IDP–CRF: Intrinsically Disordered Protein/Region Identification Based on Conditional Random Fields

Abstract
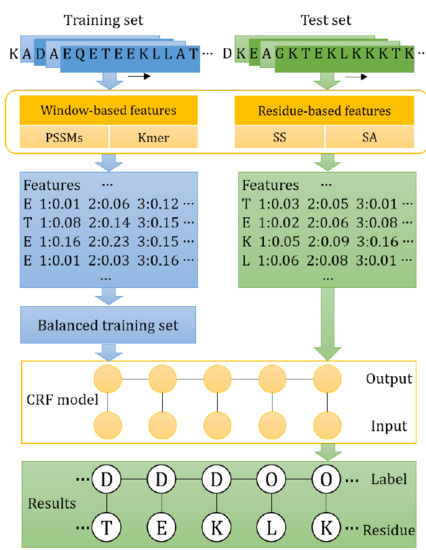
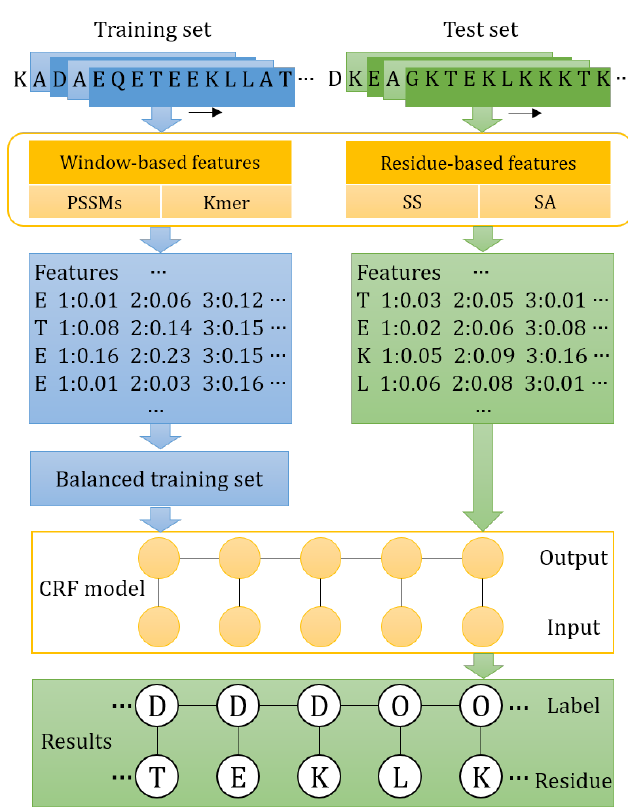
1. Introduction
2. Results and Discussion
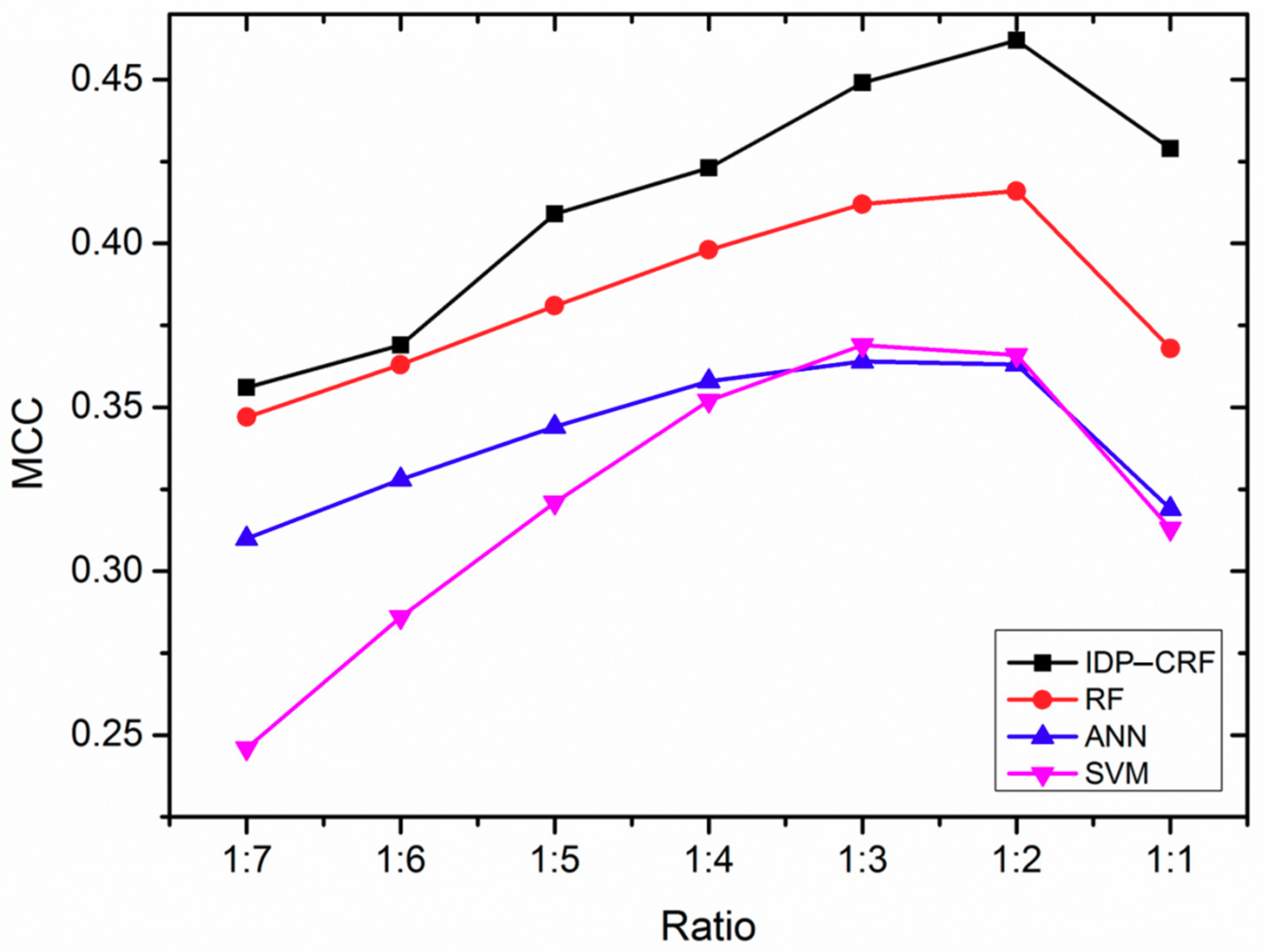
2.1. The Influence of Different Ratios of Positive and Negative Samples on the Performance of Various Predictors
2.2. IDP–CRF (Intrinsically Disordered Protein–Conditional Random Field) Outperforms Classification-Based Predictors
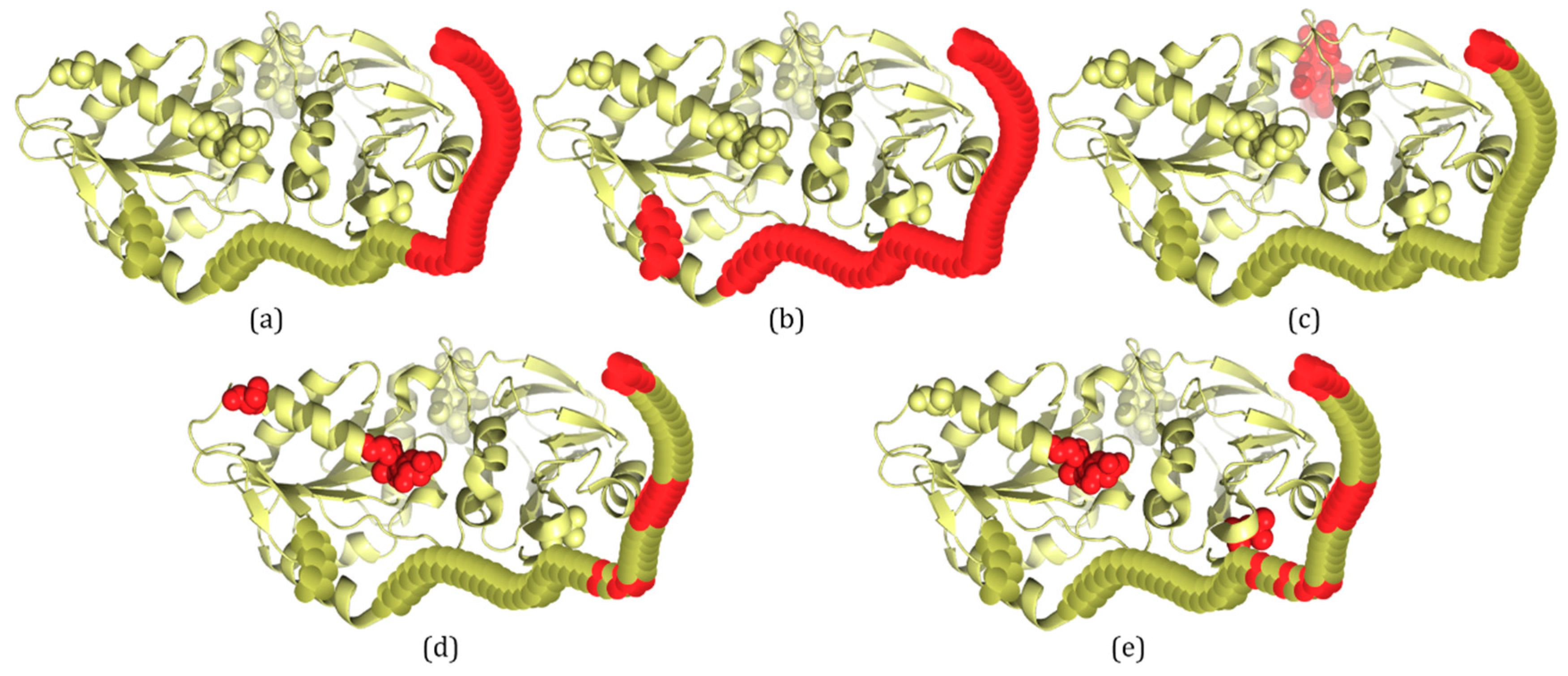
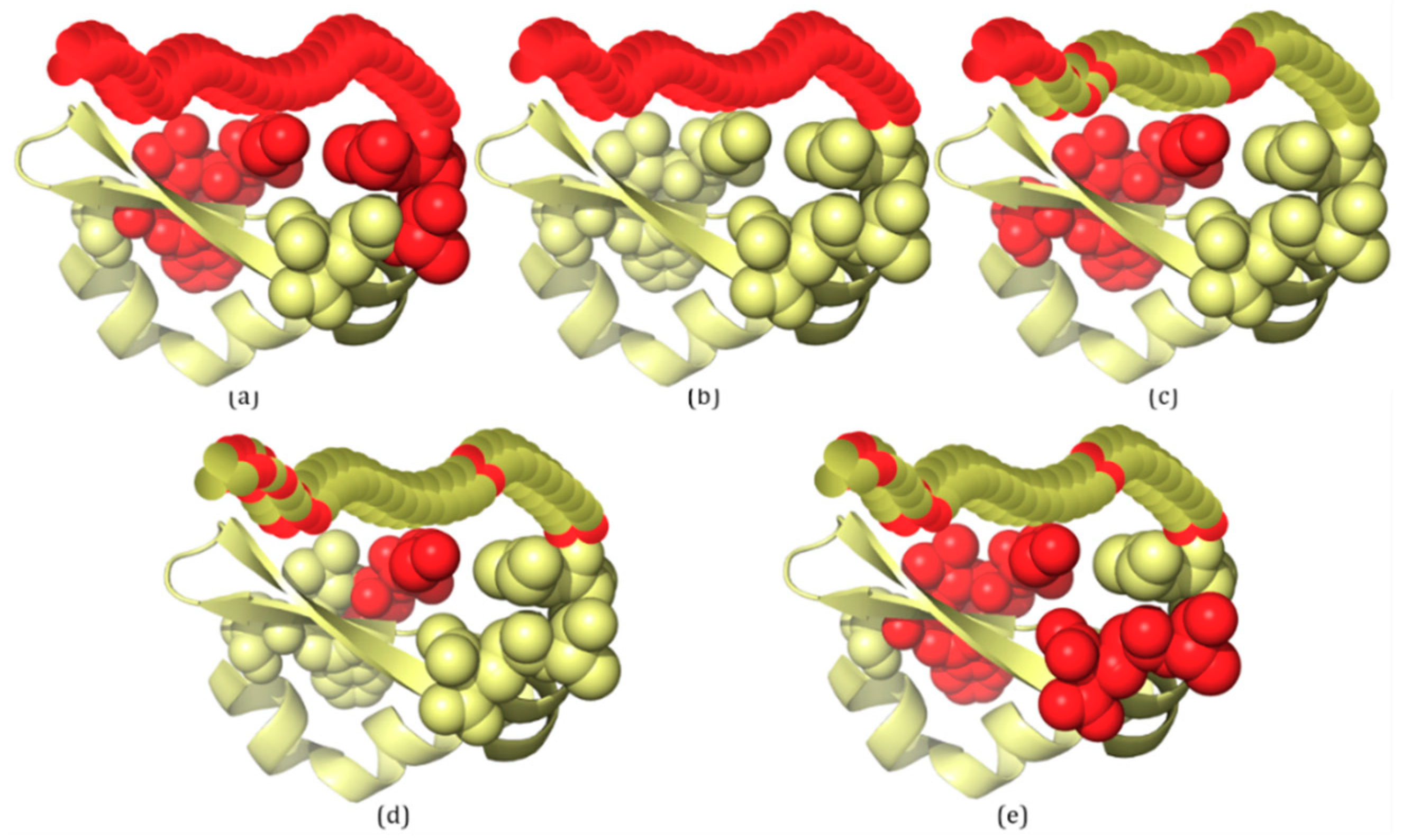
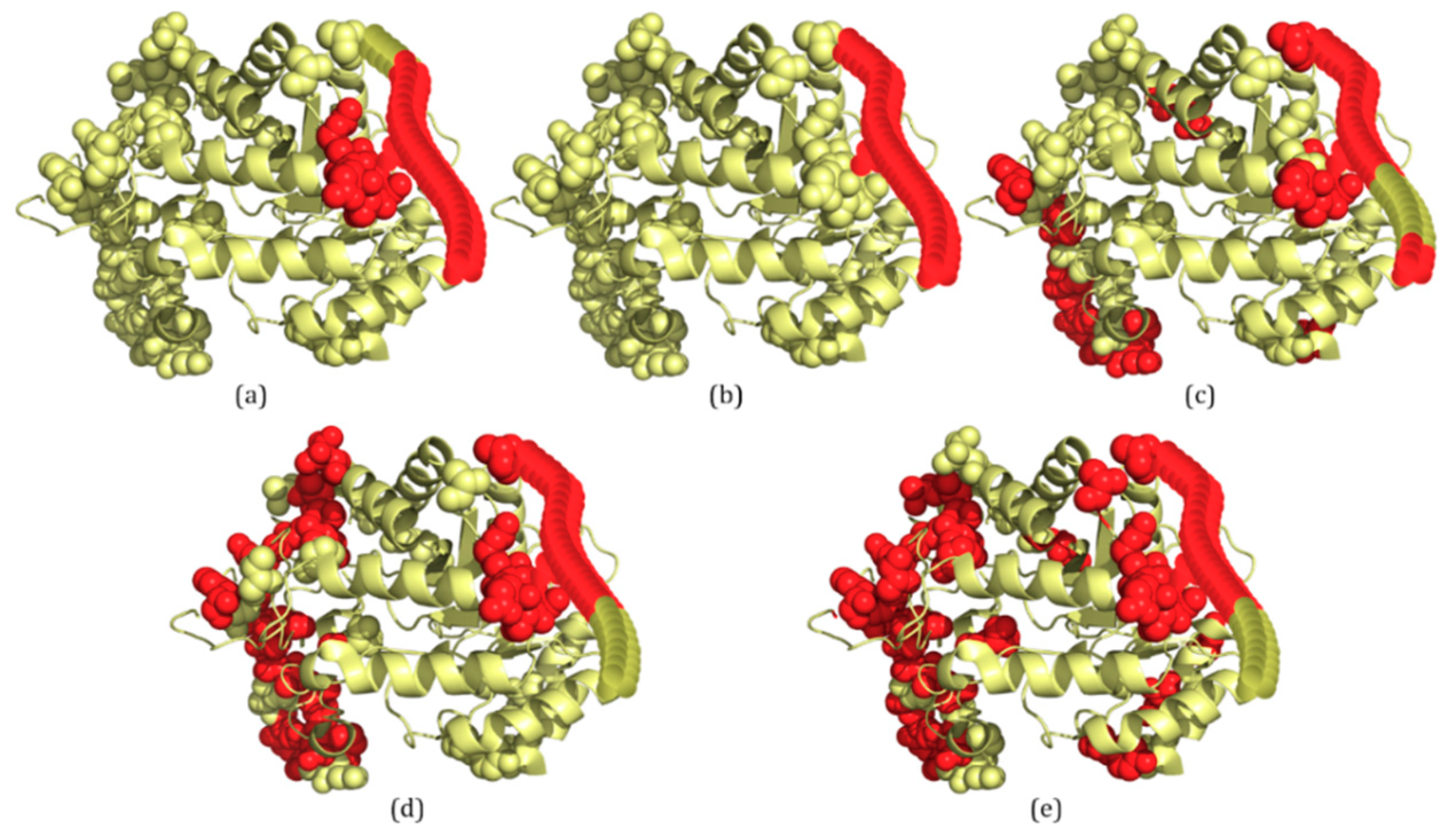
2.3. Several Examples Predicted by IDP–CRF and Three Classification-Based Predictors
2.4. Comparison with Other Related Predictors
3. Materials and Method
3.1. Benchmark Dataset
3.2. Benchmark Independent Datasets
3.3. Features
3.3.1. Transition Feature
3.3.2. PSSMs (Position-Specific Scoring Matrices)
3.3.3. Kmer
3.3.4. Secondary Structure
3.3.5. Relative Solvent Accessibility
3.4. Conditional Random Fields
3.5. Implementations
3.6. Criteria for Performance Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| IDP | intrinsically disordered protein |
| IDR | intrinsically disordered region |
| CRFs | conditional random fields |
| SVM | support vector machine |
| ANN | artificial neural network |
| RF | random forest |
| BLSTM | bidirectional long short-term memory |
| PSSM | position-specific scoring matrix |
| X-Ray | X-ray crystallography |
| NMR | nuclear magnetic resonance |
| CD | circular dichroism |
| TP | true positive |
| FP | false positive |
| TN | true negative |
| FN | false negative |
| Sn | sensitivity |
| Sp | specificity |
| ACC | balanced accuracy |
| MCC | Matthew’s correlation coefficient |
References
- Liu, Y.; Wang, X.; Liu, B. A comprehensive review and comparison of existing computational methods for intrinsically disordered protein and region prediction. Brief. Bioinform. 2017, bbx126. [Google Scholar] [CrossRef]
- Damiano, P.; Francesco, T.; Ivan, M.; Marco, N.; Federica, Q.; Christopher, J.O.; Maria, C.A.; Norman, E.D.; Radoslav, D.; Zsuzsanna, D.; et al. DisProt 7.0: A major update of the database of disordered proteins. Nucleic Acids Res. 2017, 45, D219–D227. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradović, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Uros, M.; Christopher, J.O.A.; Keith, D.; Zoran, O.; Vladimir, N.U. Protein disorder in the human diseasome: Unfoldomics of human genetic diseases. BMC Genomics 2009, 10, S12. [Google Scholar]
- Swasti, R.; Sucharita, D.; Nitai, P.B.; Debashis, M. The role of intrinsically unstructured proteins in neurodegenerative diseases. PLoS ONE 2009, 4, 5566. [Google Scholar]
