Discovery of High Affinity Receptors for Dityrosine through Inverse Virtual Screening and Docking and Molecular Dynamics

Abstract
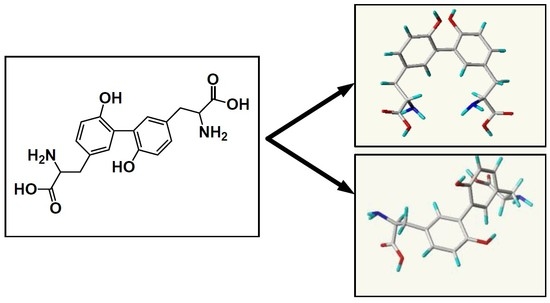
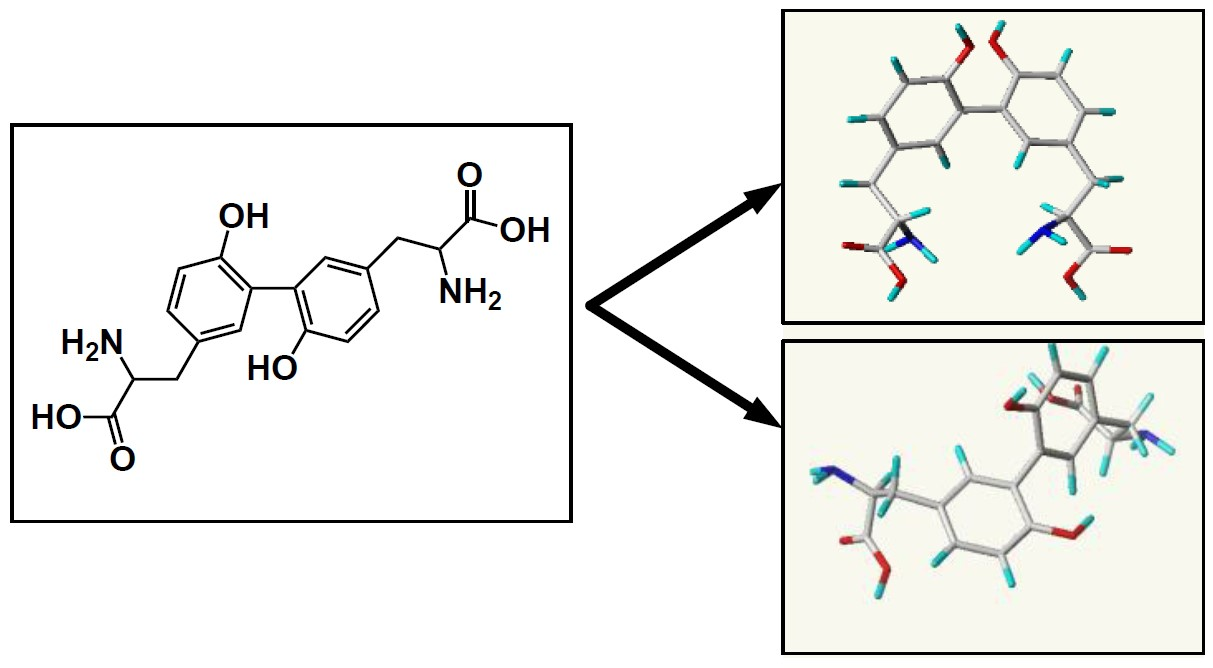{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
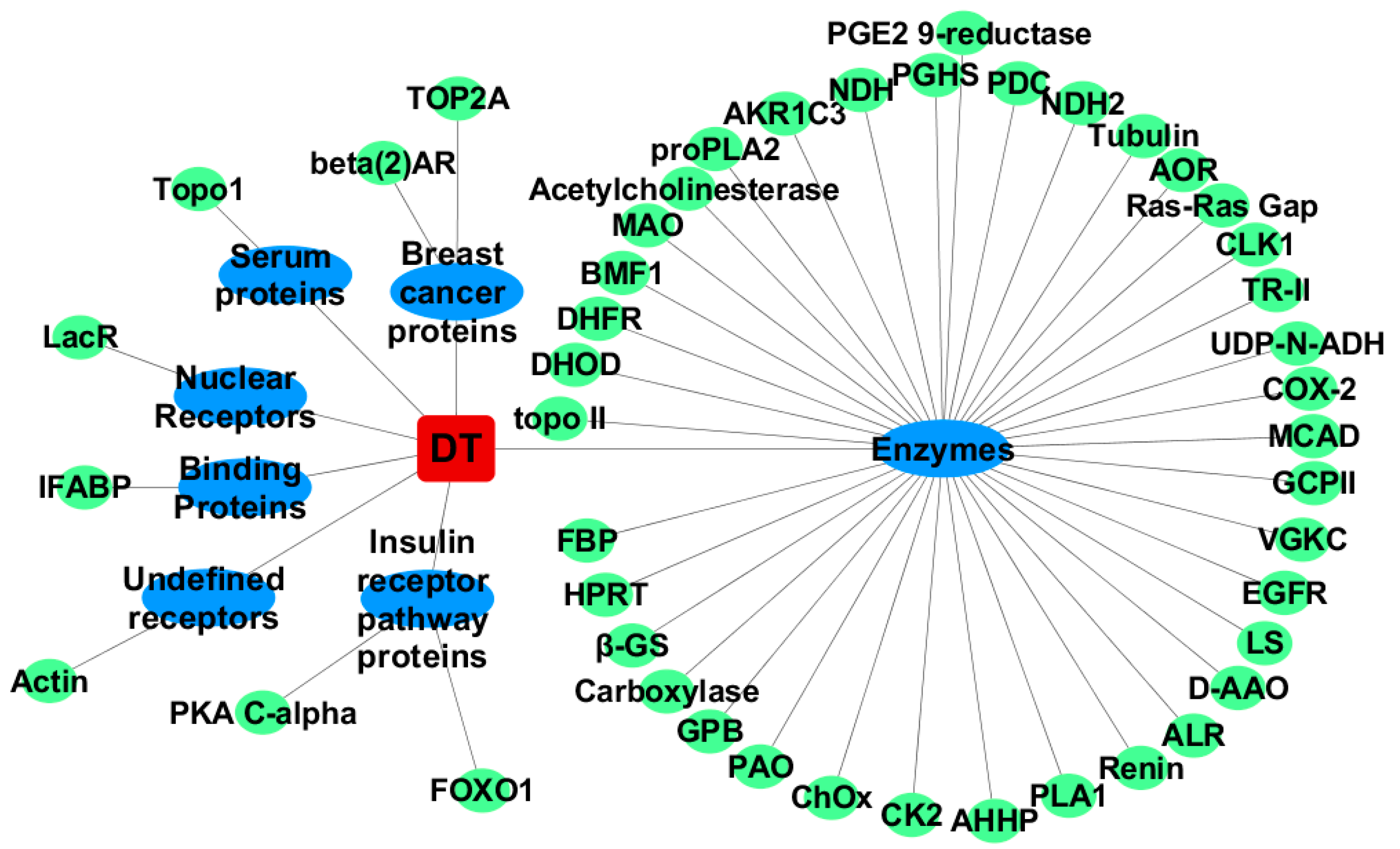
2.1. Inverse Virtual Screening
2.1.1. For Cis-Dityrosine (CDT)
2.1.2. For Trans-Dityrosine (TDT)
2.2. Refinement Docking
2.2.1. For CDT
2.2.2. For TDT
2.3. Validation
2.3.1. For CDT
2.3.2. For TDT
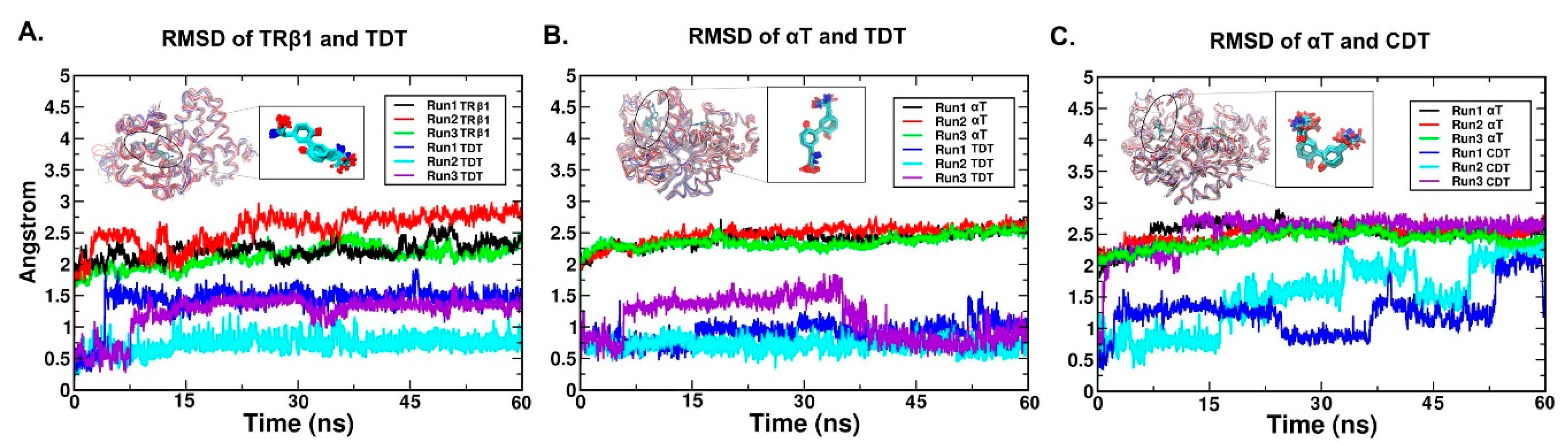
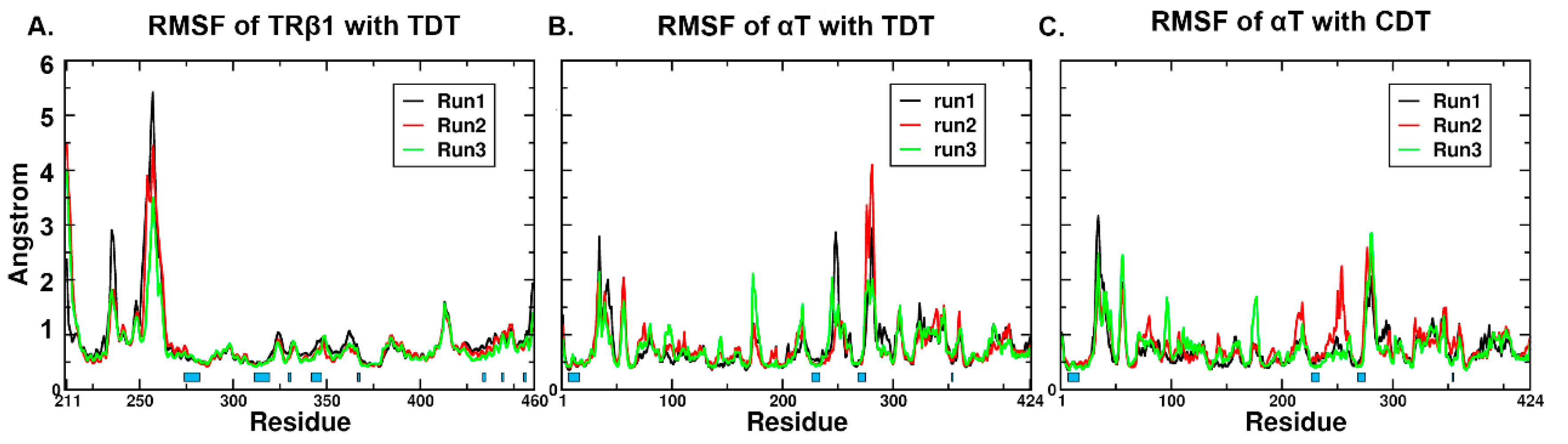
2.4. RMSD (Root Mean Square Deviation)/RMSF (Root Mean Square Fluctuation) Analysis
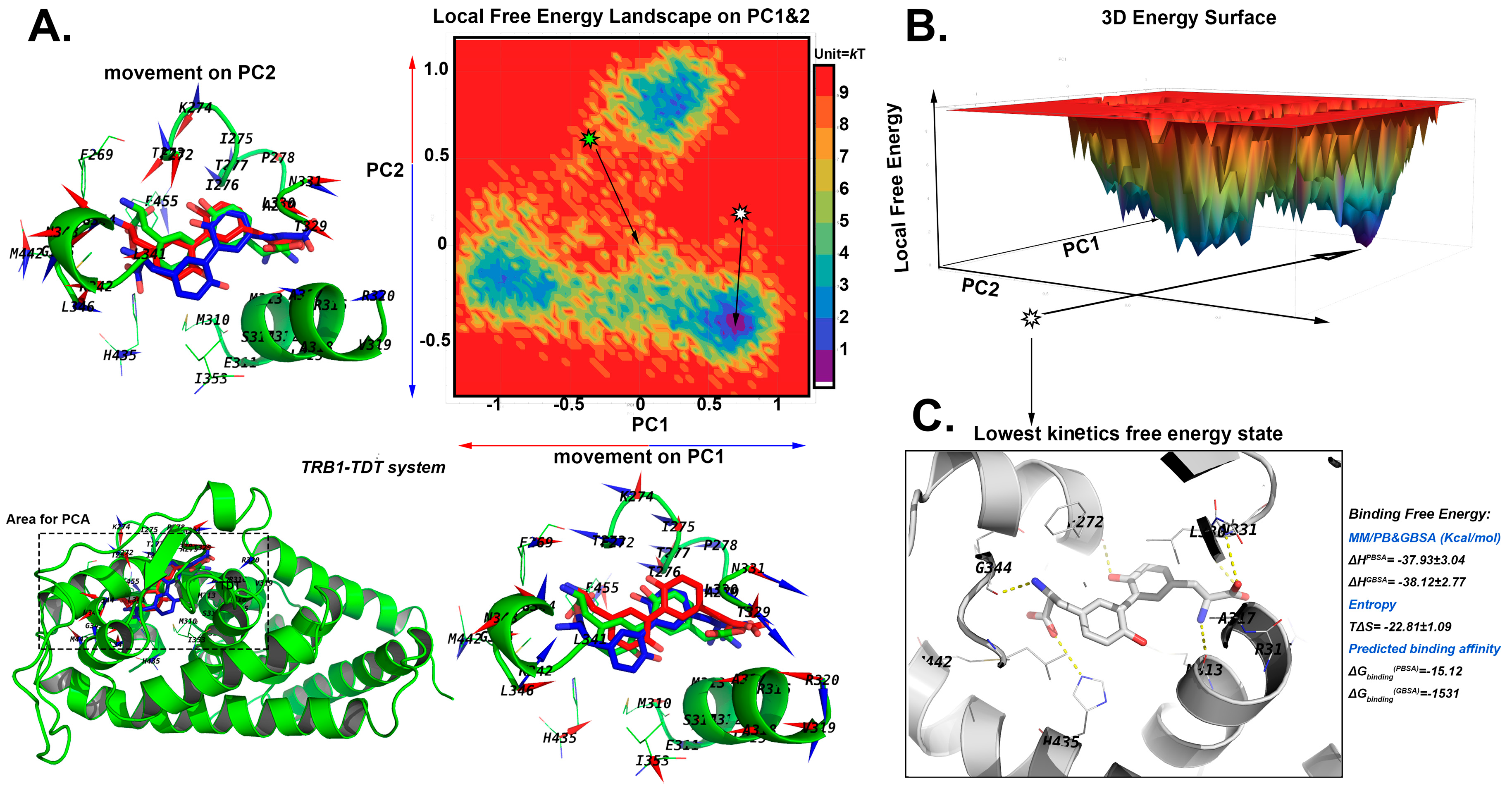
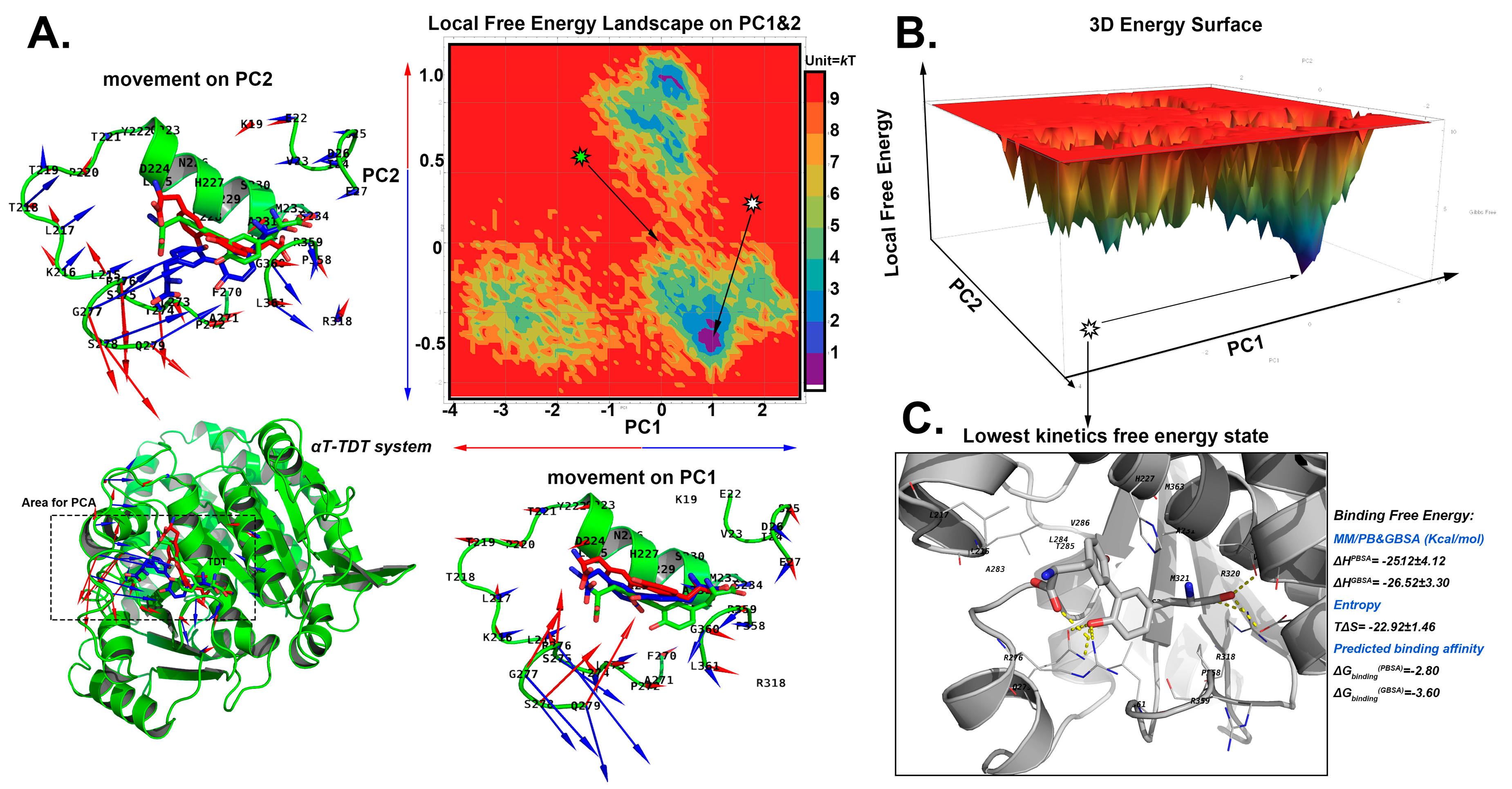
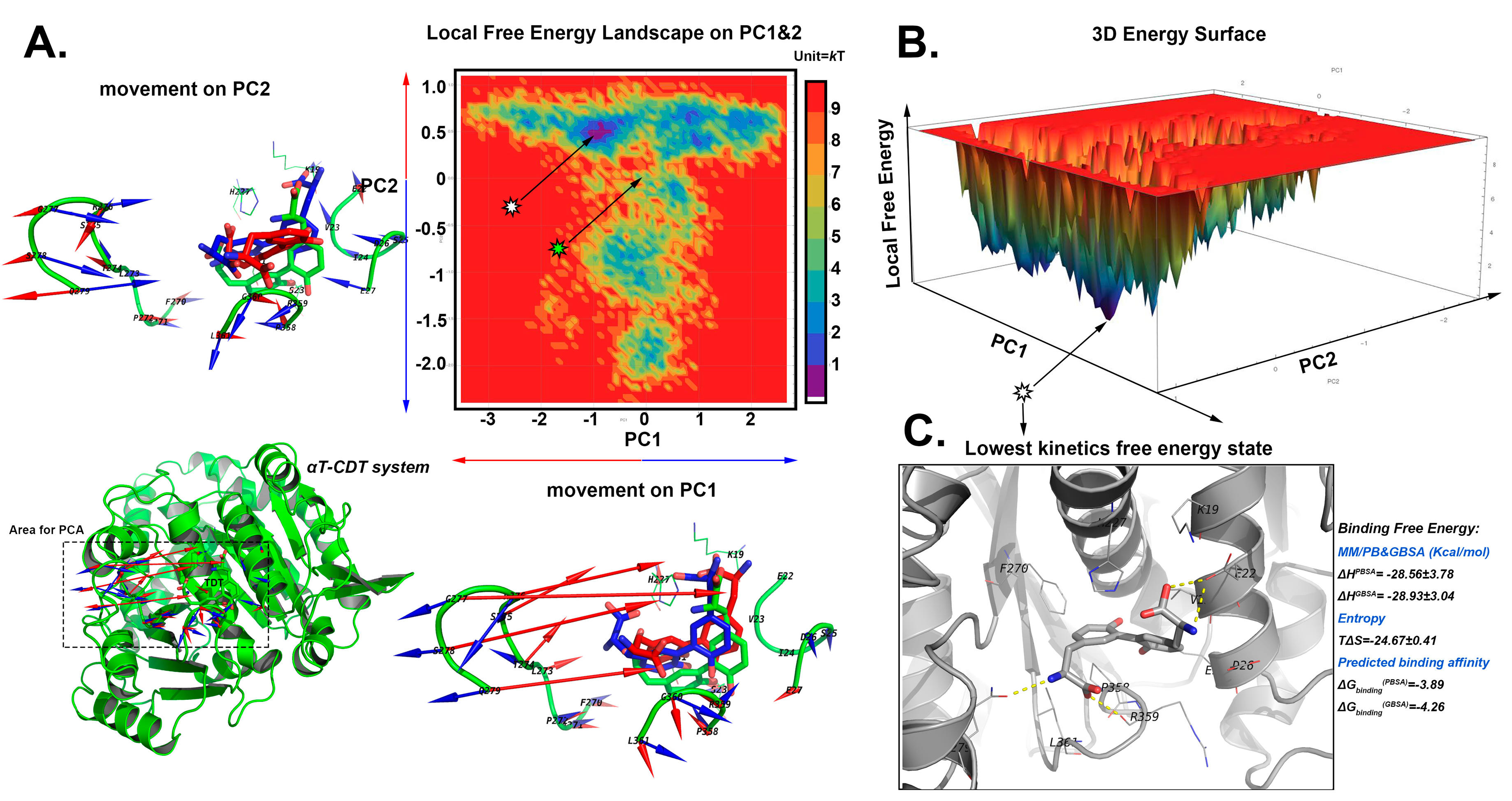
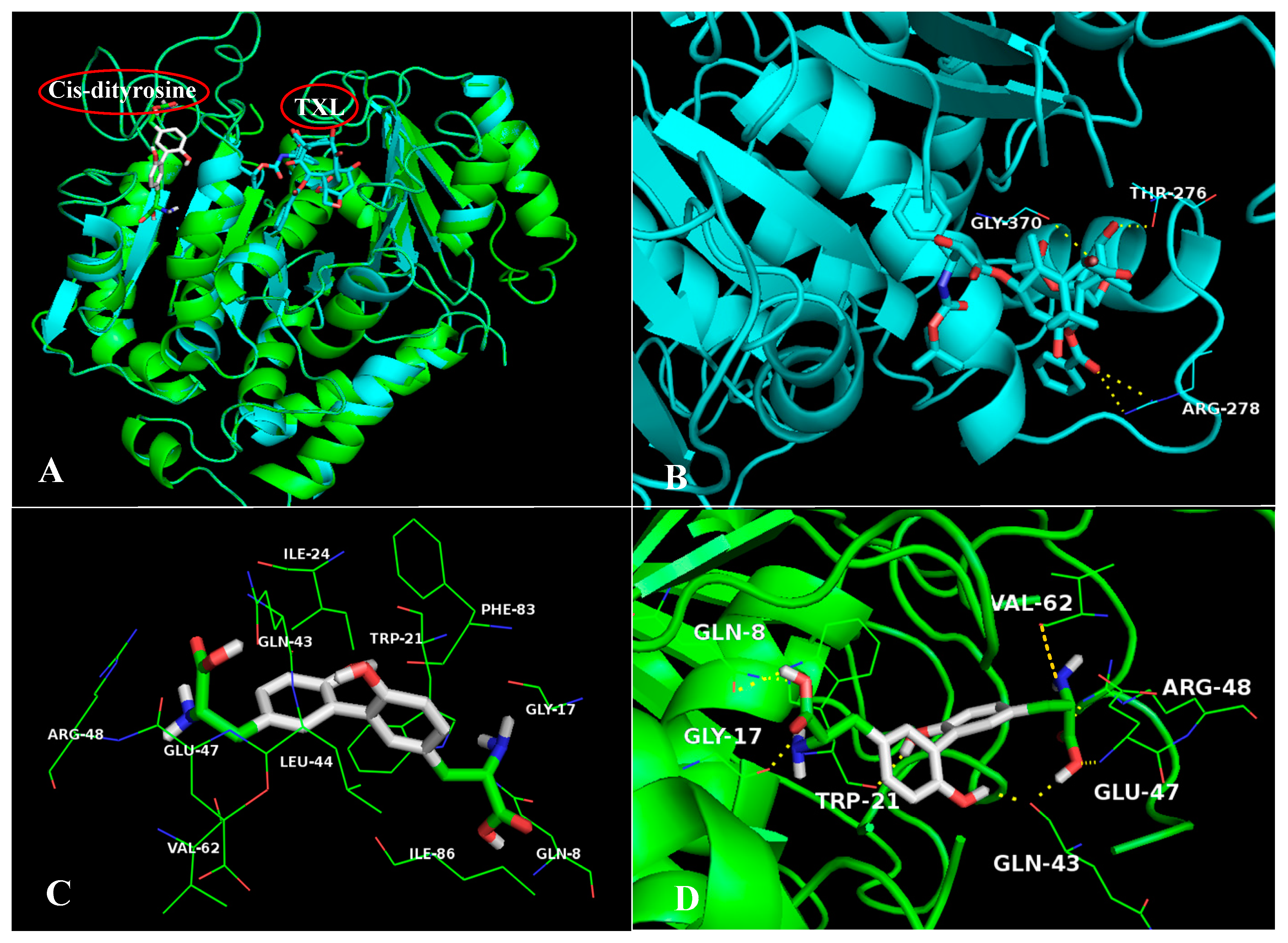
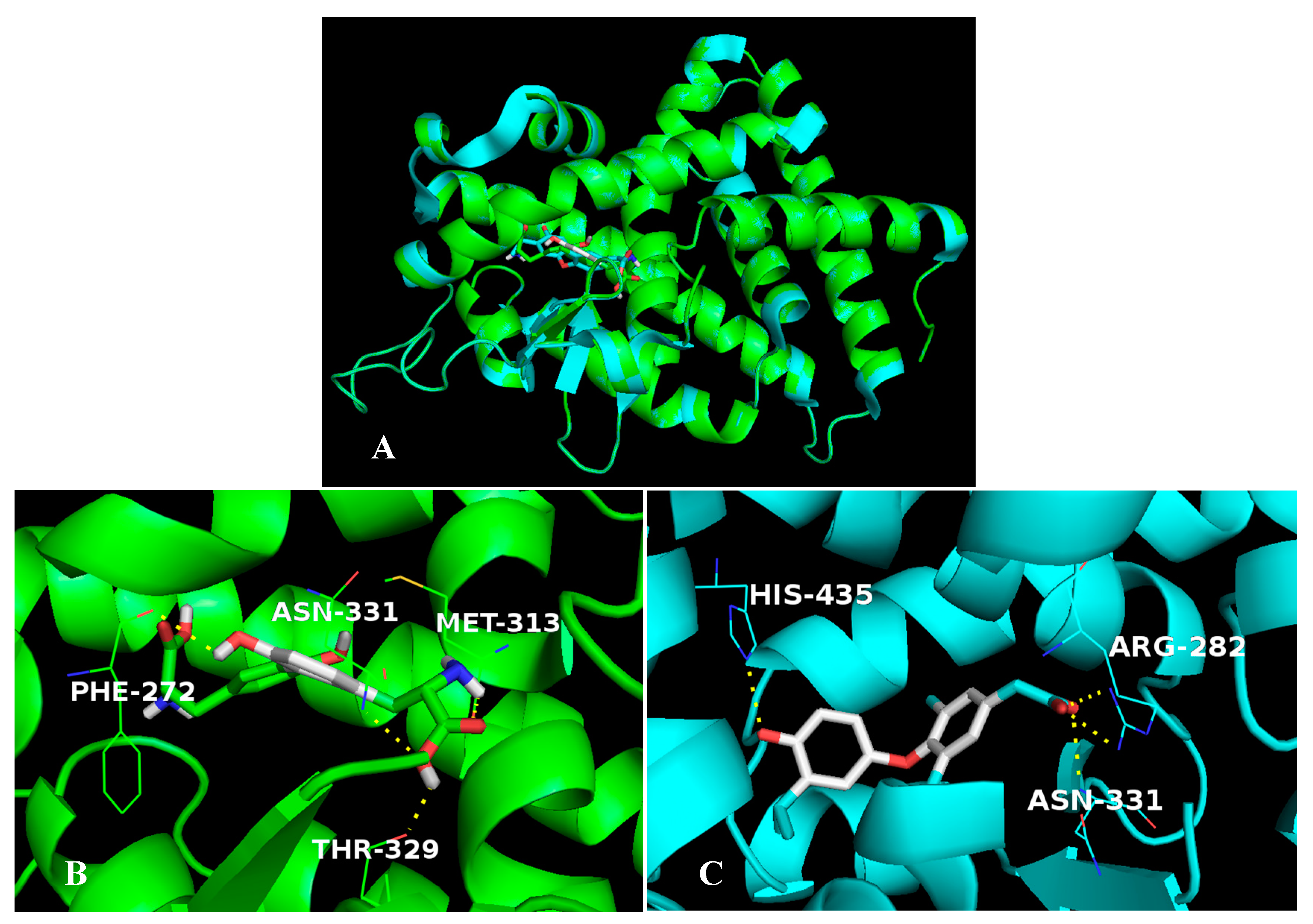
2.5. The Stable Binding Conformation Analysis
2.5.1. The Stable Binding Conformation of TRβ1-TDT
2.5.2. The Stable Binding Conformation of αT-TDT
2.5.3. The Stable Binding Conformation of αT-CDT
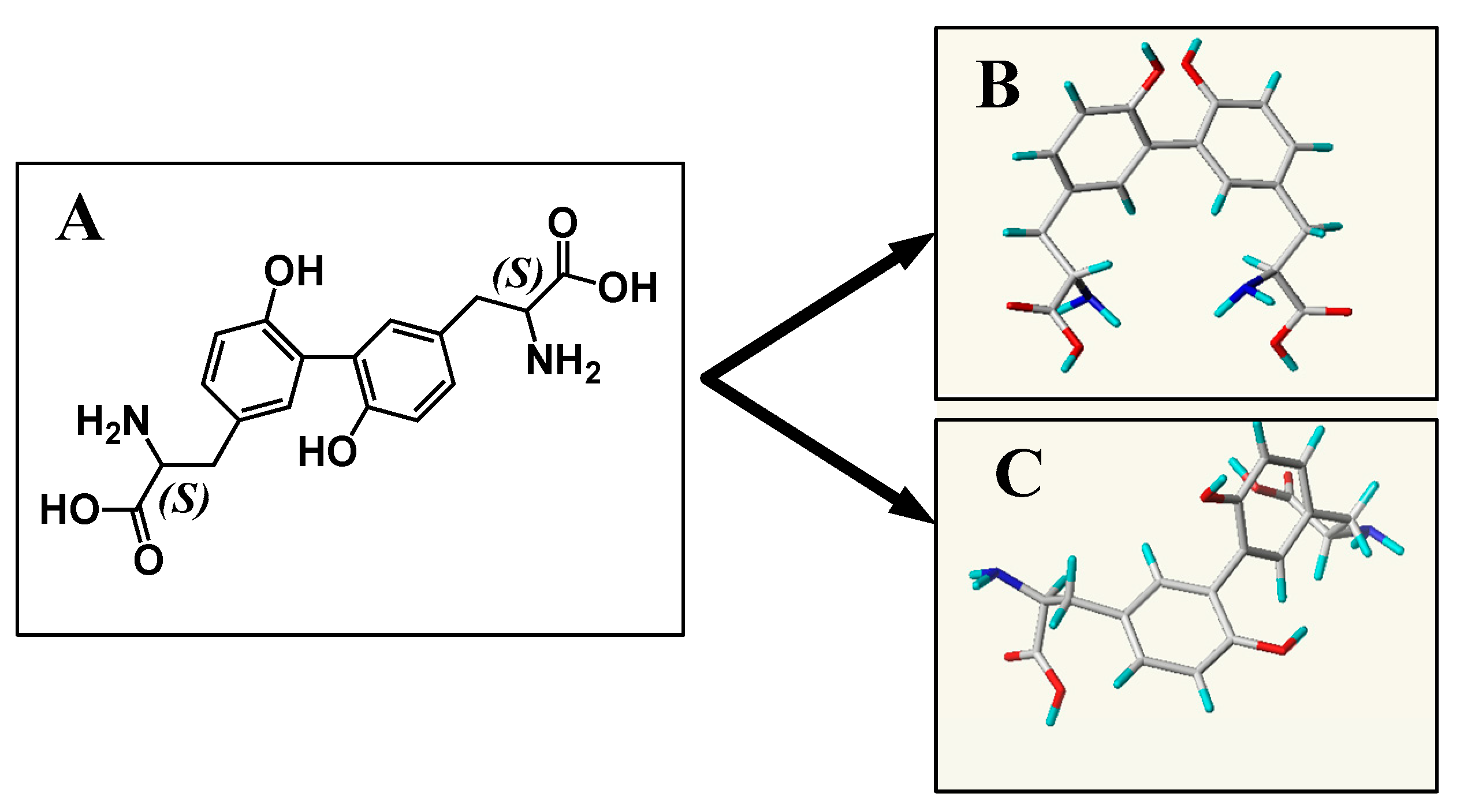
2.6. Conformational Analysis
2.6.1. For CDT
2.6.2. For TDT
3. Materials and Methods
3.1. Preparation of Ligand and Target Structures
3.2. Inverse Virtual Screening
3.3. Refinement Docking Experiments
3.4. Validation
3.5. MD Process
3.6. Stability Analysis
3.7. Local PCA
3.8. Local Free Energy Landscape
3.9. Binding Free Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dalle-Donne, I.; Scaloni, A.; Giustarini, D.; Cavarra, E.; Tell, G.; Lungarella, G.; Colombo, R.; Rossi, R.; Milzani, A. Proteins as biomarkers of oxidative/nitrosative stress in diseases: The contribution of redox proteomics. Mass Spectrom. Rev. 2005, 24, 55–99. [Google Scholar] [CrossRef] [PubMed]
