Gut Bacterial Communities of Dendroctonus valens and D. mexicanus (Curculionidae: Scolytinae): A Metagenomic Analysis across Different Geographical Locations in Mexico

 ,
,  ,
,
Abstract
1. Introduction
2. Results
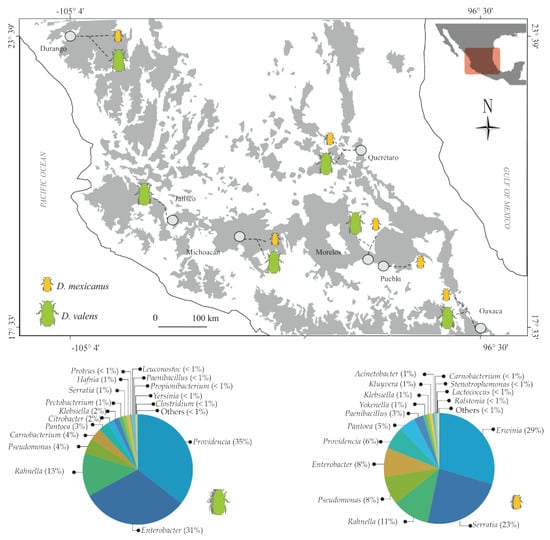
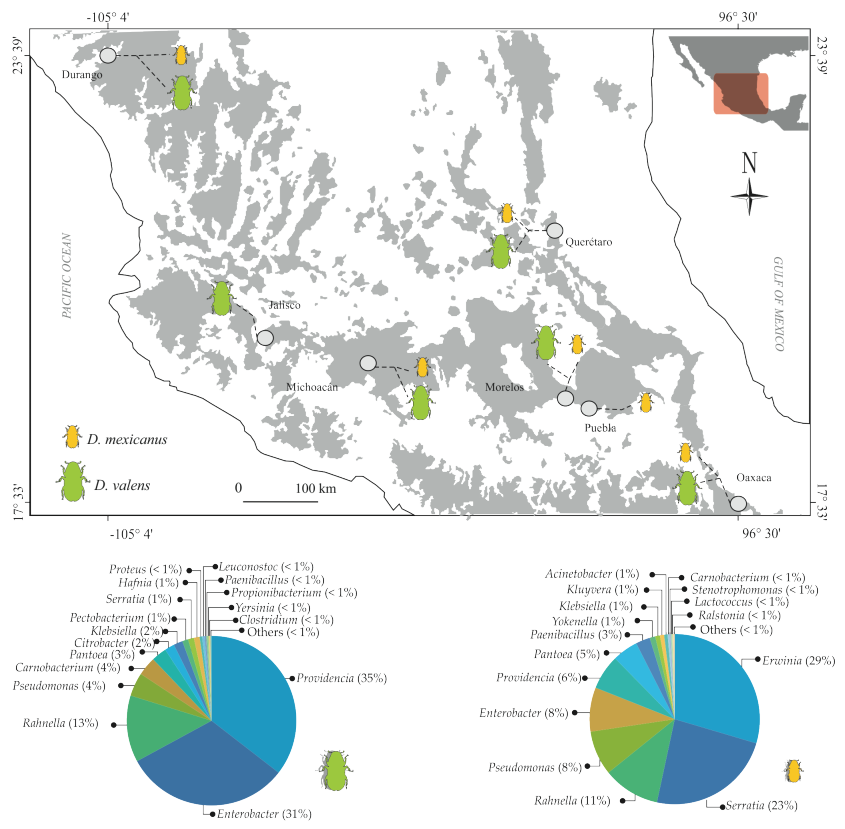
2.1. Taxonomic Analysis of Gut Bacterial Communities
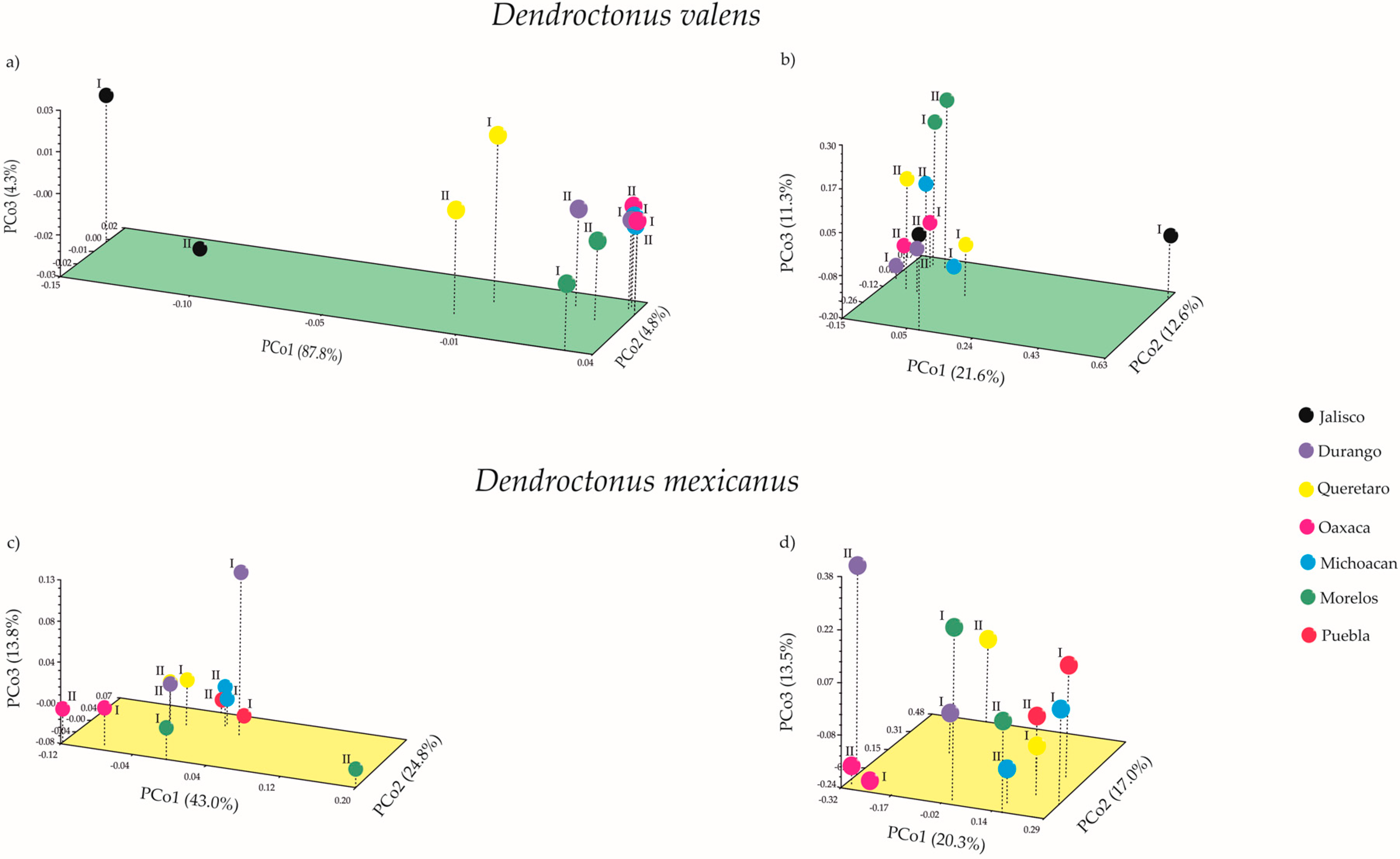
2.2. Bacterial Community Diversity
2.3. Prediction Roles of Gut Bacterial Communities
3. Discussion
4. Materials and Methods
4.1. Insect Collection, Dissection and DNA Extraction
4.2. Bacterial 16S rRNA Polymerase Chain Reaction Amplification and Pyrosequencing
4.3. Data Pyrosequencing Analysis
4.4. Bacterial Diversity Analysis
4.5. Core Bacteriome
4.6. Predictive Functional Analysis Based on Metagenomic 16S rRNA Surveys
4.7. Data Accessibility
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIC | Akaike Information Criterion |
| ANOVA | Analysis of Variance |
| DGGE | Denaturing Gradient Gel Electrophoresis |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| NGS | Next Generation Sequencing |
| NSTI | Nearest Sequenced Taxon Index |
| NTSYS-PC | Numerical Taxonomy and Multivariate Analysis |
| OTU | Operational Taxonomic Unit |
| PCoA | Principal Coordinate Analysis |
| PD | Phylogenetic Diversity |
| PICRUSt | Phylogenetic Investigation of Communities by Reconstruction of Unobserved States |
| QIIME | Quantitative Insight into Microbial Ecology |
| RDP | Ribosomal Database Project |
| uwU | Unweighted UniFrac |
| wU | Weighted UniFrac |
References
- Wood, S.L. The bark and ambrosia beetles of North and Central America (Coleoptera: Scolytidae), a taxonomic monograph. Great Basin Nat. Mem. 1982, 6, 1–1356. [Google Scholar]
- Bentz, B.J.; Régnière, J.; Fettig, C.J.; Hansen, E.M.; Hayes, J.L.; Hicke, J.A.; Seybold, S.J. Climate change and bark beetles of the western United States and Canada: Direct and indirect effects. BioScience 2010, 60, 602–613. [Google Scholar] [CrossRef]
- Six, D.L. The bark beetle holobiont: Why microbes matter. J. Chem. Ecol. 2013, 39, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.T. The ecology of yeasts in the bark beetle holobiont: A century of research revisited. Microb. Ecol. 2015, 69, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Six, D.L.; Bracewell, R. Dendroctonus. In Bark Beetles; Vega, F.E., Hofstetter, R.W., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 305–350. [Google Scholar]
