ClusterMI: Detecting High-Order SNP Interactions Based on Clustering and Mutual Information


Abstract
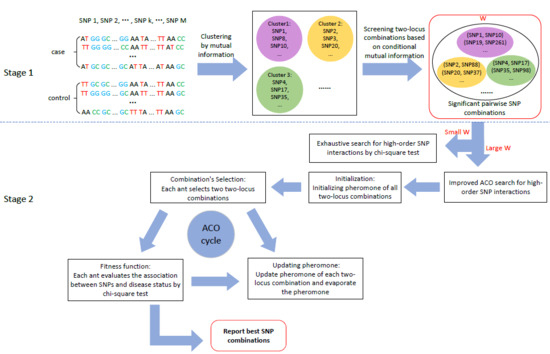
1. Introduction
2. Results
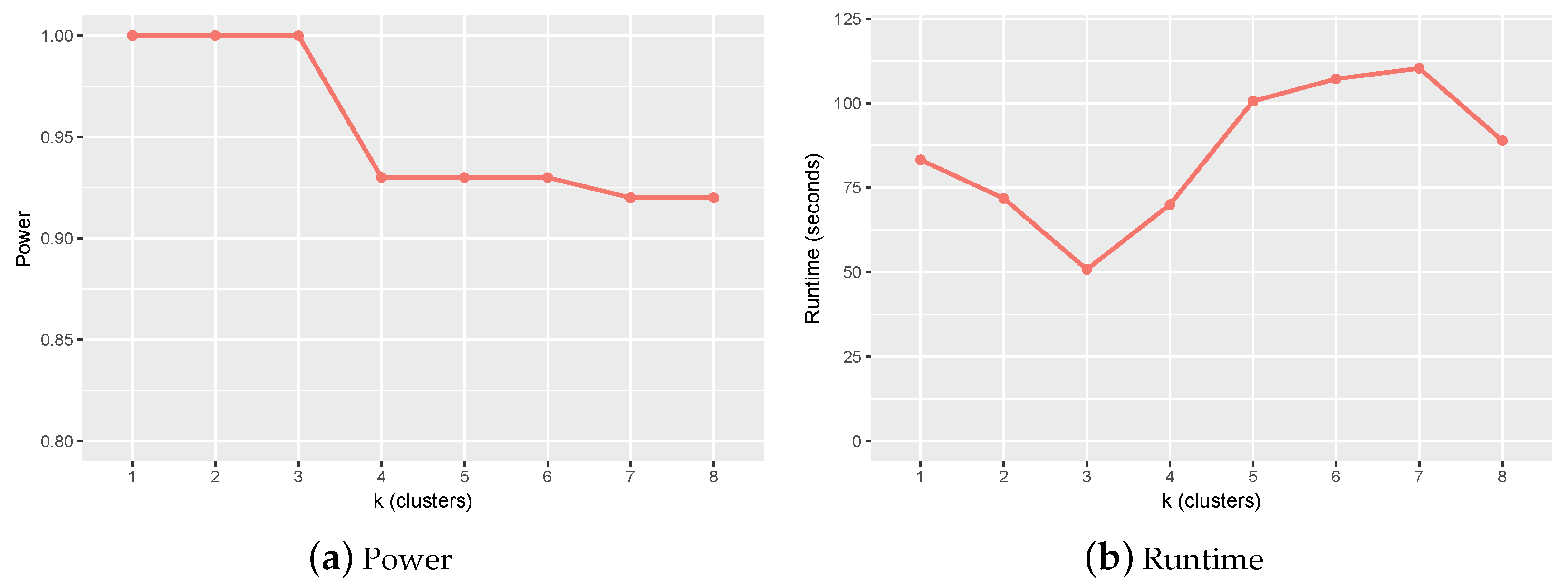
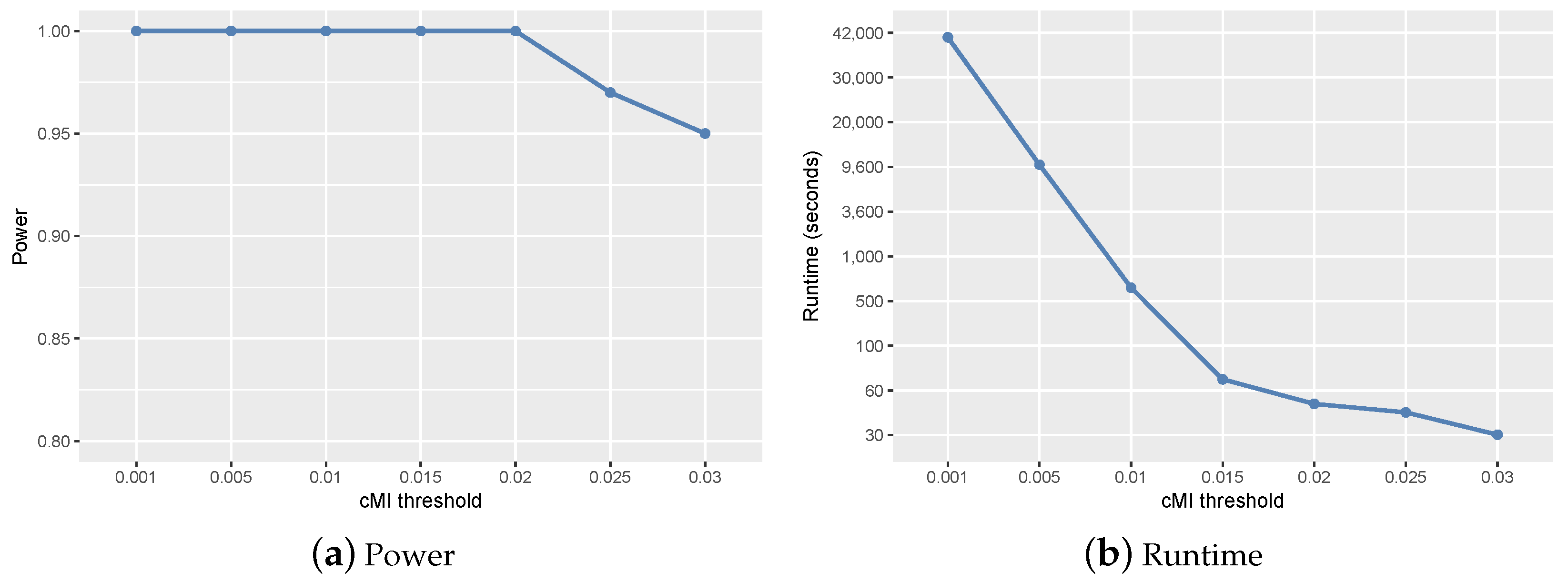
2.1. Parameter Setting and Sensitivity Analysis
- for all two-locus combinations was set to 100.
- was set to 0.6.
- ranged from 0.01–0.1 according to the size of candidate set W. In the simulation studies, we set . In the real study, was set to 0.01.
- n and were determined by W. We set and n from 500–5000.
