Bisphenol-A and Nonylphenol Induce Apoptosis in Reproductive Tract Cancer Cell Lines by the Activation of ADAM17

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
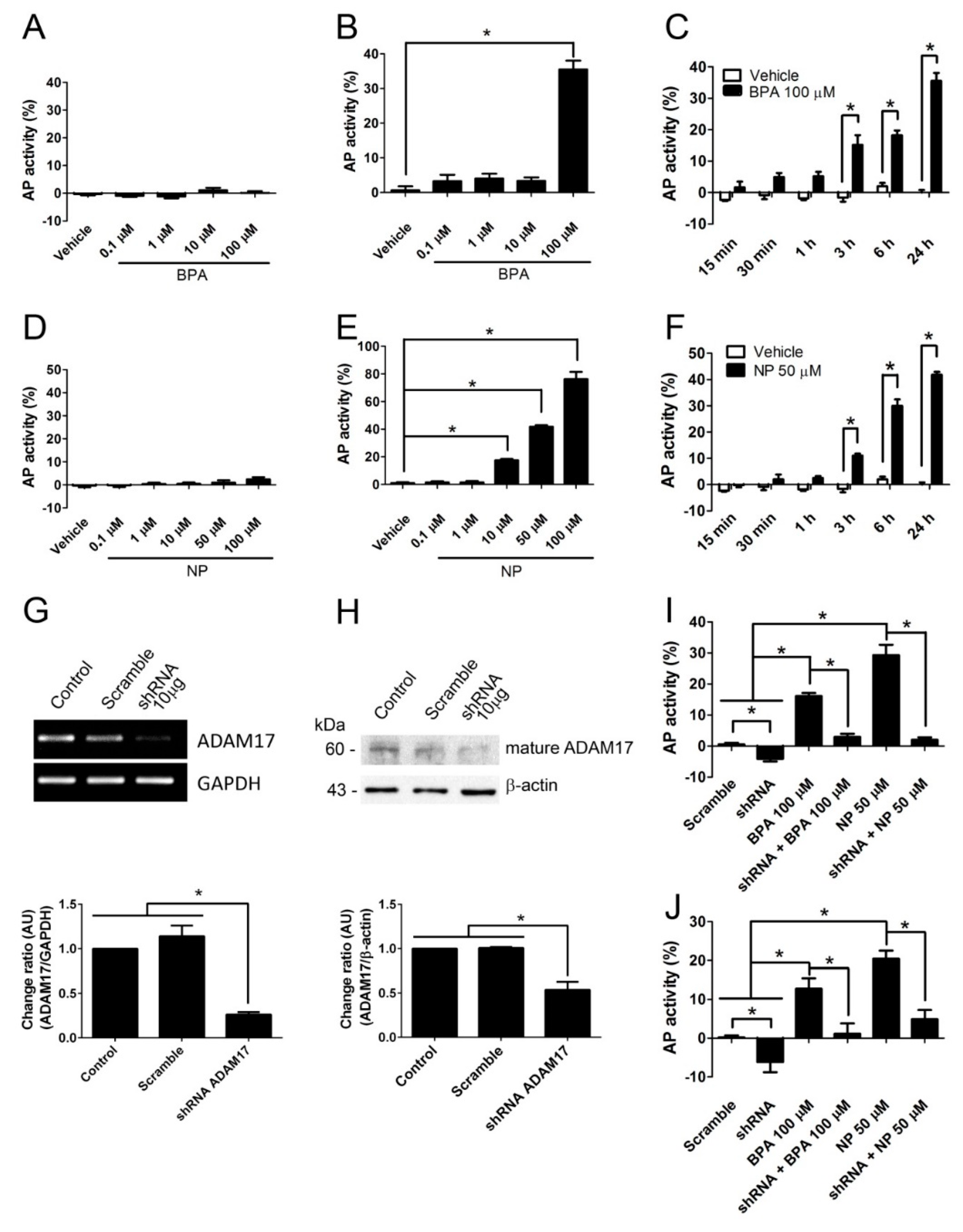
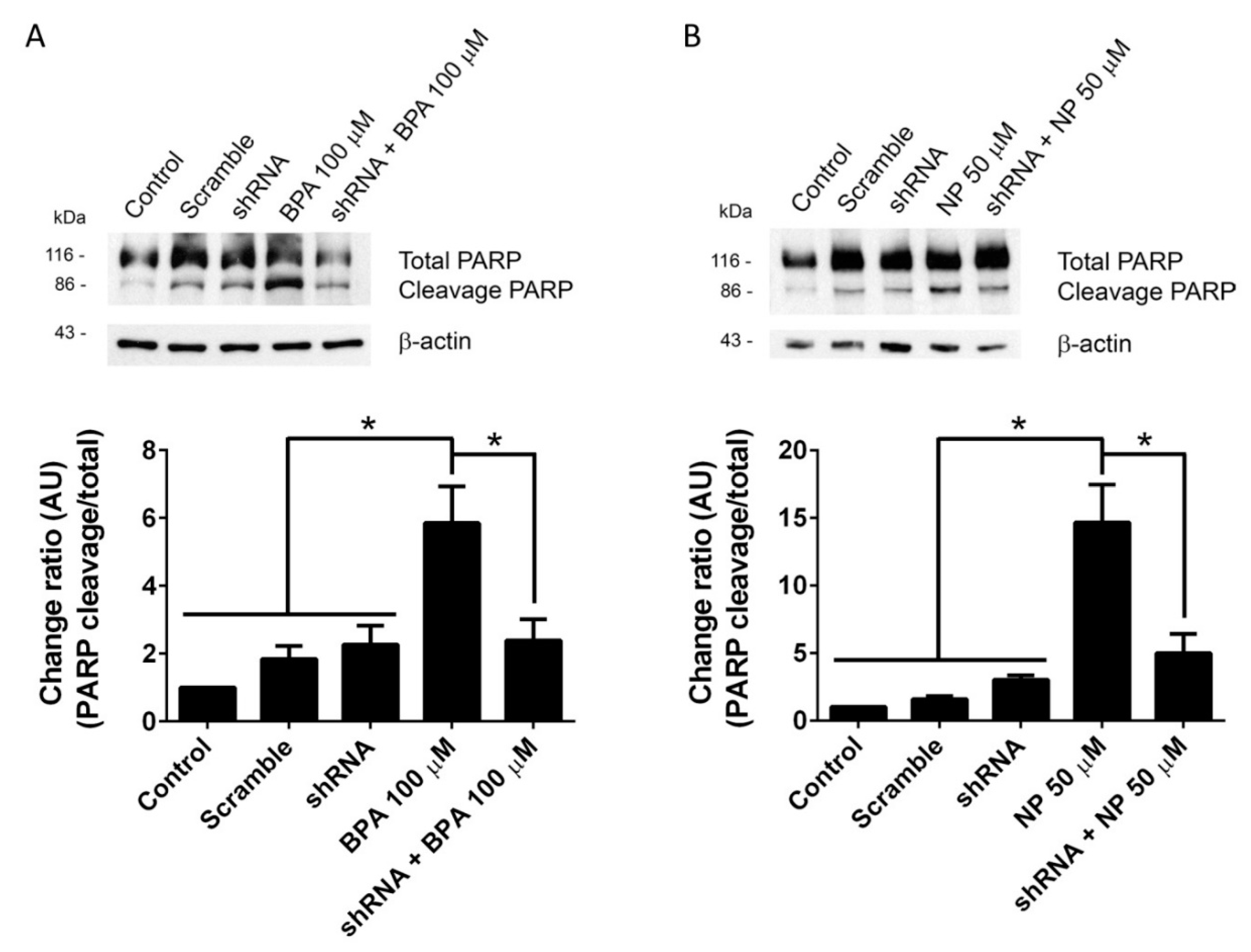