- Hofstetter, R.W.; Dinkins-Bookwalter, J.; Davis, T.S.; Klepzig, K.D. Symbiotic associations of bark beetles. In Bark Beetles; Vega, F.E., Hofstetter, R.W., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 209–245. [Google Scholar]
- Morales-Jiménez, J.; Zúñiga, G.; Ramírez-Saad, H.C.; Hernández-Rodríguez, C. Gut-associated bacteria throughout the life cycle of the bark beetle Dendroctonus rhizophagus Thomas and Bright (Curculionidae: Scolytinae) and their cellulolytic activities. Microb. Ecol. 2012, 64, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yu, J.; Wang, C.; Chen, H. Cellulolytic bacteria associated with the gut of Dendroctonus armandi larvae (Coleoptera: Curculionidae: Scolytinae). Forests 2014, 5, 455–465. [Google Scholar] [CrossRef]
- Briones-Roblero, C.I.; Rodríguez-Díaz, R.; Santiago-Cruz, J.A.; Zúñiga, G.; Rivera-Orduña, F.N. Degradation capacities of bacteria and yeasts isolated from the gut of Dendroctonus rhizophagus (Curculionidae: Scolytinae). Folia Microbiol. 2017, 62, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ayres, M.P.; Wilkens, R.T.; Ruel, J.J.; Lombardero, M.J.; Vallery, E. Nitrogen budgets of phloem-feeding bark beetles with and without symbiotic fungi. Ecology 2000, 81, 2198–2210. [Google Scholar] [CrossRef]
- Bridges, J.R. Nitrogen-fixing bacteria associated with bark beetles. Microb. Ecol. 1981, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Morales-Jiménez, J.; Zúñiga, G.; Villa-Tanaca, L.; Hernández-Rodríguez, C. Bacterial community and nitrogen fixation in the red turpentine beetle, Dendroctonus valens LeConte (Coleoptera: Curculionidae: Scolytinae). Microb. Ecol. 2009, 58, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Morales-Jiménez, J.; Vera-Ponce de León, A.; García-Domínguez, A.; Martínez-Romero, E.; Zúñiga, G.; Hernández-Rodríguez, C. Nitrogen-fixing and uricolytic bacteria associated with the gut of Dendroctonus rhizophagus and Dendroctonus valens (Curculionidae: Scolytinae). Microb. Ecol. 2013, 66, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Boone, C.K.; Keefover-Ring, K.; Mapes, A.C.; Adams, A.S.; Bohlmann, J.; Raffa, K.F. Bacteria associated with a tree-killing insect reduce concentrations of plant defense compounds. J. Chem. Ecol. 2013, 39, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.S.; Aylward, F.O.; Adams, S.M.; Erbilgin, N.; Aukema, B.H.; Currie, C.R.; Suen, G.; Raffa, K.F. Mountain pine beetles colonizing historical and naïve host trees are associated with a bacterial community highly enriched in genes contributing to terpene metabolism. Appl. Environ. Microbiol. 