Tau in Oligodendrocytes Takes Neurons in Sickness and in Health

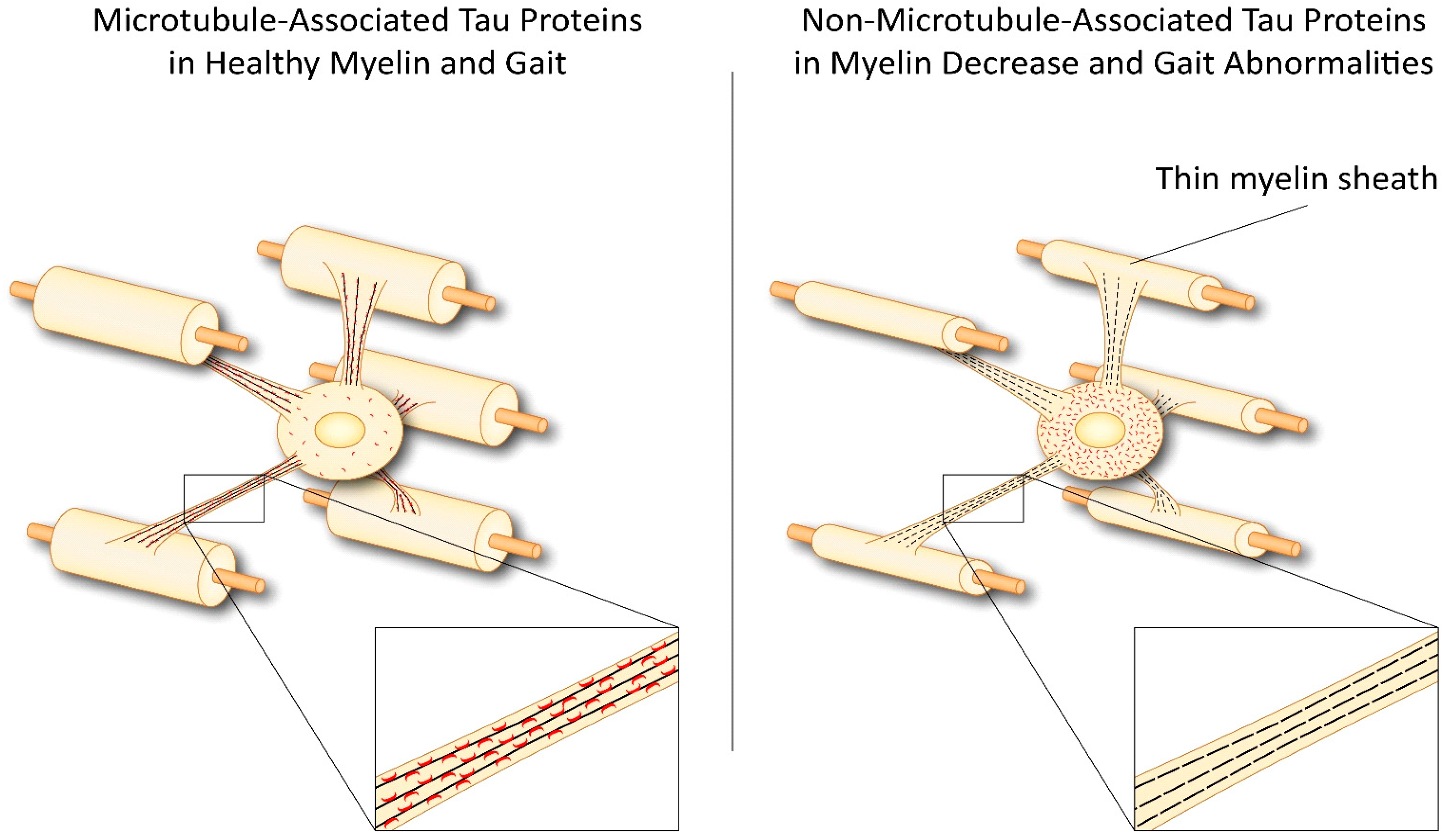{kind=link}
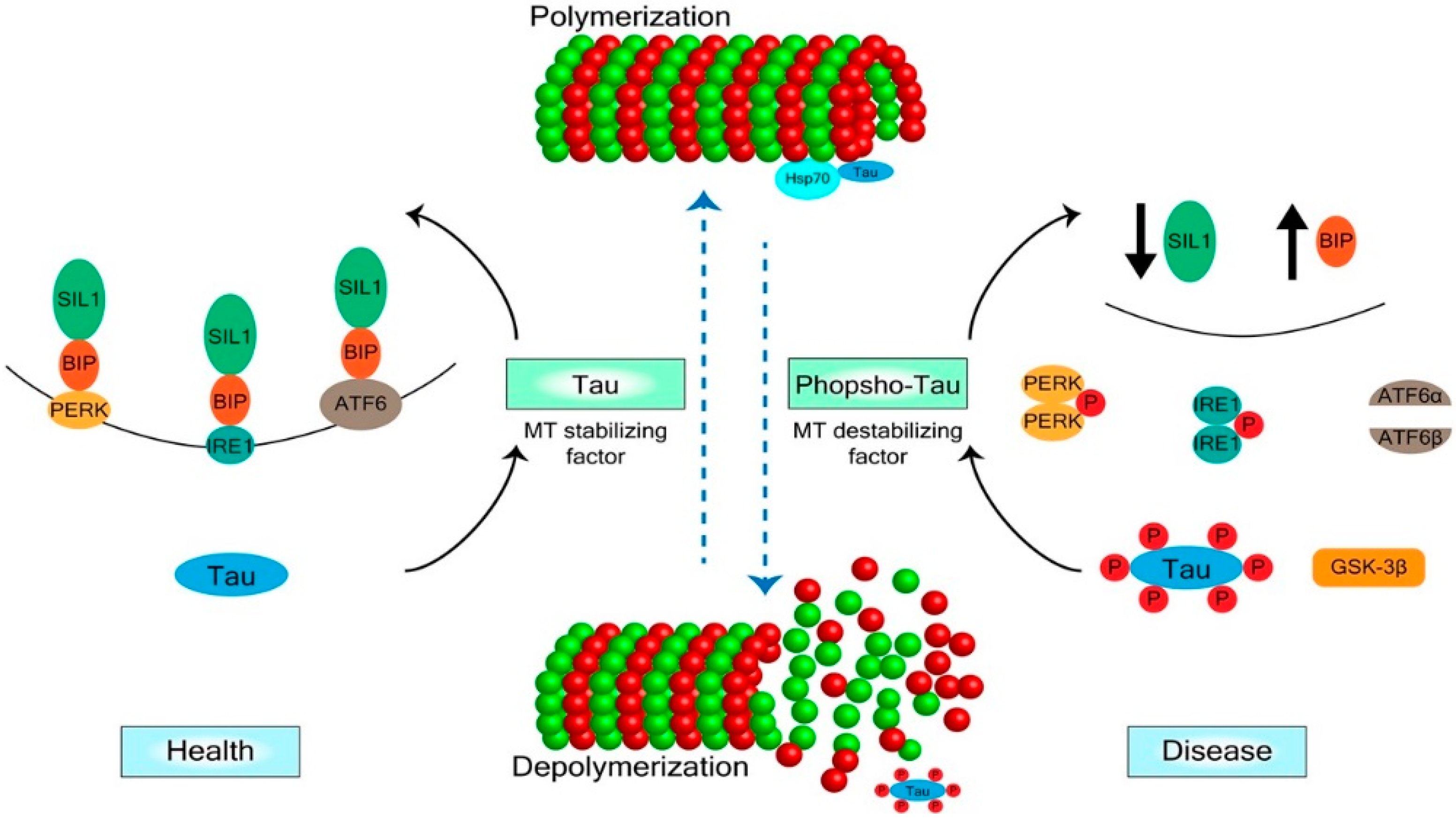{kind=link}
Abstract
1. Introduction
2. Mechanisms of Tau Regulation
3. Tau in Oligodendrocytes and CNS Degenerative Diseases
Acknowledgments
Conflicts of Interest
References
- Almeida, R.G.; Lyons, D.A. On Myelinated Axon Plasticity and Neuronal Circuit Formation and Function. J. Neurosci. 2017, 37, 10023–10034. [Google Scholar] [PubMed]
- Saab, A.S.; Nave, K.A. Myelin dynamics: Protecting and shaping neuronal functions. Curr. Opin. Neurobiol. 2017, 47, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Young, K.M. White matter plasticity in adulthood. Neuroscience 2014, 276, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Forbes, T.A.; Gallo, V. All Wrapped Up: Environmental Effects on Myelination. Trends Neurosci. 2017, 40, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Klein, M.C.; Behrens, T.E.; Johansen-Berg, H. Training induces changes in white matter architecture. Nat. Neurosci. 2009, 12, 1367–1368. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Sekiguchi, A.; Taki, Y.; Yokoyama, S.; Yomogida, Y.; Komuro, N.; Yamanouchi, T.; Suzuki, S.; Kawashima, R. Training of working memory impacts structural connectivity. J. Neurosci. 2010, 30, 3297–3303. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, I.A.; Ohayon, D.; Li, H.; de Faria, J.P.; Emery, B.; Tohyama, K.; Richardson, W.D. Motor skill learning requires active central myelination. Science 2014, 346, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.A.; Li, A.M.; Grutzendler, J. Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat. Neurosci. 2018, 21, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [PubMed]
- LoPresti, P.; Szuchet, S.; Papasozomenos, S.C.; Zinkowski, R.P.; Binder, L.I. Functional implications for the microtubule-associated protein tau: Localization in oligodendrocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 10369–10373. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Kramer, E.M.; Cardine, A.M.; Schraven, B.; Brandt, R.; Trotter, J. Process outgrowth of oligodendrocytes is promoted by interaction of Fyn kinase with the cytoskeletal protein tau. J. Neurosci. 2002, 22, 698–707. [Google Scholar] [CrossRef] [PubMed]
- LoPresti, P. Regulation and differential expression of tau mRNA isoforms as oligodendrocytes mature in vivo: Implications for myelination. Glia 2002, 37, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Seiberlich, V.; Bauer, N.G.; Schwarz, L.; Ffrench-Constant, C.; Goldbaum, O.; Richter-Landsberg, C. Downregulation of the microtubule associated protein tau impairs process outgrowth and myelin basic protein mRNA transport in oligodendrocytes. Glia 2015, 63, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- LoPresti, P. Inducible Expression of a Truncated Form of Tau in Oligodendrocytes Elicits Gait Abnormalities and a Decrease in Myelin: Implications for Selective CNS Degenerative Diseases. Neurochem. Res. 2015, 40, 2188–2199. [Google Scholar] [CrossRef] [PubMed]
- LoPresti, P. Oligodendrocyte Tau’s failed quest for microtubules results in myelin decrease and falling. Atlas of Science. 11 November 2015. Available online: http://atlasofscience.org/oligodendrocyte-taus-failed-quest-for-microtubules-results-in-myelin-decrease-and-falling/ (accessed on 14 August 2018).