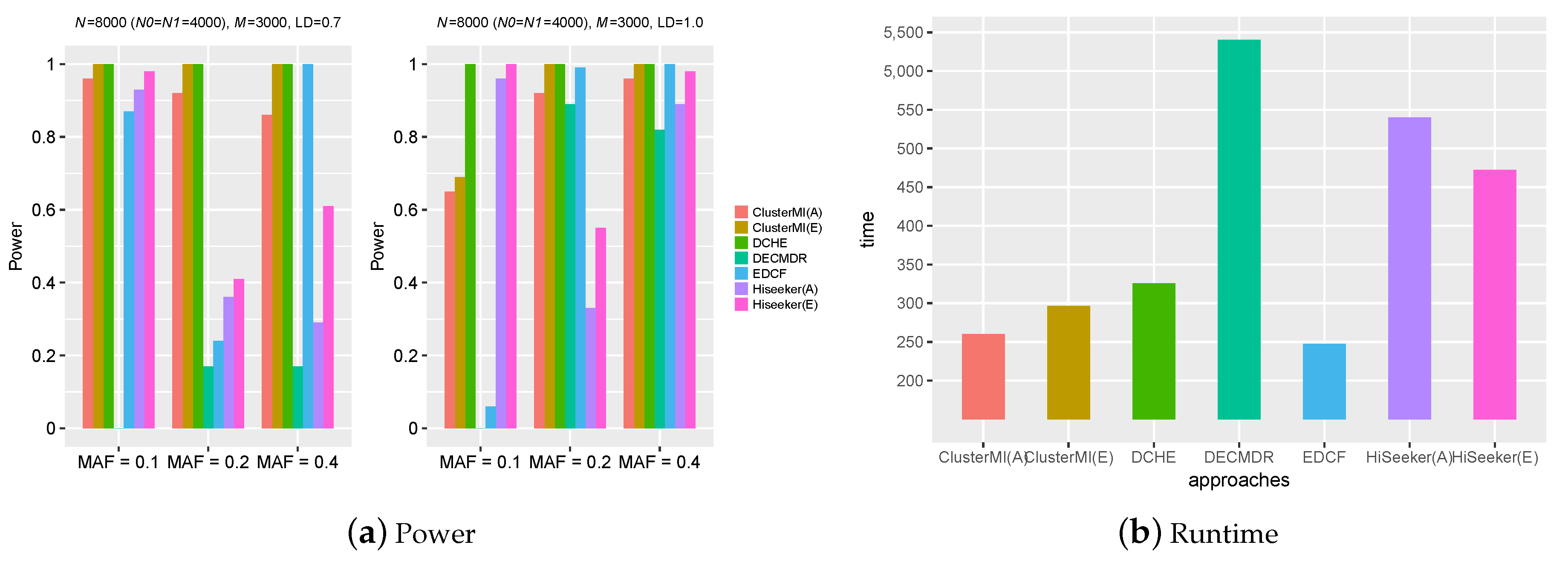
2.2. Experiments on Simulation Data
2.3. Experiments on Real Data
2.3.1. Experiments on Breast Cancer (BC) Data
2.3.2. Experiments on Celiac Disease (CD) Data
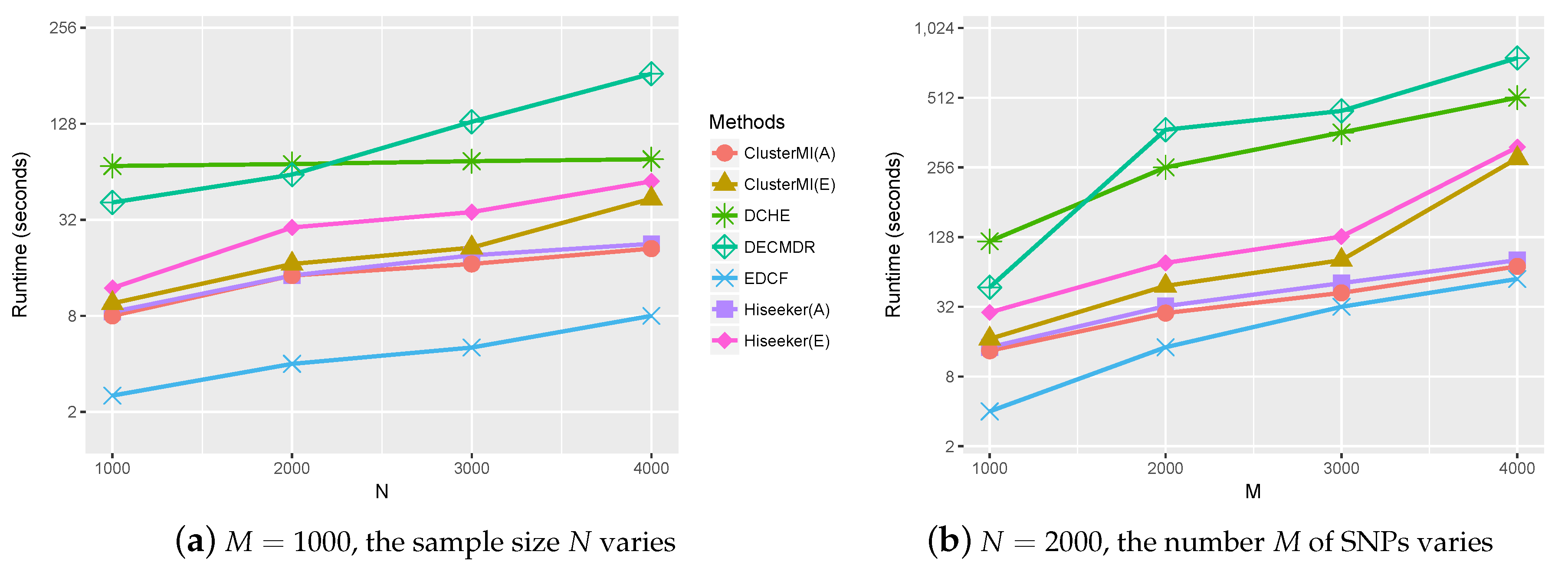
2.4. Runtime Analysis
3. Materials and Methods
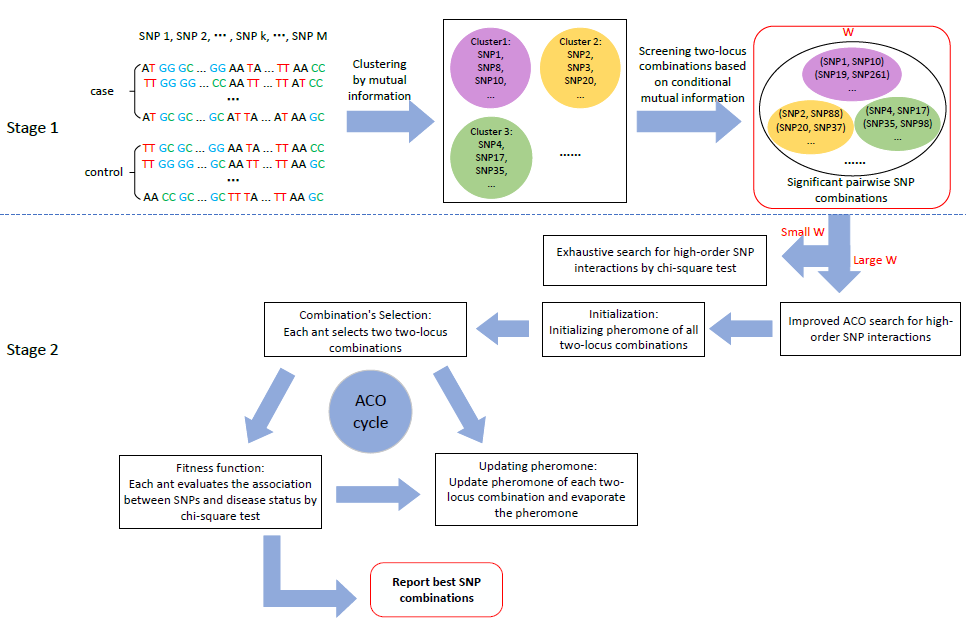
3.1. Stage 1: Candidate SNP (Single Nucleotide Polymorphisms) Combinations Selection
- (a)
- Initialization: k SNPs are randomly selected from M SNPs as initial centroids of k clusters , and k is the preset number of SNP groups.
- (b)
- Clustering: Mutual information can measure the dependency or associativity between two variables [40,41]. Given this, we take mutual information to measure the associativity between two SNPs. For an SNP pair , the mutual information can be calculated as:where u (or v) = {1,2,3} denotes the three genotypes of (or ); denotes the joint probability of and ; and are the marginal probability of and , respectively. For and the centroid of cluster , ClusterMI calculates the mutual information between and . is divided into the m-th group when .
- (c)
- Update centroids: In each iteration, ClusterMI updates each centroid after each SNP has been divided into one of the k clusters. Suppose () stores the SNPs of the m-th cluster; ClusterMI measures the sum of mutual information of centroid as:Then, for each SNP , ClusterMI also calculates the sum of mutual information of in as:If , ClusterMI renews the centroid of as . For each group, the same procedure is conducted to update the clustering centroid.