- Jiang, X.; Zhang, H.; Quan, X.; Yin, Y. A Heterogeneous Networks Fusion Algorithm Based on Local Topological Information for Neurodegenerative Disease. Curr. Bioinform. 2017, 12, 387–397. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, T.; Walsh, I.; Martin, A.J.; Tosatto, S.C. MobiDB: A comprehensive database of intrinsic protein disorder annotations. Bioinformatics 2012, 28, 2080–2081. [Google Scholar] [CrossRef] [PubMed]
- Potenza, E.; Di Domenico, T.; Walsh, I.; Tosatto, S.C. MobiDB 2.0: An improved database of intrinsically disordered and mobile proteins. Nucleic Acids Res. 2015, 43, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Prilusky, J.; Felder, C.E.; Zeev, B.-M.T.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex©: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Russell, R.B.; Neduva, V.; Gibson, T.J. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Faraggi, E.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Zhou, Y. SPINE-D: Accurate prediction of short and long disordered regions by a single neural-network based method. J. Biomol. Struct. Dyn. 2012, 29, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.; Yang, Y.; Paliwal, K.; Zhou, Y. Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Bioinformatics 2017, 33, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sauer, U.H. OnD–CRF: Predicting order and disorder in proteins using [corrected] conditional random fields. Bioinformatics 2008, 24, 1401–1402. [Google Scholar] [CrossRef] [PubMed]
- Marcin, J.; Mizianty, W.S.; Ke, C.; Kanaka, D.K.; Fatemeh, M.D.; Lukasz, K. Improved sequence-based prediction of disordered regions with multilayer fusion of multiple information sources. Bioinformatics 2010, 26, 489–496. [Google Scholar] [CrossRef]
- Liu, B.; Wu, H.; Zhang, D.; Wang, X.; Chou, K.C. Pse-Analysis: A python package for DNA, RNA and protein peptide sequence analysis based on pseudo components and kernel methods. Oncotarget 2017, 8, 13338–13343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, B. PSFM-DBT: Identifying DNA-Binding Proteins by Combing Position Specific Frequency Matrix and Distance-Bigram Transformation. Int. J. Mol. Sci. 2017, 18, 1856. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Kissinger, C.R.; Villafranca, J.E.; Garner, E.; Guilliot, S.; Dunker, A.K. Thousands of proteins likely to have long disordered regions. In Proceedings of the Pacific Symposium on Biocomputing Pacific, Maui, HI, USA, 9 January 1998; pp. 437–448. [Google Scholar]
- Lafferty, J.D.; Mccallum, A.; Pereira, F.C.N. Conditional Random Fields: Probabilistic Models for Segmenting and Labeling Sequence Data. Proceeding of the Eighteenth International Conference on Machine Learning, San Francisco, CA, USA, 28 June 2001; Morgan Kaufmann Publishers Inc.: San Francisco, CA, USA, 2001; pp. 282–289. [Google Scholar]