- LoPresti, P.; Muma, N.A.; De Vries, G.H. Neu differentiation factor regulates tau protein and mRNA in cultured neonatal oligodendrocytes. Glia 2001, 35, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Belkadi, A.; LoPresti, P. Truncated Tau with the Fyn-binding domain and without the microtubule-binding domain hinders the myelinating capacity of an oligodendrocyte cell line. J. Neurochem. 2008, 107, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Káradóttir, R.; Cavelier, P.; Bergersen, L.H.; Attwell, D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005, 438, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Micu, I.; Jiang, Q.; Coderre, E.; Ridsdale, A.; Zhang, L.; Woulfe, J.; Yin, X.; Trapp, B.D.; McRory, J.E.; Rehak, R.; et al. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 2006, 439, 988–992. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.G.; Fern, R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate. Nature 2005, 438, 1167–1171. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Komai, S.; Tezuka, T.; Hisatsune, C.; Umemori, H.; Semba, K.; Mishina, M.; Manabe, T.; Yamamoto, T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-d-aspartate receptor. J. Biol. Chem. 2001, 276, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Lu, X.; Bernard, A.; Khrestchatisky, M.; Baudry, M. Tyrosine phosphorylation of ionotropic glutamate receptors by Fyn or Src differentially modulates their susceptibility to calpain and enhances their binding to spectrin and PSD-95. J. Neurochem. 2001, 7, 382–390. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, J.S.; Freire, M.A.; Lima, R.R.; Picanço-Diniz, C.W.; Pereira, A.; Gomes-Leal, W. Minocycline treatment reduces white matter damage after excitotoxic striatal injury. Brain Res. 2010, 1329, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Irving, E.A.; McCulloch, J.; Dewar, D. Intracortical perfusion of glutamate in vivo induces alterations of tau and microtubule-associated protein 2 immunoreactivity in the rat. Acta Neuropathol. 1996, 92, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Ahrendsen, J.T.; Macklin, W. Signaling mechanisms regulating myelination in the central nervous system. Neurosci. Bull. 2013, 29, 199–215. [Google Scholar] [PubMed]
- Stevens, B.; Porta, S.; Haak, L.L.; Gallo, V.; Fields, R.D. Adenosine: A neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron 2002, 36, 855–868. [Google Scholar] [CrossRef]
- Brophy, P.J.; Boccaccio, G.L.; Colman, D.R. The distribution of myelin basic protein mRNAs within myelinating oligodendrocytes. Trends Neurosci. 1993, 16, 515–521. [Google Scholar] [CrossRef]
- Drubin, D.G.; Caput, D.; Kirschner, M.W. Studies on the expression of the microtubule-associated protein, tau, during mouse brain development, with newly isolated complementary DNA probes. J. Cell Biol. 1984, 98, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Lenk, R.; Ransom, L.; Kaufmann, Y.; Penman, S. A cytoskeletal structure with associated polyribosomes obtained from HeLa cells. Cell 1977, 10, 67–78. [Google Scholar] [CrossRef]
- Gorath, M.; Stahnke, T.; Mronga, T.; Goldbaum, O.; Richter-Landsberg, C. Developmental changes of tau protein and mRNA in cultured rat brain oligodendrocytes. Glia 2001, 36, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Niblock, M.; Gallo, J.M. Tau alternative splicing in familial and sporadic tauopathies. Biochem. Soc. Trans. 2012, 40, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Richter-Landsberg, C.; Gorath, M. Developmental regulation of alternatively spliced isoforms of mRNA encoding MAP2 and tau in rat brain oligodendrocytes during culture maturation. J. Neurosci. Res. 1999, 56, 259–270. [Google Scholar] [CrossRef]