- Bachi, A.; Dalle-Donne, I.; Scaloni, A. Redox proteomics: Chemical principles, methodological approaches and biological/biomedical promises. Chem. Rev. 2012, 113, 596–698. [Google Scholar] [CrossRef]
- Audette, M.; Blouquit, Y.; Houée-Levin, C. Oxidative dimerization of proteins: Role of tyrosine accessibility. Arch. Biochem. Biophys. 2000, 376, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Fenaille, F.; Parisod, V.; Vuichoud, J.; Tabet, J.-C.; Guy, P.A. Quantitative determination of dityrosine in milk powders by liquid chromatography coupled to tandem mass spectrometry using isotope dilution. J. Chromatogr. A 2004, 1052, 77–84. [Google Scholar] [CrossRef]
- Dalsgaard, T.K.; Nielsen, J.H.; Brown, B.E.; Stadler, N.; Davies, M.J. Dityrosine, 3, 4-dihydroxyphenylalanine (DOPA), and radical formation from tyrosine residues on milk proteins with globular and flexible structures as a result of riboflavin-mediated photo-oxidation. J. Agric. Food Chem. 2011, 59, 7939–7947. [Google Scholar] [CrossRef]
- Witko-Sarsat, V.; Friedlander, M.; Capeillère-Blandin, C.; Nguyen-Khoa, T.; Nguyen, A.T.; Zingraff, J.; Jungers, P.; Descamps-Latscha, B. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996, 49, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Barsotti, A.; Fabbi, P.; Fedele, M.; Garibaldi, S.; Balbi, M.; Bezante, G.P.; Risso, D.; Indiveri, F.; Ghigliotti, G.; Brunelli, C. Role of advanced oxidation protein products and Thiol ratio in patients with acute coronary syndromes. Clin. Biochem. 2011, 44, 605–611. [Google Scholar] [CrossRef]