- Bin, L.; Fule, L.; Wang, X.L.; Chen, J.J.; Fang, L.Y.; Chou, K.-C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, 65–71. [Google Scholar] [CrossRef]
- Liu, B. BioSeq-Analysis: A platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Giollo, M.; Di Domenico, T.; Ferrari, C.; Zimmermann, O.; Tosatto, S.C. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics 2015, 31, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Necci, M.; Piovesan, D.; Dosztányi, Z.; Tompa, P.; Tosatto, S.C.E. A comprehensive assessment of long intrinsic protein disorder from the DisProt database. Bioinformatics 2018, 34, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, X.L.; Lin, L.; Tang, B.Z.; Dong, Q.W.; Wang, X. Prediction of protein binding sites in protein structures using hidden Markov support vector machine. BMC Bioinform. 2009, 10, 381. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Zeng, J.C.; Cao, L.J.; Ji, R.R. A Novel Features Ranking Metric with Application to Scalable Visual and Bioinformatics Data Classification. Neurocomputing 2016, 173, 346–354. [Google Scholar] [CrossRef]
- PyMOL. Available online: https://pymol.org/2/ (accessed on 14 August 2018).
- Schlessinger, A.; Punta, M.; Yachdav, G.; Kajan, L.; Rost, B. Improved disorder prediction by combination of orthogonal approaches. PLoS ONE 2009, 4, 4433. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.R.; Thomson, R.; McNeil, P.; Esnouf, R.M. RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, A.; Liu, J.; Rost, B. Natively unstructured loops differ from other loops. PLoS Comput. Biol. 2007, 3, 140. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein disorder prediction: Implications for structural proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Sweredoski, M.J.; Baldi, P. Accurate Prediction of Protein Disordered Regions by Mining Protein Structure Data. Data Min. Knowl. Discov. 2005, 11, 213–222. [Google Scholar] [CrossRef]
- Hecker, J.; Yang, J.Y.; Cheng, J. Protein disorder prediction at multiple levels of sensitivity and specificity. BMC Genomics 2008, 9, S9. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, A.; Punta, M.; Rost, B. Natively unstructured regions in proteins identified from contact predictions. Bioinformatics 2007, 23, 2376–2384. [Google Scholar] [CrossRef] [PubMed]
- Vullo, A.; Bortolami, O.; Pollastri, G.; Silvio, C.E.T. Spritz: A server for the prediction of intrinsically disordered regions in protein sequences using kernel machines. Nucleic Acids Res. 2006, 34, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, A.; Yachdav, G.; Rost, B. PROFbval: Predict flexible and rigid residues in proteins. Bioinformatics 2006, 22, 891–893. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.L.; Kurgan, L. Comprehensive comparative assessment of in-silico predictors of disordered regions. Curr. Protein Pept. Sci. 2012, 13, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J. Intrinsic disorder prediction from the analysis of multiple protein fold recognition models. Bioinformatics 2008, 24, 1798–1804. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Walsh, I.; Martin, A.J.; Di Domenico, T.; Tosatto, S.C. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, S.; Long, R.; Chou, K.C. iRSpot-EL: Identify recombination spots with an ensemble learning approach. Bioinformatics 2017, 33, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Weng, F.; Huang, D.S.; Chou, K.C. iRO-3wPseKNC: Identify DNA replication origins by three-window-based PseKNC. Bioinformatics 2018. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.A.; Chan, K.C.C.; You, Z.H. Constructing prediction models from expression profiles for large scale lncRNA-miRNA interaction profiling. Bioinformatics 2018, 34, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.P.; Huang, D.S. SFAPS: An R package for structure/function analysis of protein sequences based on informational spectrum method. Methods 2014, 69, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.S. The Study of Data Mining Methods for Gene Expression Profiles; Science Press of China: Beijing, China, 2009. [Google Scholar]
- Chen, J.; Guo, M.; Wang, X.; Liu, B. A comprehensive review and comparison of different computational methods for protein remote homology detection. Brief. Bioinform. 2018, 19, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Fernanda, L.S.; Ooi, H.S.; Tobias, G.; Georg, S.; Frank, E.; Sebastian, M.S. Parameterization of disorder predictors for large-scale applications requiring high specificity by using an extended benchmark dataset. BMC Genomics 2010, 11, S15. [Google Scholar]
- Liu, B.; Zhang, D.; Xu, R.; Xu, J.; Wang, X.; Chen, Q.; Dong, Q.; Chou, K.C. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics 2014, 30, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, J.; Wang, X. Application of Learning to Rank to protein remote homology detection. Bioinformatics 2015, 31, 3492–3498. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, M.; Li, S.; Liu, B. ProtDec-LTR2.0: An improved method for protein remote homology detection by combining pseudo protein and supervised Learning to Rank. Bioinformatics 2017, 33, 3473–3476. [Google Scholar] [CrossRef] [PubMed]
- You, Z.H.; Li, X.; Chan, K.C.C. An improved sequence-based prediction protocol for protein-protein interactions using amino acids substitution matrix and rotation forest ensemble classifiers. Neurocomputing 2017, 228, 277–282. [Google Scholar] [CrossRef]