2013, 79, 3468–3475. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.T.; Lu, M.; Sun, J.H. Invasive bark beetle-associated microbes degrade a host defensive monoterpene. Insect Sci. 2016, 23, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Shi, Z.; Wang, B.; Lu, M.; Sun, J.H. Pine defensive monoterpene-pinene influences the feeding behavior of Dendroctonus valens and its gut bacterial community structure. Int. J. Mol. Sci. 2016, 17, 1734. [Google Scholar] [CrossRef] [PubMed]
- Cardoza, Y.J.; Klepzig, K.D.; Raffa, K.F. Bacteria in oral secretions of an endophytic insect inhibit antagonistic fungi. Ecol. Entomol. 2006, 31, 636–645. [Google Scholar] [CrossRef]
- Scott, J.J.; Oh, D.-C.; Yuceer, M.C.; Klepzig, K.D.; Clardy, J.; Currie, C.R. Bacterial protection of beetle-fungus mutualism. Science 2008, 322, 63. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Lou, Q.; Wang, B.; Xu, L.; Cheng, C.; Lu, M.; Sun, J. Altered carbohydrates allocation by associated bacteria-fungi interactions in a bark beetle-microbe symbiosis. Sci. Rep. 2016, 6, 20135. [Google Scholar] [CrossRef] [PubMed]
- Vasanthakumar, A.; Delalibera, I.J.; Handelsman, J.; Klepzig, K.D.; Schloss, P.D.; Raffa, K.F. Characterization of gut-associated bacteria in larvae and adults of the southern pine beetle, Dendroctonus frontalis Zimmermann. Environ. Entomol. 2006, 35, 1710–1717. [Google Scholar] [CrossRef]
- Yilmaz, H.; Sezen, K.; Kati, H.; Demirbağ, Z. The first study on the bacterial flora of the European spruce bark beetle, Dendroctonus micans (Coleoptera: Scolytidae). Biologia 2006, 61, 679–686. [Google Scholar] [CrossRef]
- Adams, A.S.; Adams, S.M.; Currie, C.R.; Gillette, N.E.; Raffa, K.F. Geographic variation in bacterial communities associated with the red turpentine beetle (Coleoptera: Curculionidae). Environ. Entomol. 2010, 39, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Yaman, M.; Ertürk, Ö.; Aslan, I. Isolation of some pathogenic bacteria from the great spruce bark beetle, Dendroctonus micans and its specific predator, Rhizophagus grandis. Folia Microbiol. 2010, 55, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Muratoglu, H.; Sezen, K.; Demirbag, Z. Determination and pathogenicity of the bacterial flora associated with the spruce bark beetle, Ips typographus (L.) (Coleoptera: Curculionidae: Scolytinae). Turk. J. Biol. 2011, 35, 9–20. [Google Scholar] [CrossRef]