- Colombo, G.; Clerici, M.; Giustarini, D.; Rossi, R.; Milzani, A.; Dalle-Donne, I. Redox albuminomics: Oxidized albumin in human diseases. Antioxid. Redox Signal. 2012, 17, 1515–1527. [Google Scholar] [CrossRef]
- Davies, M.J. The oxidative environment and protein damage. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2005, 1703, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Desiderio, D.M.; Nibbering, N.M.; Dalle-Donne, I.; Scaloni, A.; Butterfield, D.A. Redox Proteomics: From Protein Modifications to Cellular Dysfunction and Diseases; John Wiley & Sons: New York, NY, USA, 2006; Volume 9. [Google Scholar]
- Tong, M.; Longato, L.; Monte, S.M. Early limited nitrosamine exposures exacerbate high fat diet-mediated type 2 diabetes and neurodegeneration. BMC Endocr. Disord. 2010, 10, 4–19. [Google Scholar] [CrossRef]
- Abdelrahim, M.; Morris, E.; Carver, J.; Facchina, S.; White, A.; Verma, A. Liquid chromatographic assay of dityrosine in human cerebrospinal fluid. J. Chromatogr. B Biomed. Sci. Appl. 1997, 696, 175–182. [Google Scholar] [CrossRef]
- Giulivi, C.; Davies, K. Dityrosine and tyrosine oxidation products are endogenous markers for the selective proteolysis of oxidatively modified red blood cell hemoglobin by (the 19 S) proteasome. J. Boil. Chem. 1993, 268, 8752–8759. [Google Scholar]
- Giulivi, C.; Davies, K. Dityrosine: A marker for oxidatively modified proteins and selective proteolysis. Methods Enzym. 1994, 233, 363. [Google Scholar]