- Ksiezak-Reding, H.; Farooq, M.; Yang, L.S.; Dickson, D.W.; LoPresti, P. Tau protein expression in adult bovine oligodendrocytes: Functional and pathological significance. Neurochem. Res. 2003, 28, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; López-González, I.; Carmona, M.; Arregui, L.; Dalfó, E.; Torrejón-Escribano, B.; Diehl, R.; Kovacs, G.G. Glial and neuronal tau pathology in tauopathies: Characterization of disease-specific phenotypes and tau pathology progression. J. Neuropathol. Exp. Neurol. 2014, 73, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Buée, L.; Delacourte, A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol. 1999, 9, 681–693. [Google Scholar] [PubMed]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Arima, K. Ultrastructural characteristics of tau filaments in tauopathies: Immuno-electron microscopic demonstration of tau filaments in tauopathies. Neuropathology 2006, 26, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Hogg, M.; Grujic, Z.M.; Baker, M.; Demirci, S.; Guillozet, A.L.; Sweet, A.P.; Herzog, L.L.; Weintraub, S.; Mesulam, M.M.; LaPointe, N.E.; et al. The L266 V tau mutation is associated with frontotemporal dementia and Pick-like 3R and 4R tauopathy. Acta Neuropathol. 2003, 106, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Kouri, N.; Murray, M.E.; Josephs, K.A. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J. Mol. Neurosci. 2011, 45, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Ghetti, B.; Spillantini, M.G. Frontotemporal dementia: Implications for understanding Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006254. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, K.; Takio, K.; Miura, R.; Titani, K.; Ihara, Y. Fetal-type phosphorylation of the tau in paired helical filaments. J. Neurochem. 1992, 58, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- LoPresti, P.; Konat, G.W. Hydrogen peroxide induces transient dephosphorylation of tau protein in cultured rat oligodendrocytes. Neurosci. Lett. 2001, 311, 142–144. [Google Scholar] [CrossRef]
- Goldbaum, O.; Richter-Landsberg, C. Activation of PP2A-like phosphatase and modulation of tau phosphorylation accompany stress-induced apoptosis in cultured oligodendrocytes. Glia 2002, 40, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, C.A.; Egaña, J.T.; Núñez, M.T.; Maccioni, R.B.; González-Billault, C. Oxidative stress promotes tau dephosphorylation in neuronal cells: The roles of cdk5 and PP1. Free Radic. Biol. Med. 2004, 36, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Gladbach, A.; van Eersel, J.; Ittner, A.; Przybyla, M.; van Hummel, A.; Chua, S.W.; van der Hoven, J.; Lee, W.S.; Müller, J.; et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat. Commun. 2017, 8, 473. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.L.; Britton, J.W.; Blessing, M.M.; Parisi, J.E.; Cascino, G.D. Chronic traumatic encephalopathy in an epilepsy surgery cohort: Clinical and pathologic findings. Neurology 2018, 90, e474–e478. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Chen, J.; Zhang, Q.; Sun, A. Deletion of tau attenuates heat shock-induced injury in cultured cortical neurons. J. Neurosci. Res. 2010, 88, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 1993, 61, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Timm, T.; Balusamy, K.; Li, X.; Biernat, J.; Mandelkow, E.; Mandelkow, E.M. Glycogen synthase kinase (GSK) 3beta directly phosphorylates Serine 212 in the regulatory loop and inhibits microtubule affinity-regulating kinase (MARK) 2. J. Biol. Chem. 2008, 283, 18873–18882. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Biernat, J.; Wu, Y.Z.; Mandelkow, E.M.; et al. Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J. Biol. Chem. 2001, 276, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Praticò, D. Glucose deprivation increases tau phosphorylation via P38 mitogen-activated protein kinase. Aging Cell 2015, 14, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Pelech, S.L. Networking with proline-directed protein kinases implicated in tau phosphorylation. Neurobiol. Aging 1995, 16, 247–256. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Zhang, J.; Luo, F.; Herrup, K.; Bibb, J.A.; Lu, R.; Miller, R.H. Cyclin dependent kinase 5 is required for the normal development of oligodendrocytes and myelin formation. Dev. Biol. 2013, 378, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Burke, K.; Kantor, C.; Miller, R.H.; Yang, Y. Cyclin-dependent kinase 5 mediates adult OPC maturation and myelin repair through modulation of Akt and GsK-3β signaling. J. Neurosci. 2014, 34, 10415–10429. [Google Scholar] [CrossRef] [PubMed]
