AMPK Activation Reduces Hepatic Lipid Content by Increasing Fat Oxidation In Vivo



Abstract
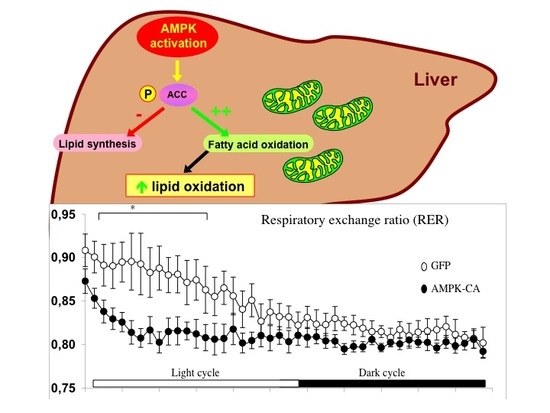
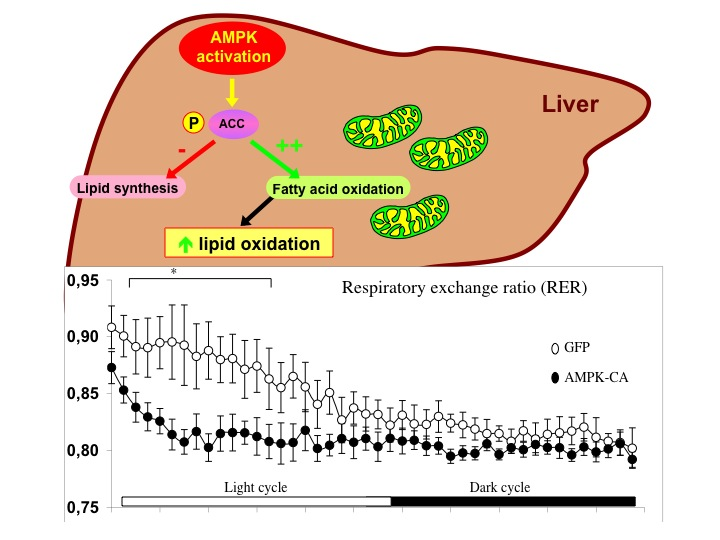{kind=link}
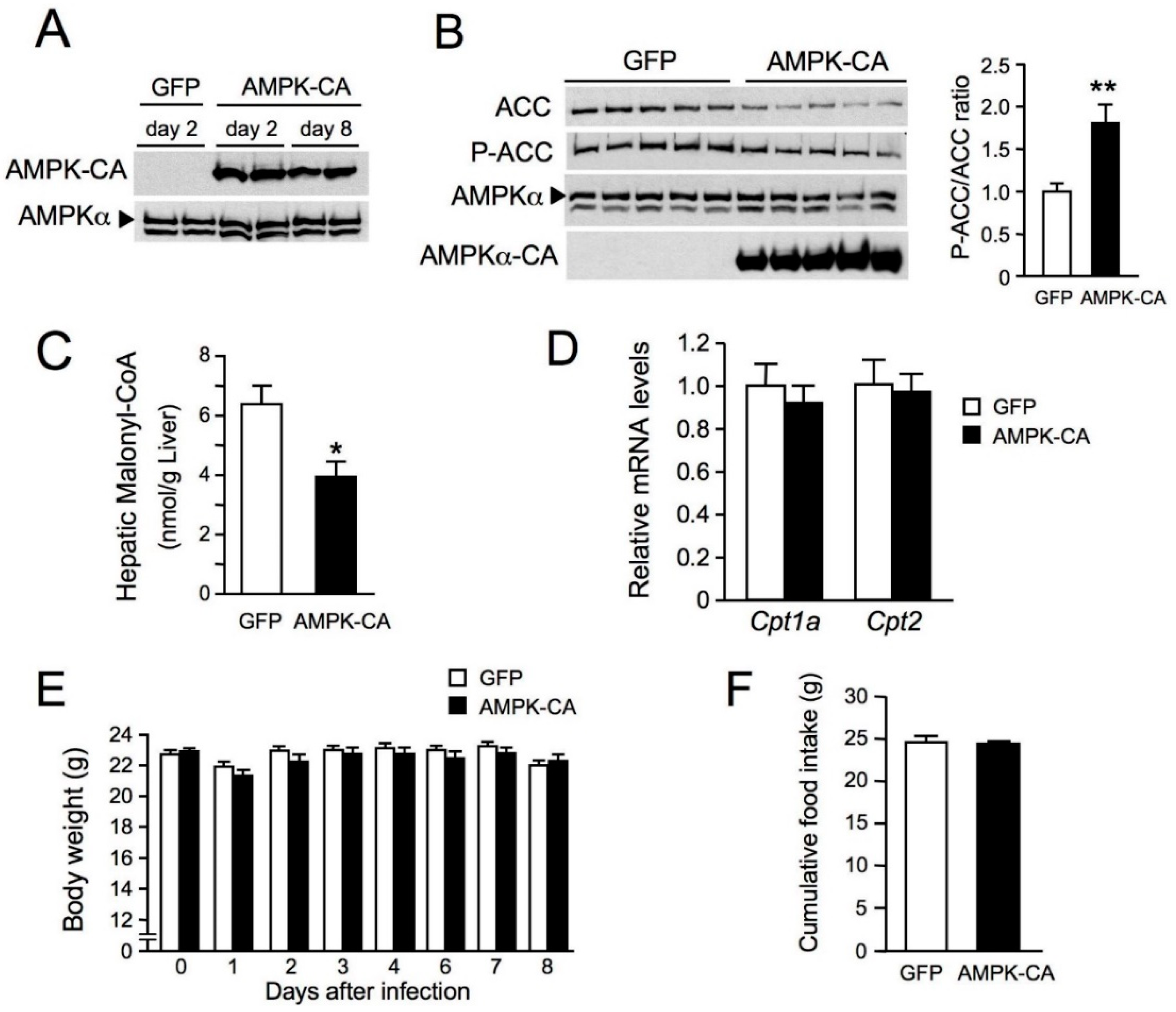{kind=link}
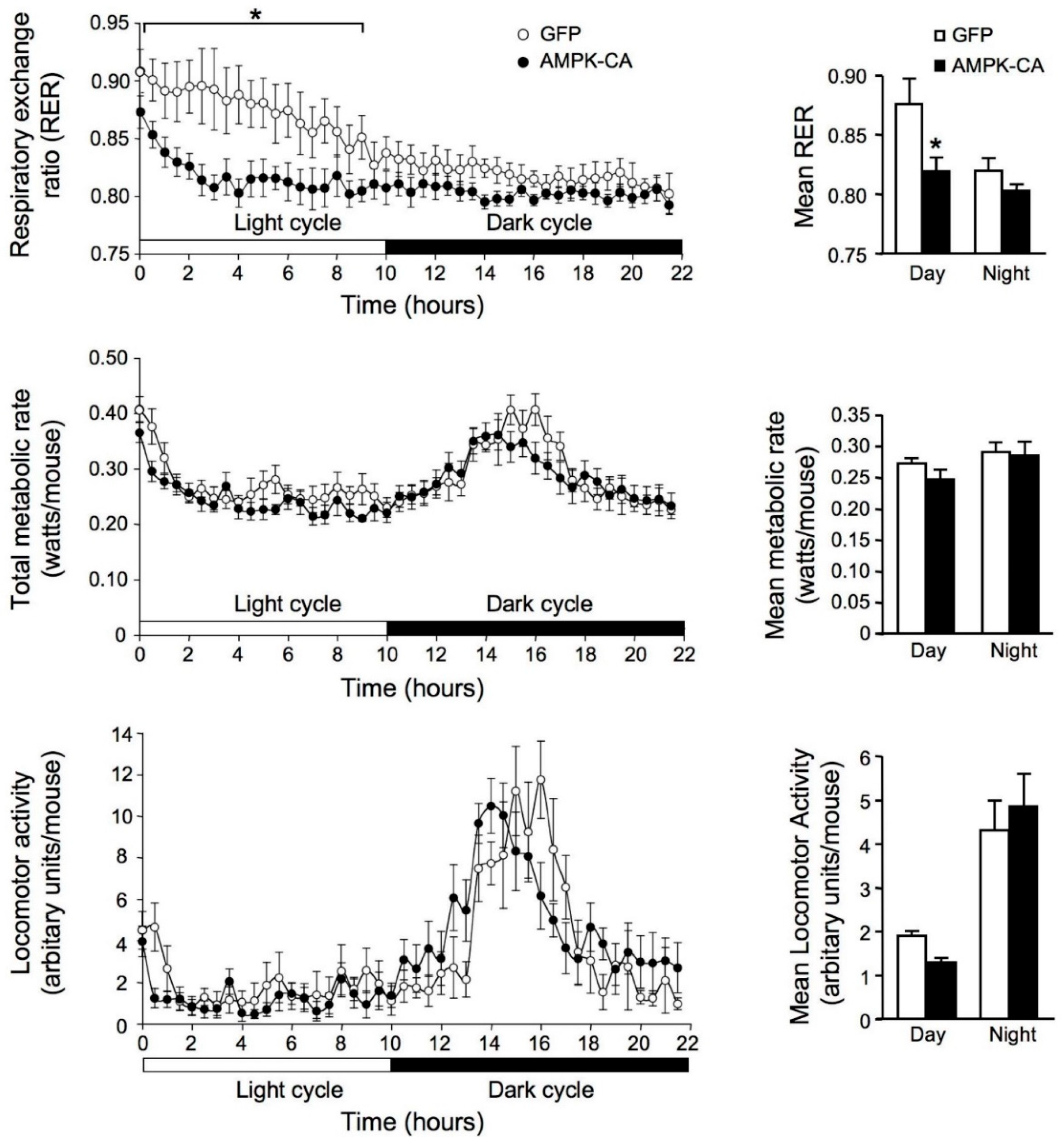{kind=link}
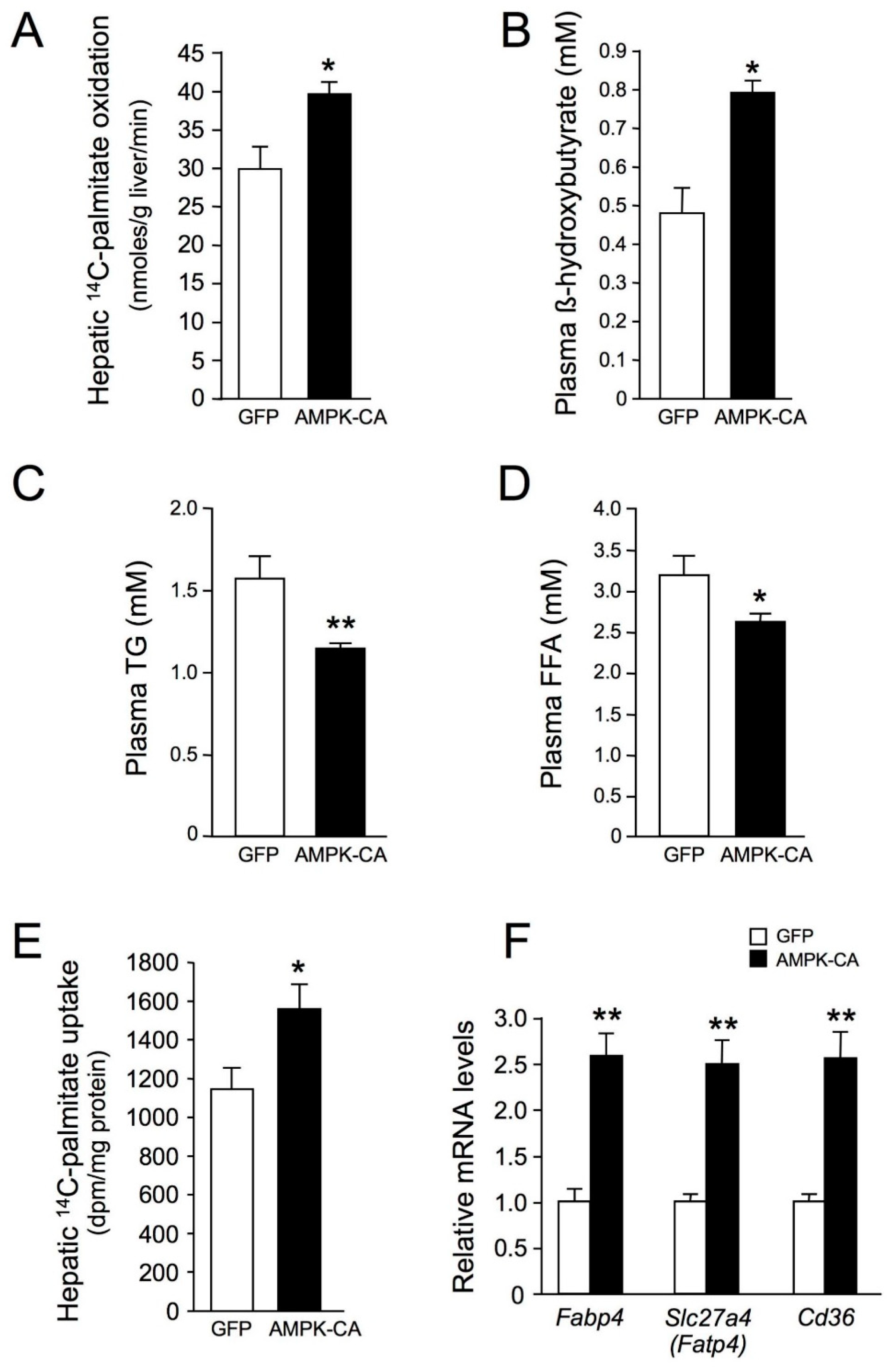{kind=link}
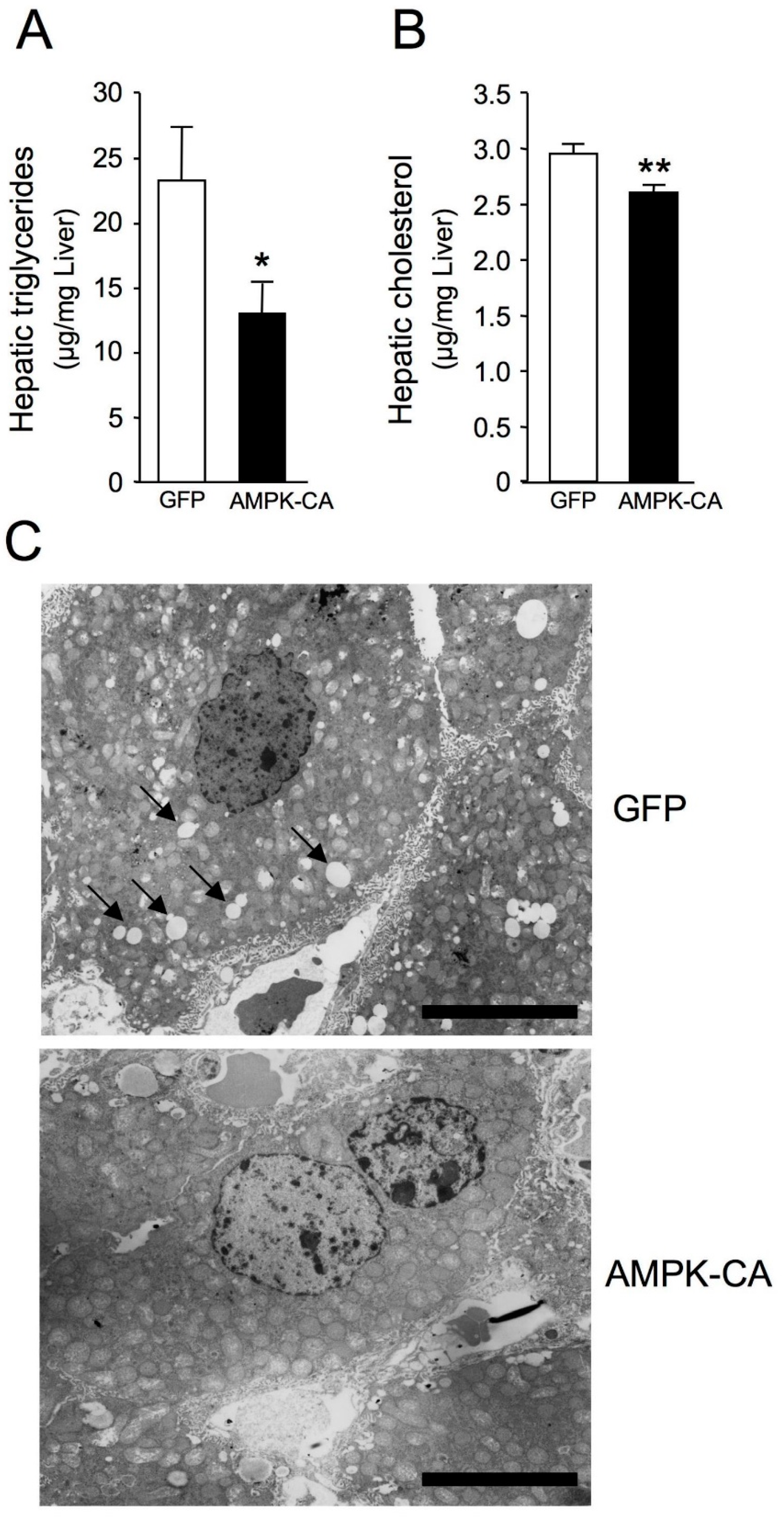{kind=link}
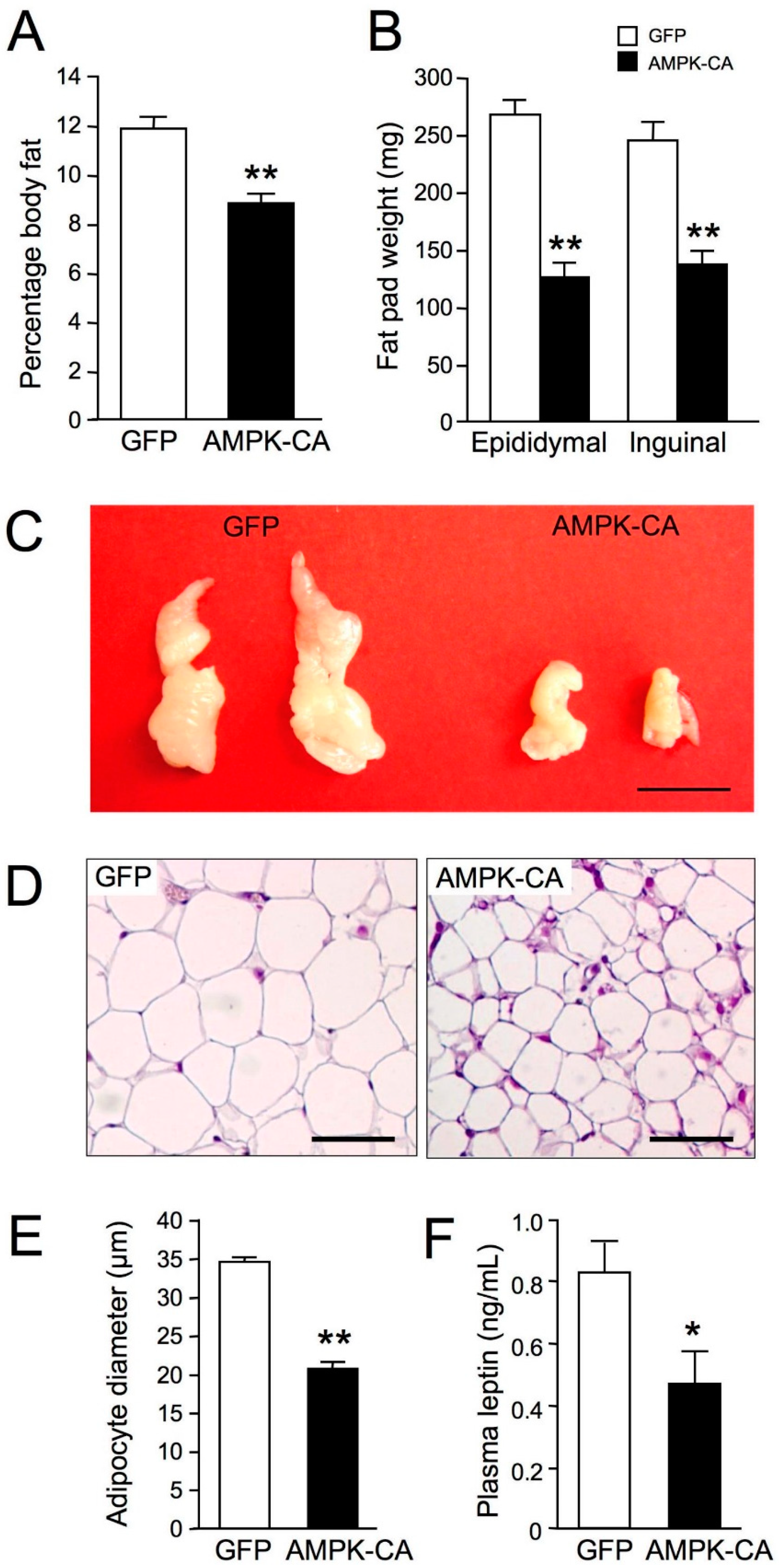{kind=link}
1. Introduction
2. Results
3. Discussion
4. Material and Methods
4.1. Reagents and Antibodies
4.2. Animals
4.3. Metabolic Parameters
4.4. Liver Triglyceride, Cholesterol, and Malonyl-CoA Contents
4.5. Indirect Calorimetry
4.6. Assessment of Fatty Acid Oxidation in Liver Homogenates
4.7. Palmitate Uptake by the Liver
4.8. Fat Mass and Histomorphometry
4.9. Injection of Recombinant Adenovirus
4.10. Isolation of Total mRNA and Quantitative RT-PCR Analysis
4.11. Western Blot Analysis
4.12. Transmission Electron Microscopy
4.13. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hardie, D.G. AMP-activated protein kinase: Maintaining energy homeostasis at the cellular and whole-body levels. Annu. Rev. Nutr. 2014, 34, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Lin, S.C. AMP-activated protein kinase—Not just an energy sensor. F1000Res 2017, 6, 1724. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Foretz, M.; Guigas, B.; Horman, S.; Dentin, R.; Bertrand, L.; Hue, L.; Andreelli, F. Activation of AMP-activated protein kinase in the liver: A new strategy for the management of metabolic hepatic disorders. J. Physiol. 2006, 574, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E730–E740. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Galeno, J.E.; Dang, Q.; Nguyen, T.H.; Boyer, S.H.; Grote, M.P.; Sun, Z.; Chen, M.; Craigo, W.A.; van Poelje, P.D.; MacKenna, D.A.; et al. A Potent and Selective AMPK Activator That Inhibits de Novo Lipogenesis. ACS Med. Chem. Lett. 2010, 1, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Esquejo, R.M.; Salatto, C.T.; Delmore, J.; Albuquerque, B.; Reyes, A.; Shi, Y.; Moccia, R.; Cokorinos, E.; Peloquin, M.; Monetti, M.; et al. Activation of Liver AMPK with PF-06409577 Corrects NAFLD and Lowers Cholesterol in Rodent and Primate Preclinical Models. EBioMedicine 2018, 31, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Boudaba, N.; Marion, A.; Huet, C.; Pierre, R.; Viollet, B.; Foretz, M. AMPK Re-Activation Suppresses Hepatic Steatosis but its Downregulation Does Not Promote Fatty Liver Development. EBioMedicine 2018, 28, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M.; Dickinson, R.; Adler, A.; Gagne, G.; Iyengar, R.; et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Williams, J.R.; Muckett, P.J.; Mayer, F.V.; Liljevald, M.; Bohlooly, Y.M.; Carling, D. Liver-Specific Activation of AMPK Prevents Steatosis on a High-Fructose Diet. Cell Rep. 2017, 18, 3043–3051. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Lindholm, C.R.; Ertel, R.L.; Bauwens, J.D.; Schmuck, E.G.; Mulligan, J.D.; Saupe, K.W. A high-fat diet decreases AMPK activity in multiple tissues in the absence of hyperglycemia or systemic inflammation in rats. J. Physiol. Biochem. 2013, 69, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Muse, E.D.; Obici, S.; Bhanot, S.; Monia, B.P.; McKay, R.A.; Rajala, M.W.; Scherer, P.E.; Rossetti, L. Role of resistin in diet-induced hepatic insulin resistance. J. Clin. Investig. 