- Andersen, S.O. Characterization of a new type of cross-linkage in resilin, a rubber-like protein. Biochim. Biophys. Acta 1963, 69, 249–262. [Google Scholar] [CrossRef]
- Smail, E.H.; Briza, P.; Panagos, A.; Berenfeld, L. Candida albicans cell walls contain the fluorescent cross-linking amino acid dityrosine. Infect. Immun. 1995, 63, 4078–4083. [Google Scholar] [PubMed]
- Briza, P.; Ellinger, A.; Winkler, G.; Breitenbach, M. Characterization of a DL-dityrosine-containing macromolecule from yeast ascospore walls. J. Boil. Chem. 1990, 265, 15118–15123. [Google Scholar]
- Foerder, C.A.; Shapiro, B.M. Release of ovoperoxidase from sea urchin eggs hardens the fertilization membrane with tyrosine crosslinks. Proc. Natl. Acad. Sci. USA 1977, 74, 4214–4218. [Google Scholar] [CrossRef]
- LaBella, F.; Keeley, F.; Vivian, S.; Thornhill, D. Evidence for dityrosine in elastin. Biochem. Biophys. Res. Commun. 1967, 26, 748–753. [Google Scholar] [CrossRef]
- Waykole, P.; Heidemann, E. Dityrosine in collagen. Connect. Tissue Res. 1976, 4, 219–222. [Google Scholar] [CrossRef]
- McNamara, M.; Augusteyn, R. 3, 3′-Dityrosine in the proteins of senile nuclear cataracts. Exp. Eye Res. 1980, 30, 319–321. [Google Scholar] [CrossRef]
- LaBella, F.; Waykole, P.; Queen, G. Formation of insoluble gels and dityrosine by the action of peroxidase on soluble collagens. Biochem. Biophys. Res. Commun. 1968, 30, 333–338. [Google Scholar] [CrossRef]
- Herzog, V.; Berndorfer, U.; Saber, Y. Isolation of insoluble secretory product from bovine thyroid: Extracellular storage of thyroglobulin in covalently cross-linked form. J. Cell Boil. 1992, 118, 1071–1083. [Google Scholar] [CrossRef]