- Maixner, D.W.; Weng, H.R. The Role of Glycogen Synthase Kinase 3 Beta in Neuroinflammation and Pain. J. Pharm. Pharmacol. 2013, 1, 1. [Google Scholar]
- Mandelkow, E.M.; Thies, E.; Trinczek, B.; Biernat, J.; Mandelkow, E. MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J. Cell Biol. 2004, 167, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Zheng-Fischhöfer, Q.; Biernat, J.; Mandelkow, E.M.; Illenberger, S.; Godemann, R.; Mandelkow, E. Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur. J. Biochem. 1998, 252, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Fujisaki, N.; Maruko-Otake, A.; Ohtake, Y.; Shimizu, S.; Saito, T.; Hisanaga, S.I.; Iijima, K.M.; Ando, K. Ca2+/calmodulin-dependent protein kinase II promotes neurodegeneration caused by tau phosphorylated at Ser262/356 in a transgenic Drosophila model of tauopathy. J. Biochem. 2017, 162, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Lee, G. Tau and src family tyrosine kinases. Biochim. Biophys. Acta 2005, 1739, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Derkinderen, P.; Scales, T.M.; Hanger, D.P.; Leung, K.Y.; Byers, H.L.; Ward, M.A.; Lenz, C.; Price, C.; Bird, I.N.; Perera, T.; et al. Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J. Neurosci. 2005, 25, 6584–6593. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, K.; Yen, S.H.; Lee, G. Disease-related modifications in tau affect the interaction between Fyn and Tau. J. Biol. Chem. 2005, 280, 35119–35125. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.H.; Garwood, C.J.; Wray, S.; Price, C.; Kellie, S.; Perera, T.; Zvelebil, M.; Yang, A.; Sheppard, P.W.; Varndell, I.M.; et al. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J. Biol. Chem. 2008, 283, 18177–18186. [Google Scholar] [CrossRef] [PubMed]
- Holleran, L.; Kim, J.H.; Gangolli, M.; Stein, T.; Alvarez, V.; McKee, A.; Brody, D.L. Axonal disruption in white matter underlying cortical sulcus tau pathology in chronic traumatic encephalopathy. Acta Neuropathol. 2017, 133, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Noack, M.; Leyk, J.; Richter-Landsberg, C. HDAC6 inhibition results in tau acetylation and modulates tau phosphorylation and degradation in oligodendrocytes. Glia 2014, 62, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Lucke-Wold, B.; Seidel, K.; Udo, R.; Omalu, B.; Ornstein, M.; Nolan, R.; Rosen, C.; Ross, J. Role of Tau Acetylation in Alzheimer’s Disease and Chronic Traumatic Encephalopathy: The Way Forward for Successful Treatment. J. Neurol. Neurosurg. 2017, 4, 140. [Google Scholar] [PubMed]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, V.; Corsetti, V.; Florenzano, F.; Atlante, A.; Bobba, A.; Ciotti, M.T.; Natale, F.; Della Valle, F.; Borreca, A.; Manca, A.; et al. NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: Implications in Alzheimer’s disease. Hum. Mol. Genet. 2015, 24, 3058–3081. [Google Scholar] [PubMed]
- Garg, S.; Timm, T.; Mandelkow, E.M.; Mandelkow, E.; Wang, Y. Cleavage of Tau by calpain in Alzheimer’s disease: The quest for the toxic 17 kDa fragment. Neurobiol. Aging 2011, 32, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.I.; Narayanan, S.P.; Morse, E.N.; Shick, H.E.; Yin, X.; Kidd, G.; Avila, R.L.; Kirschner, D.A.; Macklin, W.B. Constitutively active Akt induces enhanced myelination in the CNS. J. Neurosci. 2008, 28, 7174–7183. [Google Scholar] [CrossRef] [PubMed]
- Ebner, S.; Dunbar, M.; McKinnon, R.D. Distinct roles for PI3 K in proliferation and survival of oligodendrocyte progenitor cells. J. Neurosci. Res. 2000, 62, 336–345. [Google Scholar] [CrossRef]
- Bagayogo, I.P.; Dreyfus, C.F. Regulated release of BDNF by cortical oligodendrocytes is mediated through metabotropic glutamate receptors and the PLC pathway. ASN Neuro 2009, 1, e00001. [Google Scholar] [CrossRef] [PubMed]