- (a)
- For the m-th cluster, the association between a two-locus combination and the disease can be measured by conditional mutual information [40,41]. The conditional mutual information of a two-locus combination under case (y = 1) can be calculated as:where denotes the joint probability of and under the case; and are the marginal probability of and under the case, respectively.
- (b)
- ClusterMI takes a two-locus combination with as a significant SNP combination, and is a user-specific threshold.
3.2. Stage 2: High-Order SNP Interactions Detection
3.2.1. Exhaustive Search for a Small Candidate Set (Small W)
3.2.2. Heuristic Search for a Large Candidate Set (Large W)
- (i)
- Initialization: The pheromone value of all two-locus combinations in W is initialized as , which means the association between a combination and disease is treated with equal possibility.
- (ii)
- Combinations selection: ACO introduces ants to select two-locus combinations. An ant respectively chooses two combinations as its targeted combination set when . The probability () for an ant to select the i-th two-locus combination at iteration t can be defined as [28]:where q is a randomly-generated number with a uniform distribution in (0, 1) and is a user-defined threshold to control the rate of convergence and to avoid falling into the local optimal solution. R and T can be described as:where is the pheromone of the i-th two-locus combination at iteration t and is the heuristic information. and are the weight parameters of pheromone and heuristic information. ClusterMI sets , and as 1, indicating that each two-locus combination is treated equally before the optimization phase. is a set of two-locus combinations that are not selected by ant a at iteration t, and denotes that the ant a randomly selects a two-locus combination from .
- (iii)
- Evaluation: To search high-order SNP interactions, two selected combinations of each ant are merged into a new three-locus combination : . The fitness value of is calculated by the -test:where follows a -test with degrees of freedom, means the number of samples with the u-th joint genotype for the three-locus combination and the v-th disease status, means the number of samples with the u-th joint genotype under the case () and control () and means the number of samples with the v-th disease status for all joint genotypes.
- (iv)
- Pheromone update: In each iteration, each ant selects two two-locus combinations, the corresponding pheromone of each combination is updated as:where is the evaporating coefficient and is the variation of pheromones of the i-th two-locus combination ; it is updated aswhere is a set of ants that select the i-th two-locus combination at iteration t, is a three-locus combination identified by ant a at iteration t and is the fitness value of .
4. Conclusions
- The clustering algorithm utilized in the screening stage of ClusterMI can place SNPs with high-associativity into a cluster, in which the true SNP interactions can be more easily identified. In addition, it greatly reduces the computational complex by avoiding analyzing the whole set of two-locus combinations.
- The conditional mutual information-based evaluation strategy in each high-associativity cluster can effectively screen two-locus combinations and reduce the search space of ACO; it also can improve the power of SNP interactions’ detection and make high-order SNP interactions’ detection on genome-wide data more efficient.
Author Contributions
Funding
Conflicts of Interest
References
- Balding, D.J. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 2006, 7, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Meyer, G.; Li, W.; Yang, Y. Comparison of two-phase analyses for case-control genetic association studies. Stat. Med. 2008, 27, 5054–5075. [Google Scholar] [CrossRef] [PubMed]