2004, 114, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; McCorkle, S.; Wang, M.; Lee, Y.; Li, J.; Saha, A.K.; Unger, R.H.; Ruderman, N.B. Leptinomimetic effects of the AMP kinase activator AICAR in leptin-resistant rats: Prevention of diabetes and ectopic lipid deposition. Diabetologia 2004, 47, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Ford, R.J.; Smith, B.K.; Gowans, G.J.; Mancini, S.J.; Pitt, R.D.; Day, E.A.; Salt, I.P.; Steinberg, G.R.; Hardie, D.G. The Na+/Glucose Cotransporter Inhibitor Canagliflozin Activates AMPK by Inhibiting Mitochondrial Function and Increasing Cellular AMP Levels. Diabetes 2016, 65, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Harriman, G.; Greenwood, J.; Bhat, S.; Huang, X.; Wang, R.; Paul, D.; Tong, L.; Saha, A.K.; Westlin, W.F.; Kapeller, R.; et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl. Acad. Sci. USA 2016, 113, E1796–E1805. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Ruddy, M.; Chakravarthy, M.; et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab. 2017, 26, 576. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Ancellin, N.; Andreelli, F.; Saintillan, Y.; Grondin, P.; Kahn, A.; Thorens, B.; Vaulont, S.; Viollet, B. Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes 2005, 54, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yue, P.; Chen, Z.; Schonfeld, G. Hepatic triglyceride contents are genetically determined in mice: Results of a strain survey. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1179–G1189. [Google Scholar] [CrossRef] [PubMed]
- Borgeson, E.; Wallenius, V.; Syed, G.H.; Darshi, M.; Lantero Rodriguez, J.; Biorserud, C.; Ragnmark Ek, M.; Bjorklund, P.; Quiding-Jarbrink, M.; Fandriks, L.; et al. AICAR ameliorates high-fat diet-associated pathophysiology in mouse and ex vivo models, independent of adiponectin. Diabetologia 2017, 60, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Z.; Yang, S.Q.; Chuckaree, C.; Kuhajda, F.; Ronnet, G.; Diehl, A.M. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat. Med. 2000, 6, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, B.S.; Curtis, M.E.; Fillmore, N.; Cardon, B.R.; Thomson, D.M.; Hancock, C.R. The effects of chronic AMPK activation on hepatic triglyceride accumulation and glycerol 3-phosphate acyltransferase activity with high fat feeding. Diabetol. Metab. Syndr. 2013, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Hotta, Y.; Nakamura, H.; Konishi, M.; Murata, Y.; Takagi, H.; Matsumura, S.; Inoue, K.; Fushiki, T.; Itoh, N. Fibroblast growth factor 21 regulates lipolysis in white adipose tissue but is not required for ketogenesis and triglyceride clearance in liver. Endocrinology 2009, 150, 4625–4633. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, M.; Matsuda, M.; Kobayashi, S.; Ando, Y.; Ono, M.; Koishi, R.; Furukawa, H.; Makishima, M.; Shimomura, I. Angiopoietin-like protein 3, a hepatic secretory factor, activates lipolysis in adipocytes. Biochem. Biophys. Res. Commun. 2003, 301, 604–609. [Google Scholar] [CrossRef]
- Nygaard, E.B.; Vienberg, S.G.; Orskov, C.; Hansen, H.S.; Andersen, B. Metformin stimulates FGF21 expression in primary hepatocytes. Exp. Diabetes Res. 2012, 2012, 465282. [Google Scholar] [CrossRef] [PubMed]
- Dyck, J.R.; Barr, A.J.; Barr, R.L.; Kolattukudy, P.E.; Lopaschuk, G.D. Characterization of cardiac malonyl-CoA decarboxylase and its putative role in regulating fatty acid oxidation. Am. J. Physiol. 1998, 275, H2122–H2129. [Google Scholar] [CrossRef] [PubMed]
- Even, P.C.; Mokhtarian, A.; Pele, A. Practical aspects of indirect calorimetry in laboratory animals. Neurosci. Biobehav. Rev. 1994, 18, 435–447. [Google Scholar] [CrossRef]