- Wei, L.; Ding, Y.J.; Su, R.; Tang, J.J.; Zou, Q. Prediction of human protein subcellular localization using deep learning. J. Parallel Distrib. Comput. 2018, 117, 212–217. [Google Scholar] [CrossRef]
- Mishra, A.; Pokhrel, P.; Hoque, M.T. StackDPPred: A Stacking based Prediction of DNA-binding Protein from Sequence. Bioinformatics 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Kurgan, L. PFRES: Protein fold classification by using evolutionary information and predicted secondary structure. Bioinformatics 2007, 23, 2843–2850. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Yang, B.; Revote, J.; Leier, A.; Marquez, L.T.T.; Webb, G.; Song, J.; Chou, K.C.; Lithgow, T. POSSUM: A bioinformatics toolkit for generating numerical sequence feature descriptors based on PSSM profiles. Bioinformatics 2017, 33, 2756–2758. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, H.; Chen, K.; Shen, S.; Ruan, J.; Kurgan, L. Accurate sequence-based prediction of catalytic residues. Bioinformatics 2008, 24, 2329–2338. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Li, F.; Takemoto, K.; Haffari, G.; Akutsu, T.; Chou, K.C.; Webb, G.I. PREvaIL, an integrative approach for inferring catalytic residues using sequence, structural, and network features in a machine-learning framework. J. Theor. Biol. 2018, 443, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, C.; Revote, J.; Zhang, Y.; Webb, G.I.; Li, J.; Song, J.; Lithgow, T. GlycoMinestruct: A new bioinformatics tool for highly accurate mapping of the human N-linked and O-linked glycoproteomes by incorporating structural features. Sci. Rep. 2016, 6, 34595. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.H.; Li, J.C. Feature Extractions for Computationally Predicting Protein Post-Translational Modifications. Curr. Bioinform. 2018, 13, 387–395. [Google Scholar] [CrossRef]
- Zou, Q.; Li, X.B.; Jiang, Y.; Zhao, Y.M.; Wang, G.H. BinMemPredict: A Web Server and Software for Predicting Membrane Protein Types. Curr. Proteomics 2013, 10, 2–9. [Google Scholar] [CrossRef]
- Holm, L.; Sander, C. Removing near-neighbour redundancy from large protein sequence collections. Bioinformatics 1998, 14, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, H. Protein secondary structure prediction based on an improved support vector machines approach. Protein Eng. 2003, 16, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, X.; Lin, L.; Dong, Q.; Wang, X. A Discriminative Method for Protein Remote Homology Detection and Fold Recognition Combining Top-n-grams and Latent Semantic Analysis. BMC Bioinform. 2008, 9, 510. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, J.H.; Lan, X.; Xu, R.F.; Zhou, J.Y.; Wang, X.L.; Chou, K.C. iDNA-Prot|dis: Identifying DNA-Binding Proteins by Incorporating Amino Acid Distance-Pairs and Reduced Alphabet Profile into the General Pseudo Amino Acid Composition. PLoS ONE 2014, 9, 106691. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liao, M.H.; Gao, X.; Zou, Q. An. Improved Protein Structural Classes Prediction Method by Incorporating Both Sequence and Structure Information. IEEE Trans. Nanobiosci. 2015, 14, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liao, M.H.; Gao, X.; Zou, Q. Enhanced Protein Fold Prediction Method Through a Novel Feature Extraction Technique. IEEE Trans. Nanobiosci. 2015, 14, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Peng, Z.; Yang, J. CoABind: A novel algorithm for Coenzyme A (CoA)- and CoA derivatives-binding residues prediction. Bioinformatics 2018, 34, 2598–2604. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, C.; Wang, M.; Webb, G.I.; Zhang, Y.; Whisstock, J.C.; Song, J. GlycoMine: A machine learning-based approach for predicting N-, C- and O-linked glycosylation in the human proteome. Bioinformatics 2015, 31, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, Z.; Kurgan, L. Comprehensive review and empirical analysis of hallmarks of DNA-, RNA- and protein-binding residues in protein chains. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wang, Y.; Li, F.; Akutsu, T.; Rawlings, N.D.; Webb, G.I.; Chou, K.C. iProt-Sub: A comprehensive package for accurately mapping and predicting protease-specific substrates and cleavage sites. Brief. Bioinform. 2018, 97. [Google Scholar] [CrossRef] [PubMed]
- Adamczak, R.; Porollo, A.; Meller, J. Accurate prediction of solvent accessibility using neural networks–based regression. Proteins Struct. Funct. Bioinf. 2004, 56, 753–767. [Google Scholar] [CrossRef]
- Wagner, M.; Adamczak, R.; Porollo, A.; Meller, J. Linear regression models for solvent accessibility prediction in proteins. J. Comput. Biol. 2005, 12, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Wang, K.; Dang, T.K.; Gültas, M.; Welter, M.; Wierschin, T.; Stanke, M.; Waack, S. CRF-based models of protein surfaces improve protein-protein interaction site predictions. BMC Bioinform. 2014, 15, 277. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, M.; Kamada, M.; Song, J.N.; Akutsu, T. Conditional random field approach to prediction of protein-protein interactions using domain information. BMC Syst. Biol. 2011, 5, S8. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.H.; Van, L.K.; Verschoren, A.; Laukens, K. Prediction of kinase-specific phosphorylation sites using conditional random fields. Bioinformatics 2008, 24, 2857–2864. [Google Scholar] [CrossRef] [PubMed]
- Meysman, P.; Dang, T.H.; Laukens, K.; de Smet, R.; Wu, Y.; Marchal, K.; Engelen, K. Use of structural DNA properties for the prediction of transcription-factor binding sites in Escherichia coli. Nucleic Acids Res. 2011, 39, 6. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, M.; Kamada, K.; Song, J.N.; Akutsu, T. Prediction of protein-RNA residue-base contacts using two-dimensional conditional random field with the lasso. BMC Syst. Biol. 2013, 7, S15. [Google Scholar] [CrossRef] [PubMed]