- Pennathur, S.; Jackson-Lewis, V.; Przedborski, S.; Heinecke, J.W. Mass spectrometric quantification of 3-nitrotyrosine, ortho-tyrosine, and o, o′-dityrosine in brain tissue of 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-treated mice, a model of oxidative stress in Parkinson′s disease. J. Boil. Chem. 1999, 274, 34621–34628. [Google Scholar] [CrossRef]
- Balestrieri, M.; Spagnuolo, M.S.; Cigliano, L.; Storti, G.; Ferrara, L.; Abrescia, P.; Fedele, E. Evaluation of oxidative damage in mozzarella cheese produced from bovine or water buffalo milk. Food Chem. 2002, 77, 293–299. [Google Scholar] [CrossRef]
- Ruth, H.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar]
- Mane, J.Y.; Mariusz, K.; Huzil, J.T.; Jack, T. Free energy calculations on the binding of colchicine and its derivatives with the alpha/beta-tubulin isoforms. J. Chem. Inf. Model. 2008, 48, 1824–1832. [Google Scholar] [CrossRef]
- Ren, Y.; Zhao, J.; Feng, J. Parkin binds to α/β tubulin and increases their ubiquitination and degradation. J. Neurosci. 2003, 23, 3316–3324. [Google Scholar] [CrossRef]
- Brent, G.A. The molecular basis of thyroid hormone action. N. Engl. J. Med. 1994, 331, 847–853. [Google Scholar]
- Tsai, M.J.; O′Malley, B.W. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Ann. Rev. Biochem. 1994, 63, 451–486. [Google Scholar] [CrossRef]
- Baxter, J.D.; Dillmann, W.H.; West, B.L.; Huber, R.; Furlow, J.D.; Fletterick, R.J.; Webb, P.; Apriletti, J.W.; Scanlan, T.S. Selective modulation of thyroid hormone receptor action. J. Steroid Biochem. Mol. Biol. 2001, 76, 31–42. [Google Scholar] [CrossRef]