- Maher, B. Personal genomes: The case of the missing heritability. Nature 2008, 456, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Cardon, L.R.; Bell, J.I. Association study designs for complex diseases. Nat. Rev. Genet. 2001, 2, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.H.; Hemani, G.; Haley, C.S. Detecting epistasis in human complex traits. Nat. Rev. Genet. 2014, 15, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.C. Epistasis–the essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 2008, 9, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Cordell, H.J. Detecting gene-gene interactions that underlie human diseases. Nat. Rev. Genet. 2009, 10, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, Y.; Wang, G.; Wang, Z.; Gao, M. ELM-Based Large-Scale Genetic Association Study via Statistically Significant Pattern. IEEE Trans. Syst. Man Cybern. Syst. 2017, 1–14, 1–14. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Toddbrown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Yang, C.; Yang, Q.; Xue, H.; Fan, X.; Tang, N.L.; Yu, W. Boost: A fast approach to detecting gene-gene interactions in genome-wide case-control studies. Am. J. Hum. Genet. 2010, 87, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Herold, C.; Steffens, M.; Brockschmidt, F.F.; Baur, M.P.; Becker, T. INTERSNP: Genome-wide interaction analysis guided by a priori information. Bioinformatics 2009, 25, 3275–3281. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Liu, Q.; Zhang, Z.; Li, Z.; Wang, S.; He, L.; Shi, Y. SHEsisEpi, a GPU-enhanced genome-wide SNP-SNP interaction scanning algorithm, efficiently reveals the risk genetic epistasis in bipolar disorder. Cell. Res. 2010, 20, 854–857. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Hahn, L.W.; Roodi, N.; Bailey, L.R.; Dupont, W.D.; Parl, F.F.; Moore, J.H. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am. J. Hum. Genet. 2001, 69, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.S. Bayesian inference of epistatic interactions in case-control studies. Nat. Genet. 2007, 39, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Wu, X.; Jiang, R.; Li, Y. Epistatic Module Detection for Case-Control Studies: A Bayesian Model with a Gibbs Sampling Strategy. PLoS Genet. 2009, 5, e1000464. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Chuang, L.Y.; Lin, Y.D. CMDR based differential evolution identifies the epistatic interaction in genome-wide association studies. Bioinformatics 2017, 33, 2354–2362. [Google Scholar] [CrossRef] [PubMed]
- Niel, C.; Sinoquet, C.; Dina, C.; Rocheleau, G. A survey about methods dedicated to epistasis detection. Front. Genet. 2015, 6, 285. [Google Scholar] [CrossRef] [PubMed]
- Motsingerreif, A.A.; Dudek, S.M.; Hahn, L.W.; Ritchie, M.D. Comparison of approaches for machine-learning optimization of neural networks for detecting gene-gene interactions in genetic epidemiology. Genet. Epidemiol. 2010, 32, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.F.; Konig, I.R.; Ziegler, A. On safari to Random Jungle: A fast implementation of Random Forests for high-dimensional data. Bioinformatics 2010, 26, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Bennett, K.; Embrechts, M.; Breneman, C.; Song, M. Dimensionality Reduction via Sparse Support Vector Machines. J. Mach. Learn. Res. 2003, 3, 1229–1243. [Google Scholar]
- Bureau, A.; Dupuis, J.; Falls, K.; Lunetta, K.L.; Hayward, B.; Keith, T.P.; Van, E.P. Identifying SNPs predictive of phenotype using random forests. Genet. Epidemiol. 2005, 28, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Uppu, S.; Krishna, A.; Gopalan, R.P. Towards Deep Learning in Genome-Wide Association Interaction Studies. In Proceedings of the 20th Pacific Asia Conference on Information Systems, PACIS 2016, Chiayi, Taiwan, 27 June–1 July 2016; Volume 20. [Google Scholar]
- Wang, Y.; Liu, G.; Feng, M.; Wong, L. An empirical comparison of several recent epistatic interaction detection methods. Bioinformatics 2011, 27, 2936–2943. [Google Scholar] [CrossRef] [PubMed]