- Röhl, A.; Rohrberg, J.; Buchner, J. The chaperone Hsp90: Changing partners for demanding clients. Trends Biochem. Sci. 2013, 38, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.S.; Yang, X.; Lau, J.C.; Hung, C.H.; Wuwongse, S.; Zhang, Q.; Wang, J.; Baum, L.; So, K.F.; Chang, R.C. Endoplasmic reticulum stress induces tau pathology and forms a vicious cycle: Implication in Alzheimer’s disease pathogenesis. J. Alzheimers Dis. 2012, 28, 839–854. [Google Scholar] [CrossRef] [PubMed]
- Dou, F.; Netzer, W.J.; Tanemura, K.; Li, F.; Hartl, F.U.; Takashima, A.; Gouras, G.K.; Greengard, P.; Xu, H. Chaperones increase association of tau protein with microtubules. Proc. Natl. Acad. Sci. USA 2003, 100, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Araújo, G.W.; Trajkovic, K.; Herrmann, M.M.; Merkler, D.; Mandelkow, E.M.; Weissert, R.; Simons, M. Hyperphosphorylation and aggregation of Tau in experimental autoimmune encephalomyelitis. J. Biol. Chem. 2004, 279, 55833–55839. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Hampton, D.W.; Patani, R.; Pryce, G.; Crowther, R.A.; Reynolds, R.; Franklin, R.J.; Giovannoni, G.; Compston, D.A.; Baker, D.; et al. Abnormally phosphorylated Tau is associated with neuronal and axonal loss in experimental autoimmune encephalomyelitis and multiple sclerosis. Brain 2008, 131, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Patani, R.; Reynolds, R.; Nicholas, R.; Compston, A.; Spillantini, M.G.; Chandran, S. Evidence for abnormal Tau phosphorylation in early aggressive multiple sclerosis. Acta Neuropathol. 2009, 117, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Buchkremer, S.; González Coraspe, J.A.; Weis, J.; Roos, A. Sil1-Mutant Mice Elucidate Chaperone Function in Neurological Disorders. J. Neuromuscul. Dis. 2016, 3, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Voss, K.; Combs, B.; Patterson, K.R.; Binder, L.I.; Gamblin, T.C. Hsp70 alters tau function and aggregation in an isoform specific manner. Biochemistry 2012, 51, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Mori, K.; Ma, W.; Gething, M.J.; Sambrook, J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993, 74, 743–756. [Google Scholar] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Okada, T.; Yoshida, H.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem. J. 2001, 355, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Clayton, B.L.L.; Popko, B. Endoplasmic reticulum stress and the unfolded protein response in disorders of myelinating glia. Brain Res. 2016, 1648, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Hussien, Y.; Cavener, D.R.; Popko, B. Genetic inactivation of PERK signaling in mouse oligodendrocytes: Normal developmental myelination with increased susceptibility to inflammatory demyelination. Glia 2014, 62, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Hussien, Y.; Podojil, J.R.; Robinson, A.P.; Lee, A.S.; Miller, S.D.; Popko, B. ER Chaperone BiP/GRP78 Is Required for Myelinating Cell Survival and Provides Protection during Experimental Autoimmune Encephalomyelitis. J. Neurosci. 2015, 35, 15921–15933. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, A.; Danley, M.M.; LeVine, S.M. Immunohistochemical localization of phosphorylated protein kinase R and phosphorylated eukaryotic initiation factor-2 alpha in the central nervous system of SJL mice with experimental allergic encephalomyelitis. J. Neurosci. Res. 2004, 76, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Kahlson, M.A.; Colodner, K.J. Glial Tau Pathology in Tauopathies: Functional Consequences. J. Exp. Neurosci. 