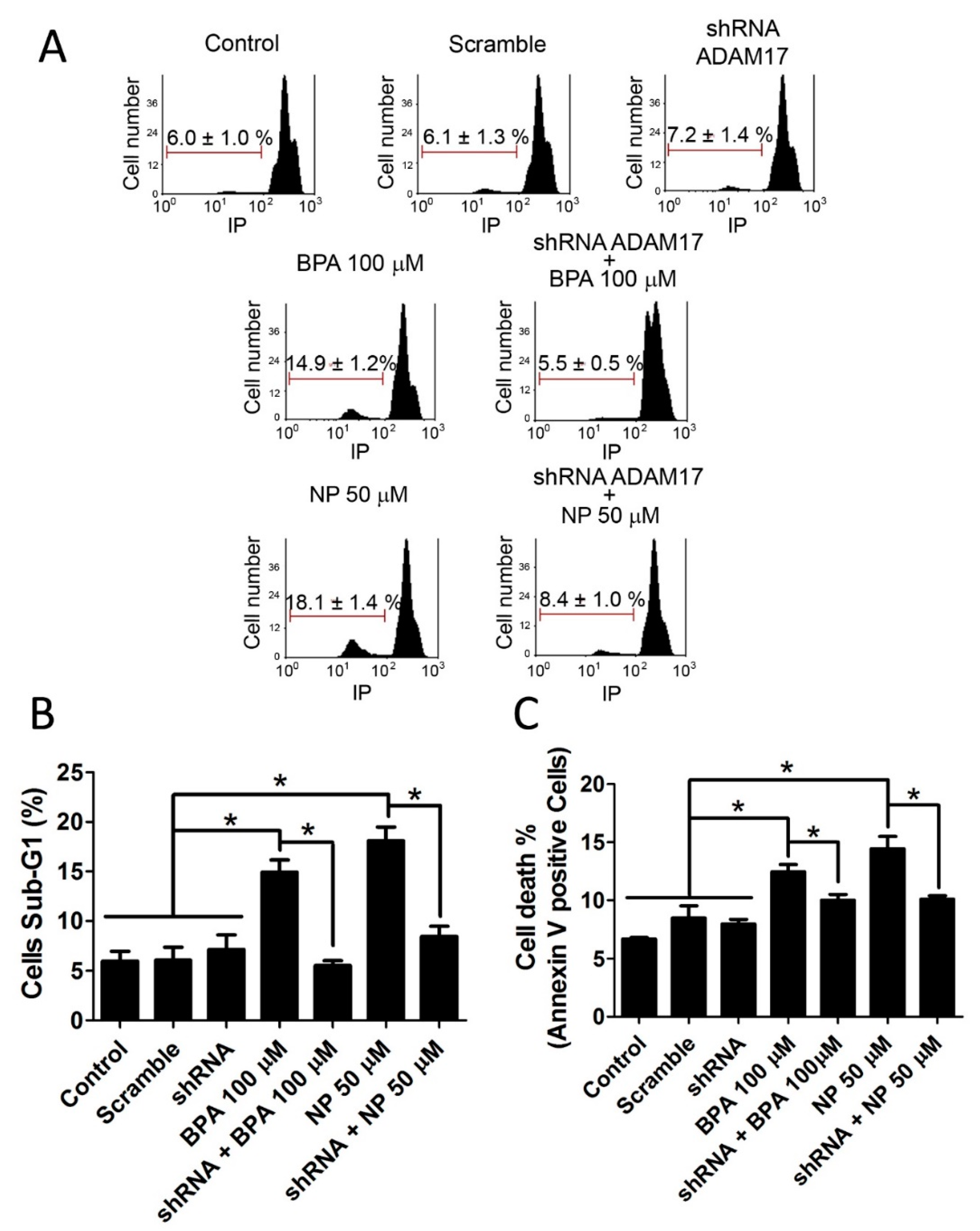
2.1. BPA and NP Induced Apoptosis in LNCaP Requires ADAM17