- Upstill-Goddard, R.; Eccles, D.; Fliege, J.; Collins, A. Machine learning approaches for the discovery of gene-gene interactions in disease data. Brief. Bioinform. 2013, 14, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.Z.; Li, J.; Jiang, T. Detecting genome-wide epistases based on the clustering of relatively frequent items. Bioinformatics 2012, 28, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Meng, Y.; Yu, N.; Pan, Y. Cloud computing for detecting high-order genome-wide epistatic interaction via dynamic clustering. BMC Bioinform. 2014, 15, 102. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yu, G.; Jiang, Y.; Wang, J. Hiseeker: Detecting high-order snp interactions based on pairwise snp combinations. Genes 2017, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shang, J.; Liu, J.; Li, S. An Improved Ant Colony Optimization Algorithm for the Detection of SNP-SNP Interactions. In International Conference on Intelligent Computing; Springer: Berlin, Germany, 2016; pp. 21–32. [Google Scholar]
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; Mccarthy, M.I.; Ouwehand, W.H. Association scan of 14,500 nonsynonymous snps in four diseases identifies autoimmunity variants. Nat. Genet. 2007, 39, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Jing, P.J.; Shen, H.B. MACOED: A multi-objective ant colony optimization algorithm for SNP epistasis detection in genome-wide association studies. Bioinformatics 2015, 31, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Marchini, J.; Donnelly, P.; Cardon, L.R. Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat. Genet. 2005, 37, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; He, Z.; Wan, X.; Yang, Q.; Xue, H.; Yu, W. SNPHarvester: A filtering-based approach for detecting epistatic interactions in genome-wide association studies. Bioinformatics 2009, 25, 504. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Geng, S.; Jin, F.; Liu, J.; Qu, C.; Chen, B. POU5F1/Oct-4 expression in breast cancer tissue is significantly associated with non-sentinel lymph node metastasis. BMC Cancer 2016, 16, 175. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Ishikawa, T.; Ota, I.; Kimura, M.; Shimizu, D.; Tanabe, M.; Chishima, T.; Sasaki, T.; Ichikawa, Y.; Morita, S. High expression of ATP-binding cassette transporter ABCC11 in breast tumors is associated with aggressive subtypes and low disease-free survival. Breast Cancer Res. Trans. 2013, 137, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, S.A.; Settasatian, C.; Crawford, J.; Lower, K.M.; Mccallum, B.; Seshadri, R.; Cornelisse, C.J.; Moerland, E.W.; Cleton-Jansen, A.M.; Tipping, A.J. Characterization and Screening for Mutations of the Growth Arrest-Specific 11 (GAS11) and C16orf3 Genes at 16q24.3 in Breast Cancer. Genomics 1998, 52, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Hicklin, D.J.; Marincola, F.M.; Ferrone, S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol. Med. Today 1999, 5, 178–186. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Corazza, G.R. Coeliac disease. Lancet 2009, 373, 1480–1493. [Google Scholar] [CrossRef]
- Dubois, P.C.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.; Adany, R.; Aromaa, A. Multiple Common Genetic Variants for Celiac Disease Influencing Immune Gene Expression-Gastroenterology. Gastroenterology 2010, 138, S-54. [Google Scholar] [CrossRef]
- Jakulin, A.; Bratko, I.; Smrke, D.; Demsar, J.; Zupan, B. Attribute Interactions in Medical Data Analysis. Lect. Notes Artif. Intell. 2003, 2780, 229–238. [Google Scholar]
- Fan, R.; Zhong, M.; Wang, S.; Zhang, Y.; Andrew, A.; Karagas, M.; Chen, H.; Amos, C.I.; Xiong, M.; Moore, J.H. Entropy-based information gain approaches to detect and to characterize gene-gene and gene-environment interactions/correlations of complex diseases. Genet. Epidemiol. 2011, 35, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Kelly, R.; Romdhane, R. Antepiseeker: Detecting epistatic interactions for case-control studies using a two-stage ant colony optimization algorithm. BMC Res. Notes. 2010, 3, 117. [Google Scholar]