- Grover, G.J.; Mellström, K.; Ye, L.; Malm, J.; Li, Y.L.; Bladh, L.G.; Sleph, P.G.; Smith, M.A.; George, R.; Vennström, B.; et al. Selective thyroid hormone receptor-beta activation: A strategy for reduction of weight, cholesterol, and lipoprotein (a) with reduced cardiovascular liability. Proc. Natl. Acad. Sci. USA 2003, 100, 10067–10072. [Google Scholar] [CrossRef]
- Brenta, G.; Danzi, S.; Klein, I. Potential therapeutic applications of thyroid hormone analogs. Nat. Clin. Pract. Endocrinol. Met. 2007, 3, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Bryzgalova, G.; Effendic, S.; Khan, A.; Rehnmark, S.; Barbounis, P.; Boulet, J.; Gao, D.; Singh, R.; Shapses, S.; Malm, J. Anti-obesity, anti-diabetic, and lipid lowering effects of the thyroid receptor β subtype selective agonist KB-141. J. Steroid Biochem. Mol. Biol. 2008, 111, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Baudry, N.; Lejeune, P.-J.; Niccoli, P.; Vinet, L.; Carayon, P.; Mallet, B. Dityrosine bridge formation and thyroid hormone synthesis are tightly linked and are both dependent on N-glycans. FEBS Lett. 1996, 396, 223–226. [Google Scholar] [CrossRef]
- Li, Z.L.; Mo, L.; Le, G.; Shi, Y. Oxidized casein impairs antioxidant defense system and induces hepatic and renal injury in mice. Food Chem. Toxicol. 2014, 64, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A.; Wang, J.; Kollman, P.A.; Case, D.A. Antechamber, An Accessory Software Package for Molecular Mechanical Calculations. Abstr. Pap. Am. Chem. Soc. 2001, 222, U403–U443. [Google Scholar]