- FlexCRFs: Flexible Conditional Random Fields. Available online: http://flexcrfs.sourceforge.net/documents.html (accessed on 14 August 2018).
- Li, M.H.; Lin, L.; Wang, X.L.; Liu, T. Protein-protein interaction site prediction based on conditional random fields. Bioinformatics 2007, 23, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Fan, R.E.; Chang, K.W.; Hsieh, C.J.; Wang, X.R.; Lin, C.J. LIBLINEAR: A Library for Large Linear Classification. J. Mach. Learn. Res. 2008, 9, 1871–1874. [Google Scholar]
- Monastyrskyy, B.; Kryshtafovych, A.; Moult, J.; Tramontano, A.; Fidelis, K. Assessment of protein disorder region predictions in CASP10. Proteins 2014, 82, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, J.; Fan, S.; Xu, R.; Zhou, J.; Wang, X. PseDNA-Pro: DNA-Binding Protein Identification by Combining Chou’s PseAAC and Physicochemical Distance Transformation. Mol. Inform. 2015, 34, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Huang, Z.H.; Yuan, C.A.; Huang, D.S. Pupylation sites prediction with ensemble classification model. Int. J. Data Min. Bioinform. 2017, 18, 91–104. [Google Scholar] [CrossRef]
- Huang, D.S.; Du, J.X. A constructive hybrid structure optimization methodology for radial basis probabilistic neural networks. IEEE Trans. Neural Netw. 2008, 19, 2099–2115. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.S. A constructive approach for finding arbitrary roots of polynomials by neural networks. IEEE Trans. Neural Netw. 2004, 15, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.S. Radial basis probabilistic neural networks: Model and application. Intern. J. Pattern Recognit. Artif. Intell. 1999, 13, 1083–1101. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Ratio a | Sn b | Sp c | ACC d | MCC e |
|---|---|---|---|---|---|
| IDP–CRF | 1:2 | 0.637 | 0.910 | 0.774 | 0.462 |
| RF | 1:2 | 0.524 | 0.928 | 0.726 | 0.416 |
| SVM | 1:2 | 0.543 | 0.896 | 0.720 | 0.366 |
| ANN | 1:2 | 0.537 | 0.897 | 0.717 | 0.363 |
| Predictor a | Sn | Sp | ACC | MCC | Rank | |
|---|---|---|---|---|---|---|
| ACC | MCC | |||||
| IDP–CRF | 0.680 | 0.821 | 0.750 | 0.460 | 2 | 1 |
| MFDp [15] | 0.746 | 0.768 | 0.757 | 0.451 | 1 | 2 |
| MD [27] | 0.673 | 0.813 | 0.743 | 0.444 | 3 | 3 |
| PONDR-FIT [28] | 0.631 | 0.821 | 0.726 | 0.419 | 6 | 4 |
| DISOPRED2 [29] | 0.647 | 0.800 | 0.724 | 0.406 | 7 | 5 |
| IUPred-long [30] | 0.581 | 0.841 | 0.711 | 0.405 | 8 | 6 |
| PONDR VSL2B [31] | 0.774 | 0.698 | 0.736 | 0.401 | 4 | 7 |
| OnD–CRF b [14] | 0.752 | 0.711 | 0.732 | 0.396 | 5 | 8 |
| IUPred-short [30] | 0.522 | 0.866 | 0.694 | 0.389 | 10 | 9 |