- Durand, A.A.; Bergeron, A.; Constant, P.; Buffet, J.P.; Déziel, E.; Guertin, C. Surveying the endomicrobiome and ectomicrobiome of bark beetles: The case of Dendroctonus simplex. Sci. Rep. 2015, 5, 17190. [Google Scholar] [CrossRef] [PubMed]
- Dohet, L.; Grégoire, J.C.; Berasategui, A.; Kaltenpoth, M.; Biedermann, P.H. Bacterial and fungal symbionts of parasitic Dendroctonus bark beetles. FEMS Microbiol. Ecol. 2016, 92, fiw129. [Google Scholar] [CrossRef] [PubMed]
- Hernández-García, J.A.; Briones-Roblero, C.I.; Rivera-Orduña, F.N.; Zúñiga, G. Revealing the gut bacteriome of Dendroctonus bark beetles (Curculionidae: Scolytinae): Diversity, core members and co-evolutionary patterns. Sci. Rep. 2017, 7, 13864. [Google Scholar] [CrossRef] [PubMed]
- Mason, C.J.; Hanshew, A.S.; Raffa, K.F. Contributions by host trees and insect activity to bacterial communities in Dendroctonus valens (Coleoptera: Curculionidae) galleries, and their high overlap with other microbial assemblages of bark beetles. Environ. Entomol. 2015, 45, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Briones-Roblero, C.I.; Hernández-García, J.A.; Gonzalez-Escobedo, R.; Soto-Robles, L.V.; Rivera-Orduña, F.N.; Zúñiga, G. Structure and dynamics of the gut bacterial microbiota of the bark beetle, Dendroctonus rhizophagus (Curculionidae: Scolytinae) across their life stages. PLoS ONE 2017, 12, e0175470. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, C.; Chen, H.; Ma, J. Differences in the structure of the gut bacteria communities in development stages of the Chinese white pine beetle (Dendroctonus armandi). Int. J. Mol. Sci. 2013, 14, 21006–21020. [Google Scholar] [CrossRef] [PubMed]
- Berasategui, A.; Axelsson, K.; Nordlander, G.; Schmidt, A.; Borg-Karlson, A.K.; Gershenzon, J.; Terenius, O.; Kaltenpoth, M. The gut microbiota of the pine weevil is similar across Europe and resembles that of other conifer-feeding beetles. Mol. Ecol. 2016, 25, 4014–4031. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, S.; Pilhofer, M.; Höll, W. Microbial associations in gut systems of wood-and bark-inhabiting longhorned beetles [Coleoptera: Cerambycidae]. Syst. Appl. Microbiol. 2010, 33, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.A.; Moreau, C.S.; Goldman-Huertas, B.; Fujiwara, M.; Lohman, D.J.; Pierce, N.E. Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. Proc. Nat. Acad. Sci. USA 2009, 106, 21236–21241. [Google Scholar] [CrossRef] [PubMed]