2016, 9, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Rossor, M.N.; Tyrrell, P.J.; Warrington, E.K.; Thompson, P.D.; Marsden, C.D.; Lantos, P. Progressive frontal gait disturbance with atypical Alzheimer’s disease and corticobasal degeneration. J. Neurol. Neurosurg. Psychiatry 1999, 67, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Majtenyi, K.; Spina, S.; Murrell, J.R.; Gelpi, E.; Hoftberger, R.; Fraser, G.; Crowther, R.A.; Goedert, M.; Budka, H.; et al. White matter tauopathy with globular glial inclusions: A distinct sporadic frontotemporal lobar degeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Allali, G.; Dubois, B.; Assal, F.; Lallart, E.; de Souza, L.C.; Bertoux, M.; Annweiler, C.; Herrmann, F.R.; Levy, R.; Beauchet, O. Frontotemporal dementia: Pathology of gait? Mov. Disord. 2010, 25, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Facheris, M.F.; Maniak, S.; Scaravilli, F.; Schüle, B.; Klein, C.; Pramstaller, P.P. Pure akinesia as initial presentation of PSP: A clinicopathological study. Parkinsonism Relat. Disord. 2008, 14, 517–519. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Sakowski, L.; Sperle, K.; Banser, L.; Landel, C.P.; Bessert, D.A.; Skoff, R.P.; Hobson, G.M. Gait abnormalities and progressive myelin degeneration in a new murine model of Pelizaeus-Merzbacher disease with tandem genomic duplication. J. Neurosci. 2013, 33, 11788–11799. [Google Scholar] [CrossRef] [PubMed]
- Harrington, E.P.; Zhao, C.; Fancy, S.P.; Kaing, S.; Franklin, R.J.; Rowitch, D.H. Oligodendrocyte PTEN is required for myelin and axonal integrity, not remyelination. Ann. Neurol. 2010, 68, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Arai, T.; Masuda-Suzukake, M.; Kondo, H.; Matsuwaki, T.; Nishihara, M.; Hasegawa, M.; Akiyama, H. Progranulin reduction is associated with increased tau phosphorylation in P301L tau transgenic mice. J. Neuropathol. Exp. Neurol. 2015, 74, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Kondo, H.; Serrano, G.E.; Beach, T.G.; Robinson, A.C.; Mann, D.M.; Akiyama, H.; Hasegawa, M.; Arai, T. Accumulation of multiple neurodegenerative disease-related proteins in familial frontotemporal lobar degeneration associated with granulin mutation. Sci. Rep. 2017, 7, 1513. [Google Scholar] [CrossRef] [PubMed]
- Sottejeau, Y.; Bretteville, A.; Cantrelle, F.X.; Malmanche, N.; Demiaute, F.; Mendes, T.; Delay, C.; Dos Alves, H.A.; Flaig, A.; Davies, P.; et al. Tau phosphorylation regulates the interaction between BIN1’s SH3 domain and Tau’s proline-rich domain. Acta Neuropathol. Commun. 2015, 3, 58. [Google Scholar] [CrossRef] [PubMed]
- De Rossi, P.; Buggia-Prévot, V.; Clayton, B.L.; Vasquez, J.B.; van Sanford, C.; Andrew, R.J.; Lesnick, R.; Botté, A.; Deyts, C.; Salem, S.; et al. Predominant expression of Alzheimer’s disease-associated BIN1 in mature oligodendrocytes and localization to white matter tracts. Mol. Neurodegener. 2016, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol. Aging 2011, 32, 1341–1371. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Lu, P.H.; Mintz, J. Quantifying age-related myelin breakdown with MRI: novel therapeutic targets for preventing cognitive decline and Alzheimer’s disease. J. Alzheimers Dis. 2004, 6, S53–S59. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, Y.; Liu, Z.; Geng, Q.; Chen, Z.; Zhang, Y. Alterations of myelin morphology and oligodendrocyte development in early stage of Alzheimer’s disease mouse model. Neurosci. Lett. 2017, 642, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Xiao, M. Oligodendrocytes and Alzheimer’s disease. Int. J. Neurosci. 2016, 126, 97–104. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
LoPresti, P. Tau in Oligodendrocytes Takes Neurons in Sickness and in Health. Int. J. Mol. Sci. 2018, 19, 2408. https://doi.org/10.3390/ijms19082408
LoPresti P. Tau in Oligodendrocytes Takes Neurons in Sickness and in Health. International Journal of Molecular Sciences. 2018; 19(8):2408. https://doi.org/10.3390/ijms19082408
Chicago/Turabian StyleLoPresti, Patrizia. 2018. "Tau in Oligodendrocytes Takes Neurons in Sickness and in Health" International Journal of Molecular Sciences 19, no. 8: 2408. https://doi.org/10.3390/ijms19082408
APA StyleLoPresti, P. (2018). Tau in Oligodendrocytes Takes Neurons in Sickness and in Health. International Journal of Molecular Sciences, 19(8), 2408. https://doi.org/10.3390/ijms19082408