- Sanner, M.F.; Stoffler, D.; Olson, A.J. ViPEr, a visual programming environment for Python. In Proceedings of the 10th International Python Conference, Jaiper, India, 13–15 October 2017; pp. 103–115. [Google Scholar]
- Gao, Z.; Li, H.; Zhang, H.; Liu, X.; Kang, L.; Luo, X.; Zhu, W.; Chen, K.; Wang, X.; Jiang, H. PDTD: A web-accessible protein database for drug target identification. BMC Bioinform. 2008, 9, 104–110. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Ranjan, N.; Andreasen, K.F.; Kumar, S.; Hyde-Volpe, D.; Arya, D.P. Aminoglycoside binding to Oxytricha nova telomeric DNA. Biochemistry 2010, 49, 9891–9903. [Google Scholar] [CrossRef] [PubMed]
- Lauro, G.; Masullo, M.; Piacente, S.; Riccio, R.; Bifulco, G. Inverse Virtual Screening allows the discovery of the biological activity of natural compounds. Bioorg. Med. Chem. 2012, 20, 3596–3602. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Stephens, P.; Devlin, F.; Chabalowski, C.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Nogales, E.; Wolf, S.G.; Downing, K.H. Correction: Structure of the αβ tubulin dimer by electron crystallography. Nature 1998, 393, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Marinelli, F.; Pietrucci, F.; Laio, A.; Piana, S. A kinetic model of trp-cage folding from multiple biased molecular dynamics simulations. PLoS Comput. Biol. 2009, 5, e1000452. [Google Scholar] [CrossRef] [PubMed]
- Wolfram Research, Inc. Mathematica; Wolfram Research, Inc.: Champaign, IL, USA, 2018. [Google Scholar]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. Py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar] [CrossRef]