- Yu, X.X.; Drackley, J.K.; Odle, J. Rates of mitochondrial and peroxisomal beta-oxidation of palmitate change during postnatal development and food deprivation in liver, kidney and heart of pigs. J. Nutr. 1997, 127, 1814–1821. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foretz, M.; Even, P.C.; Viollet, B. AMPK Activation Reduces Hepatic Lipid Content by Increasing Fat Oxidation In Vivo. Int. J. Mol. Sci. 2018, 19, 2826. https://doi.org/10.3390/ijms19092826
Foretz M, Even PC, Viollet B. AMPK Activation Reduces Hepatic Lipid Content by Increasing Fat Oxidation In Vivo. International Journal of Molecular Sciences. 2018; 19(9):2826. https://doi.org/10.3390/ijms19092826
Chicago/Turabian StyleForetz, Marc, Patrick C. Even, and Benoit Viollet. 2018. "AMPK Activation Reduces Hepatic Lipid Content by Increasing Fat Oxidation In Vivo" International Journal of Molecular Sciences 19, no. 9: 2826. https://doi.org/10.3390/ijms19092826
APA StyleForetz, M., Even, P. C., & Viollet, B. (2018). AMPK Activation Reduces Hepatic Lipid Content by Increasing Fat Oxidation In Vivo. International Journal of Molecular Sciences, 19(9), 2826. https://doi.org/10.3390/ijms19092826