| RONN [32] | 0.664 | 0.754 | 0.709 | 0.368 | 9 | 10 |
| NORSnet [33] | 0.532 | 0.829 | 0.681 | 0.347 | 11 | 11 |
| DisEMBL-R [34] | 0.316 | 0.936 | 0.626 | 0.323 | 15 | 12 |
| DISpro [35,36] | 0.303 | 0.940 | 0.622 | 0.318 | 16 | 13 |
| Ucon [37] | 0.554 | 0.787 | 0.671 | 0.313 | 12 | 14 |
| Spritz [38] | 0.494 | 0.812 | 0.653 | 0.293 | 14 | 15 |
| FoldIndex [10] | 0.602 | 0.717 | 0.660 | 0.278 | 13 | 16 |
| DisEMBL-H [34] | 0.435 | 0.792 | 0.614 | 0.216 | 17 | 17 |
| PROFbval [39] | 0.835 | 0.387 | 0.611 | 0.196 | 18 | 18 |
| GlobPlot [11] | 0.353 | 0.826 | 0.590 | 0.182 | 19 | 19 |
| DisEMBL-C [34] | 0.760 | 0.414 | 0.587 | 0.150 | 20 | 20 |
| Predictor a | Sn | Sp | ACC | MCC | Rank | |
|---|---|---|---|---|---|---|
| ACC | MCC | |||||
| IDP–CRF | 0.75 | 0.88 | 0.817 | 0.64 | 1 | 2 |
| SPOT-disorder [13] | 0.67 | 0.96 | 0.815 | 0.67 | 2 | 1 |
| SPINE-D [12] | 0.78 | 0.85 | 0.815 | 0.63 | 2 | 3 |
| DISOPRED3 [41] | - | - | 0.795 | 0.61 | 4 | 4 |
| DISOPRED2 [29] | 0.69 | 0.90 | 0.795 | 0.59 | 4 | 5 |
| OnD–CRF b [14] | 0.79 | 0.80 | 0.793 | 0.58 | 6 | 6 |
| MD [27] | 0.66 | 0.89 | 0.775 | 0.58 | 7 | 6 |
| PONDR-FIT [28] | 0.61 | 0.91 | 0.760 | 0.55 | 8 | 8 |
| IUPred-long [30] | 0.60 | 0.92 | 0.760 | 0.55 | 8 | 8 |
| MFDp [15] | 0.88 | 0.62 | 0.750 | 0.51 | 11 | 10 |
| DISOClust [42] | 0.81 | 0.70 | 0.755 | 0.51 | 10 | 10 |
| NORSnet [33] | 0.54 | 0.92 | 0.730 | 0.51 | 12 | 10 |
| IUPred-short [30] | 0.50 | 0.94 | 0.720 | 0.50 | 13 | 13 |
| Ucon [37] | 0.59 | 0.81 | 0.700 | 0.42 | 14 | 14 |
| DisEMBL [34] | - | - | 0.660 | 0.40 | 16 | 15 |
| Dispro [35,36] | 0.28 | 0.99 | 0.635 | 0.40 | 18 | 15 |
| PONDR VL-XT [43] | 0.59 | 0.78 | 0.685 | 0.38 | 15 | 17 |
| Espritz [44] | - | - | 0.605 | 0.35 | 19 | 18 |
| PROFbval [39] | - | - | 0.648 | 0.30 | 17 | 19 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Wang, X.; Liu, B. IDP–CRF: Intrinsically Disordered Protein/Region Identification Based on Conditional Random Fields. Int. J. Mol. Sci. 2018, 19, 2483. https://doi.org/10.3390/ijms19092483
Liu Y, Wang X, Liu B. IDP–CRF: Intrinsically Disordered Protein/Region Identification Based on Conditional Random Fields. International Journal of Molecular Sciences. 2018; 19(9):2483. https://doi.org/10.3390/ijms19092483
Chicago/Turabian StyleLiu, Yumeng, Xiaolong Wang, and Bin Liu. 2018. "IDP–CRF: Intrinsically Disordered Protein/Region Identification Based on Conditional Random Fields" International Journal of Molecular Sciences 19, no. 9: 2483. https://doi.org/10.3390/ijms19092483
APA StyleLiu, Y., Wang, X., & Liu, B. (2018). IDP–CRF: Intrinsically Disordered Protein/Region Identification Based on Conditional Random Fields. International Journal of Molecular Sciences, 19(9), 2483. https://doi.org/10.3390/ijms19092483