- Hou, T.; Chen, K.; McLaughlin, W.A.; Lu, B.; Wang, W. Computational analysis and prediction of the binding motif and protein interacting partners of the Abl SH3 domain. PLoS Comput. Biol. 2006, 2, e1. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the generalized Born model suitable for macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Hermann, R.B. Theory of hydrophobic bonding. II. Correlation of hydrocarbon solubility in water with solvent cavity surface area. J. Phys. Chem. 1972, 76, 2754–2759. [Google Scholar] [CrossRef]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Yang, W.; Hu, X. Discovery of High Affinity Receptors for Dityrosine through Inverse Virtual Screening and Docking and Molecular Dynamics. Int. J. Mol. Sci. 2019, 20, 115. https://doi.org/10.3390/ijms20010115
Wang F, Yang W, Hu X. Discovery of High Affinity Receptors for Dityrosine through Inverse Virtual Screening and Docking and Molecular Dynamics. International Journal of Molecular Sciences. 2019; 20(1):115. https://doi.org/10.3390/ijms20010115
Chicago/Turabian StyleWang, Fangfang, Wei Yang, and Xiaojun Hu. 2019. "Discovery of High Affinity Receptors for Dityrosine through Inverse Virtual Screening and Docking and Molecular Dynamics" International Journal of Molecular Sciences 20, no. 1: 115. https://doi.org/10.3390/ijms20010115
APA StyleWang, F., Yang, W., & Hu, X. (2019). Discovery of High Affinity Receptors for Dityrosine through Inverse Virtual Screening and Docking and Molecular Dynamics. International Journal of Molecular Sciences, 20(1), 115. https://doi.org/10.3390/ijms20010115