G. (2019). Translatomics: The Global View of Translation. International Journal of Molecular Sciences, 20(1), 212. https://doi.org/10.3390/ijms20010212