- Ishak, H.D.; Plowes, R.; Sen, R.; Kellner, K.; Meyer, E.; Estrada, D.A.; Dowd, S.E.; Mueller, U.G. Bacterial diversity in Solenopsis invicta and Solenopsis geminata ant colonies characterized by 16S amplicon 454 pyrosequencing. Microb. Ecol. 2011, 61, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Waite, D.W.; Dsouza, M.; Biswas, K.; Ward, D.F.; Deines, P.; Taylor, M.W. Microbial community structure in the gut of the New Zealand insect Auckland tree weta (Hemideina thoracica). Arch. Microbiol. 2015, 197, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Köhler, T.; Dietrich, C.; Scheffrahn, R.H.; Brune, A. High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation of the gut in wood-feeding higher termites (Nasutitermes spp.). Appl. Environ. Microbiol. 2012, 78, 4691–4701. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Escobedo, R.; Briones-Roblero, C.I.; Pineda-Mendoza, R.M.; Rivera-Orduña, F.N.; Zúñiga, G. Bacteriome from Pinus arizonica and P. durangensis: Diversity, comparison of assemblages, and overlapping degree with the gut bacterial community of a bark beetle that kills pines. Front. Microbiol. 2018, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Moreno, Y.; Mendoza, G.; Barrios, M.A.; Cisneros, R.; Macías-Sámano, J.; Zúñiga, G. Areography of the genus Dendroctonus (Coleoptera: Curculionidae: Scolytinae) in Mexico. J. Biogeogr. 2004, 31, 1163–1177. [Google Scholar] [CrossRef]
- Owen, D.R.; Smith, S.L.; Seybold, S.J. Red turpentine beetle. In Forest Insect and Disease Leaflet 55; US Department of Agriculture: Washington, DC, USA, 2010; p. 8. [Google Scholar]
- Martiny, J.B.H.; Bohannan, B.J.M.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.A.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Krumins, J.A.; Kuske, C.R.; et al. Microbial biogeography: Putting microorganisms on the map. Nat. Rev. Microbiol. 2006, 4, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Hanson, C.A. Microbial biogeography. In Encyclopedia of Biodiversity, 2nd ed.; Academic Press: San Diego, CA, USA, 2013; pp. 271–279. [Google Scholar]
- Toju, H.; Fukatsu, T. Diversity and infection prevalence of endosymbionts in natural populations of the chestnut weevil: Relevance of local climate and host plants. Mol. Ecol. 2011, 20, 853–868. [Google Scholar] [CrossRef] [PubMed]