- Greene, C.S.; White, B.C.; Moore, J.H. Ant colony optimization for genome-wide genetic analysis. In International Conference on Ant Colony Optimization and Swarm Intelligence; Springer: Berlin, Germany, 2008; pp. 37–47. [Google Scholar]
- Sapin, E.; Keedwell, E.; Frayling, T. An ant colony optimization and tabu list approach to the detection of gene-gene interactions in genome-wide association studies. IEEE Comput. Intell. Mag. 2015, 10, 54–65. [Google Scholar] [CrossRef]
- Cai, Z.; Gong, W.; Ling, C.X.; Zhang, H. A clustering-based differential evolution for global optimization. Appl. Soft. Comput. 2011, 11, 1363–1379. [Google Scholar] [CrossRef]
- Liu, G.; Li, Y.; Nie, X.; Zheng, H. A novel clustering-based differential evolution with 2 multi-parent crossovers for global optimization. Appl. Soft Comput. 2012, 12, 663–681. [Google Scholar] [CrossRef]
- Karaboga, D.; Ozturk, C. A novel clustering approach: Artificial Bee Colony (ABC) algorithm. Appl. Soft Comput. 2011, 11, 652–657. [Google Scholar] [CrossRef]
- Jiang, L.; Li, C.; Zhang, H.; Cai, Z. A novel distance function: Frequency difference metric. Int. J. Pattern Recogn. 2014, 2, 1451002. [Google Scholar] [CrossRef]
- Li, C.; Jiang, L.; Li, H.; Wu, J.; Jiang, P. Toward value difference metric with attribute weighting. Knowl. Inf. Syst. 2017, 3, 795–825. [Google Scholar] [CrossRef]
- Gong, W.; Cai, Z.; Liang, D. Engineering optimization by means of an improved constrained differential evolution. Comput. Meth. Appl. Mech. 2014, 268, 884–904. [Google Scholar] [CrossRef]
- Yang, M.; Cai, Z.; Li, C.; Guan, J. An improved JADE algorithm for global optimization. In Proceedings of the 2014 IEEE Congress on Evolutionary Computation (CEC), Beijing, China, 6–11 July 2014; pp. 806–812. [Google Scholar]
- Gong, W.; Cai, Z.; Liang, D. Adaptive ranking mutation operator based differential evolution for constrained optimization. IEEE Trans. Cybern. 2015, 45, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, C.; Cai, Z.; Guan, J. Differential evolution with auto-enhanced population diversity. IEEE Trans. Cybern. 2017, 45, 302–315. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | SNP Combinations | Related Genes | Single-Locus p-Value | Combination p-Value |
|---|---|---|---|---|
| chr3 | (rs13100173, rs1108842) | (HYAL3, GNL3) | () | |
| chr6 | (rs9257694, rs879882) | (LOC105375005, POU5F1) | () | |
| chr6 | (rs3094576, rs644827) | (*, SLC44A4) | () | |
| chr16 | (rs17822931, rs3785181) | (ABCC11, GAS11) | () | |
| chr16 | (rs7190823, rs4408545) | (FANCA, AFG3L1P) | () | |
| chr6 | (rs9257694, rs2523608, rs11244) | (LOC105375005, HLA-B, HLA-DOB) | () |
| Chromosome | SNP Combinations | Related Genes | Single-Locus p-Value | Combination p-Value |
|---|---|---|---|---|
| chr1 | (rs3748816, rs3795263) | (MMEL1, ACTRT2) | () | |
| chr2 | (rs3816281, rs4973588) | (PLEK, NGEF) | () | |
| chr6 | (rs3823418, rs4151664) | (PSORS1C1, NELFE) | () | |
| chr6 | (rs2021723, rs3093662) | (TRIM40, TNF) | () | |
| chr22 | (rs2298428, rs1321, rs5771069) | (YDJC, ALG12, IL17REL) | () |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, X.; Yu, G.; Liu, J.; Jia, L.; Wang, J. ClusterMI: Detecting High-Order SNP Interactions Based on Clustering and Mutual Information. Int. J. Mol. Sci. 2018, 19, 2267. https://doi.org/10.3390/ijms19082267
Cao X, Yu G, Liu J, Jia L, Wang J. ClusterMI: Detecting High-Order SNP Interactions Based on Clustering and Mutual Information. International Journal of Molecular Sciences. 2018; 19(8):2267. https://doi.org/10.3390/ijms19082267
Chicago/Turabian StyleCao, Xia, Guoxian Yu, Jie Liu, Lianyin Jia, and Jun Wang. 2018. "ClusterMI: Detecting High-Order SNP Interactions Based on Clustering and Mutual Information" International Journal of Molecular Sciences 19, no. 8: 2267. https://doi.org/10.3390/ijms19082267
APA StyleCao, X., Yu, G., Liu, J., Jia, L., & Wang, J. (2018). ClusterMI: Detecting High-Order SNP Interactions Based on Clustering and Mutual Information. International Journal of Molecular Sciences, 19(8), 2267. https://doi.org/10.3390/ijms19082267