- Zouache, K.; Raharimalala, F.N.; Raquin, V.; Tran-Van, V.; Raveloson, L.H.R.; Ravelonandro, P.; Mavingui, P. Bacterial diversity of field-caught mosquitoes, Aedes albopictus and Aedes aegypti, from different geographic regions of Madagascar. FEMS Microbiol. Ecol. 2011, 75, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Ceja-Navarro, J.A.; Vega, F.E.; Karaoz, U.; Hao, Z.; Jenkins, S.; Lim, H.C.; Kosina, P.; Infante, F.; Northen, T.R.; Brodie, E.L. Gut microbiota mediate caffeine detoxification in the primary insect pest of coffee. Nat. Commun. 2015, 6, 7618. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lou, Q.; Cheng, C.; Lu, M.; Sun, J. Gut-associated bacteria of Dendroctonus valens and their involvement in verbenone production. Microb. Ecol. 2015, 70, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Redford, A.J.; Bowers, R.M.; Knight, R.; Linhart, Y.; Fierer, N. The ecology of the phyllosphere: Geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ. Microbiol. 2010, 12, 2885–2893. [Google Scholar] [CrossRef] [PubMed]
- Carrell, A.A.; Carper, D.L.; Frank, A.C. Subalpine conifers in different geographical locations host highly similar foliar bacterial endophyte communities. FEMS Microbiol. Ecol. 2016, 92, fiw124. [Google Scholar] [CrossRef] [PubMed]
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef] [PubMed]
- Winder, R.S.; Macey, D.E.; Cortese, J. Dominant bacteria associated with broods of mountain pine beetle, Dendroctonus ponderosae (Coleoptera: Curculionidae, Scolytinae). J. Entomol. Soc. BC 2010, 107, 43–56. [Google Scholar]
- Smith, R.H. Toxicity of pine resin vapors to three species of Dendroctonus bark beetles. J. Econ. Entomol. 1963, 56, 827–831. [Google Scholar] [CrossRef]
- López, M.F.; Cano-Ramírez, C.; Shibayama, M.G.; Zúñiga, G. α-pinene and myrcene induce ultrastructural change in the midgut of Dendroctonus valens (Coleoptera: Curculionidae: Scolytinae). Ann. Entomol. Soc. Am. 2011, 104, 553–561. [Google Scholar] [CrossRef]
- Chiu, C.C.; Keeling, C.I.; Bohlmann, J. Toxicity of pine monoterpenes to mountain pine beetle. Sci. Rep. 2017, 7, 8858. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Noya, Y.E.; Suárez-Arriaga, M.C.; Rojas-Valdes, A.; Montoya-Ciriaco, N.M.; Gómez-Acata, S.; Fernández-Luqueño, F.; Dendooven, L. Pyrosequencing analysis of the bacterial community in drinking water wells. Microb. Ecol. 2013, 66, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Reeder, J.; Knight, R. Rapid denoising of pyrosequencing amplicon data: Exploiting the rank-abundance distribution. Nat. Methods 2010, 7, 668–669. [Google Scholar] [CrossRef] [PubMed]
- Navas-Molina, J.A.; Peralta-Sánchez, J.M.; González, A.; McMurdie, P.J.; Vázquez-Baeza, Y.; Xu, Z.; Ursell, L.K.; Lauber, C.; Zhou, H.; Song, S.J.; et al. Advancing our understanding of the human microbiome using QIIME. Meth. Enzymol. 2013, 531, 371–444. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gilbreath, T.M.; Kukutla, P.; Yan, G.; Xu, J. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS ONE 2011, 6, e24767. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.; Lee, S.M.; Chen, T.C.A. A generalized Good’s nonparametric coverage estimator. Chin. J. Math. 1988, 16, 189–199. [Google Scholar]
- Magurran, A.E. Ecological Diversity and Its Measurement; Princeton University Press: Princeton, NJ, USA, 1988. [Google Scholar]
- Faith, D.P.; Baker, A.M. Phylogenetic diversity (PD) and biodiversity conservation: Some bioinformatics challenges. Evol. Bioinform. 2007, 2, 121–128. [Google Scholar] [CrossRef]
- Zar, J.H. Biostatistical Analysis; Prentice-Hall: Upper Saddle River, NJ, USA, 2010. [Google Scholar]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.R.; Curtis, J.T. An Ordination of the upland forest community of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYSpc Numerical Taxonomy and Multivariate Analysis System Version 2.0 User Guide; Applied Biostatistics Inc.: East Setauket, NY, USA, 1997. [Google Scholar]
- Manly, B.F.J. Randomization, Bootstrap and Monte Carlo Methods in Biology 2; Chapman & Hall: London, UK, 1997. [Google Scholar]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011, 40, 109–114. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | No. of High Quality Reads | Observed OTUs97 | Richness and Diversity Index | Good’s Coverage | ||||
|---|---|---|---|---|---|---|---|---|
| Chao1 | PD * | Shannon | Simpson | Simpson−1 | ||||
| D. valens | ||||||||
| Oaxaca I | 8300 | 118 | 232 | 5.20 | 3.6 | 0.866 | 7.5 | 0.973 |
| Oaxaca II | 2703 | 115 | 234 | 4.73 | 3.7 | 0.872 | 7.8 | 0.974 |
| Queretaro I | 6403 | 173 | 371 | 5.64 | 4.7 | 0.930 | 14.3 | 0.960 |
| Queretaro II | 13,305 | 158 | 364 | 5.79 | 4.9 | 0.944 | 17.8 | 0.965 |
| Morelos I | 11,589 | 174 | 390 | 6.51 | 4.6 | 0.916 | 11.9 | 0.961 |
| Morelos II | 5815 | 153 | 327 | 6.50 | 4.2 | 0.896 | 9.6 | 0.966 |
| Jalisco I | 13,459 | 87 | 118 | 3.11 | 3.6 | 0.851 | 6.7 | 0.986 |
| Jalisco II | 3713 | 164 | 314 | 4.94 | 4.5 | 0.899 | 9.9 | 0.965 |
| Durango I | 8409 | 109 | 224 | 4.68 | 3.4 | 0.834 | 6.0 | 0.975 |
| Durango II | 4723 | 158 | 251 | 5.41 | 4.2 | 0.898 | 9.8 | 0.968 |
| Michoacan I | 3461 | 100 | 268 | 4.13 | 3.4 | 0.859 | 7.1 | 0.975 |
| Michoacan II | 4469 | 117 | 196 | 5.59 | 3.5 | 0.861 | 7.2 | 0.975 |
| D. mexicanus | ||||||||
| Oaxaca I | 7310 | 95 | 143 | 3.68 | 4.0 | 0.899 | 9.9 | 0.981 |
| Oaxaca II | 6836 | 60 | 95 | 2.27 | 2.5 | 0.748 | 4.0 | 0.988 |
| Queretaro I | 4436 | 91 | 166 | 3.19 | 2.8 | 0.649 | 2.8 | 0.981 |
| Queretaro II | 3589 | 73 | 104 | 1.95 | 3.3 | 0.789 | 4.7 | 0.989 |
| Morelos I | 37,466 | 73 | 184 | 2.82 | 2.5 | 0.623 | 2.7 | 0.982 |
| Morelos II | 4444 | 95 | 163 | 4.47 | 3.7 | 0.828 | 5.8 | 0.980 |
| Puebla I | 4681 | 87 | 120 | 2.91 | 3.0 | 0.741 | 3.9 | 0.983 |
| Puebla II | 2906 | 72 | 89 | 2.66 | 3.0 | 0.743 | 3.9 | 0.989 |
| Durango I | 16,022 | 82 | 139 | 2.95 | 3.0 | 0.775 | 4.4 | 0.984 |
| Durango II | 5786 | 65 | 83 | 2.10 | 3.2 | 0.798 | 4.9 | 0.990 |
| Michoacan I | 3589 | 102 | 154 | 3.42 | 2.9 | 0.641 | 2.8 | 0.980 |
| Michoacan II | 4447 | 97 | 125 | 4.14 | 3.5 | 0.770 | 4.3 | 0.985 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-García, J.A.; Gonzalez-Escobedo, R.; Briones-Roblero, C.I.; Cano-Ramírez, C.; Rivera-Orduña, F.N.; Zúñiga, G. Gut Bacterial Communities of Dendroctonus valens and D. mexicanus (Curculionidae: Scolytinae): A Metagenomic Analysis across Different Geographical Locations in Mexico. Int. J. Mol. Sci. 2018, 19, 2578. https://doi.org/10.3390/ijms19092578
Hernández-García JA, Gonzalez-Escobedo R, Briones-Roblero CI, Cano-Ramírez C, Rivera-Orduña FN, Zúñiga G. Gut Bacterial Communities of Dendroctonus valens and D. mexicanus (Curculionidae: Scolytinae): A Metagenomic Analysis across Different Geographical Locations in Mexico. International Journal of Molecular Sciences. 2018; 19(9):2578. https://doi.org/10.3390/ijms19092578
Chicago/Turabian StyleHernández-García, Juan Alfredo, Roman Gonzalez-Escobedo, Carlos Iván Briones-Roblero, Claudia Cano-Ramírez, Flor N. Rivera-Orduña, and Gerardo Zúñiga. 2018. "Gut Bacterial Communities of Dendroctonus valens and D. mexicanus (Curculionidae: Scolytinae): A Metagenomic Analysis across Different Geographical Locations in Mexico" International Journal of Molecular Sciences 19, no. 9: 2578. https://doi.org/10.3390/ijms19092578
APA StyleHernández-García, J. A., Gonzalez-Escobedo, R., Briones-Roblero, C. I., Cano-Ramírez, C., Rivera-Orduña, F. N., & Zúñiga, G. (2018). Gut Bacterial Communities of Dendroctonus valens and D. mexicanus (Curculionidae: Scolytinae): A Metagenomic Analysis across Different Geographical Locations in Mexico. International Journal of Molecular Sciences, 19(9), 2578. https://doi.org/10.3390/ijms19092578