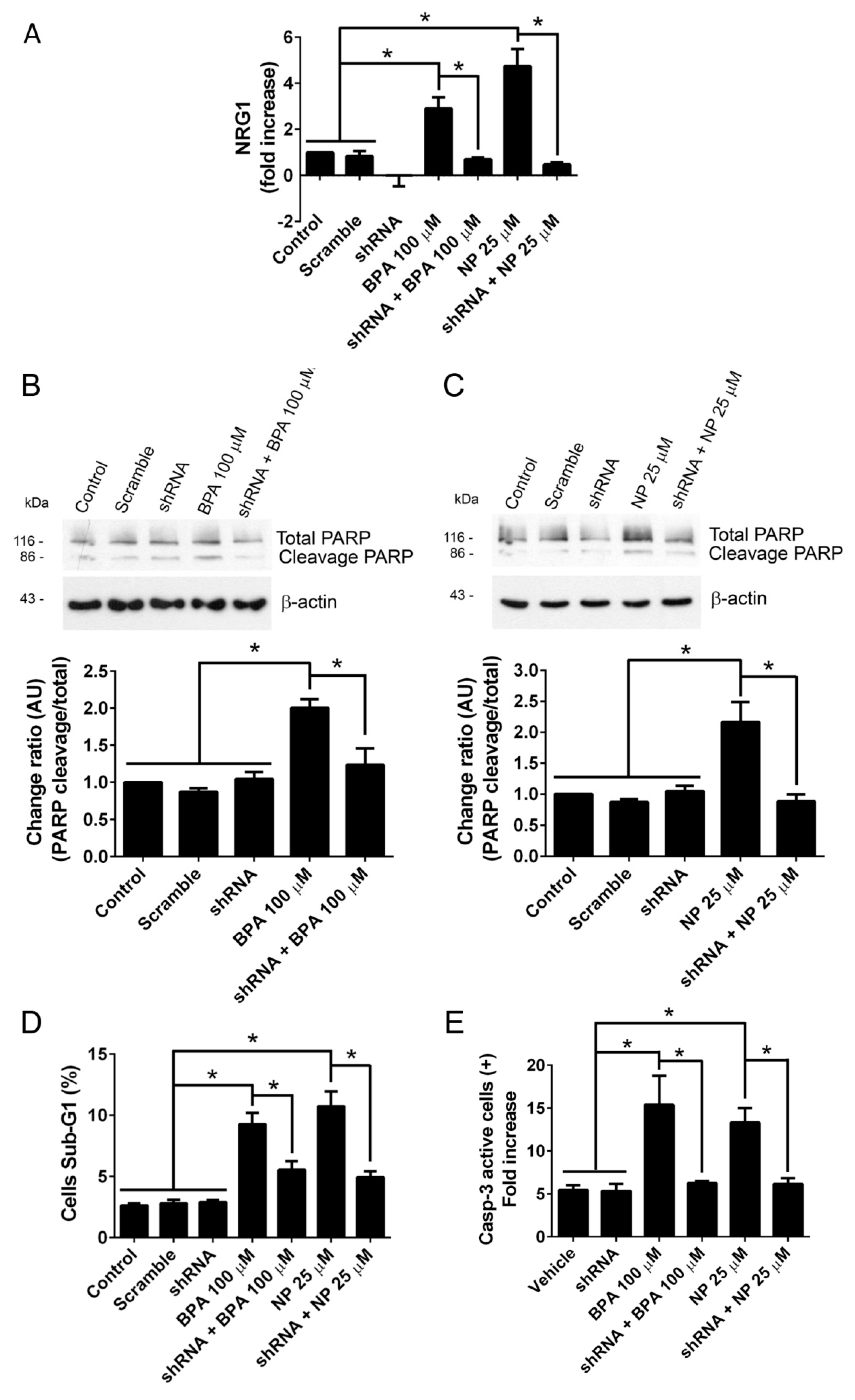
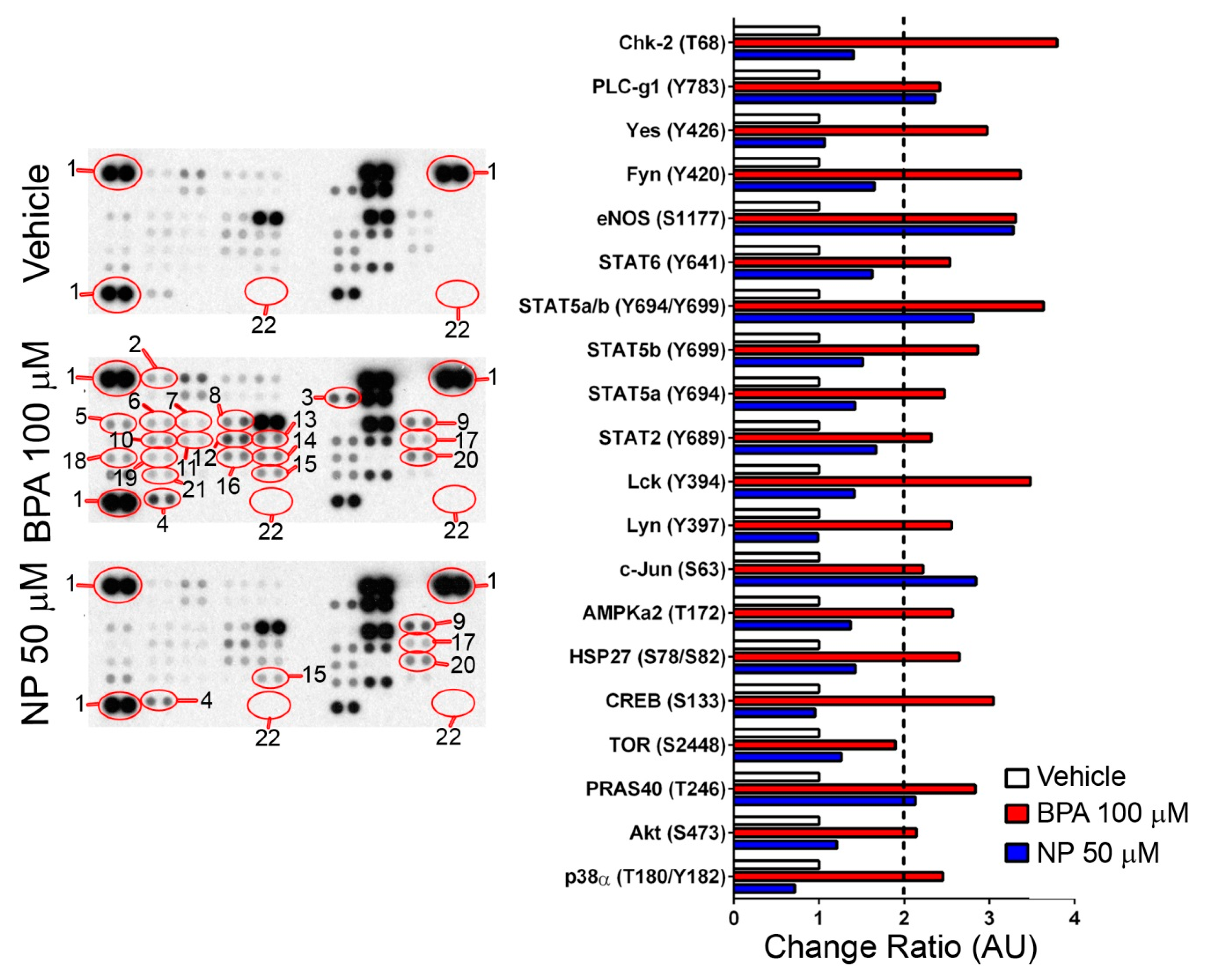
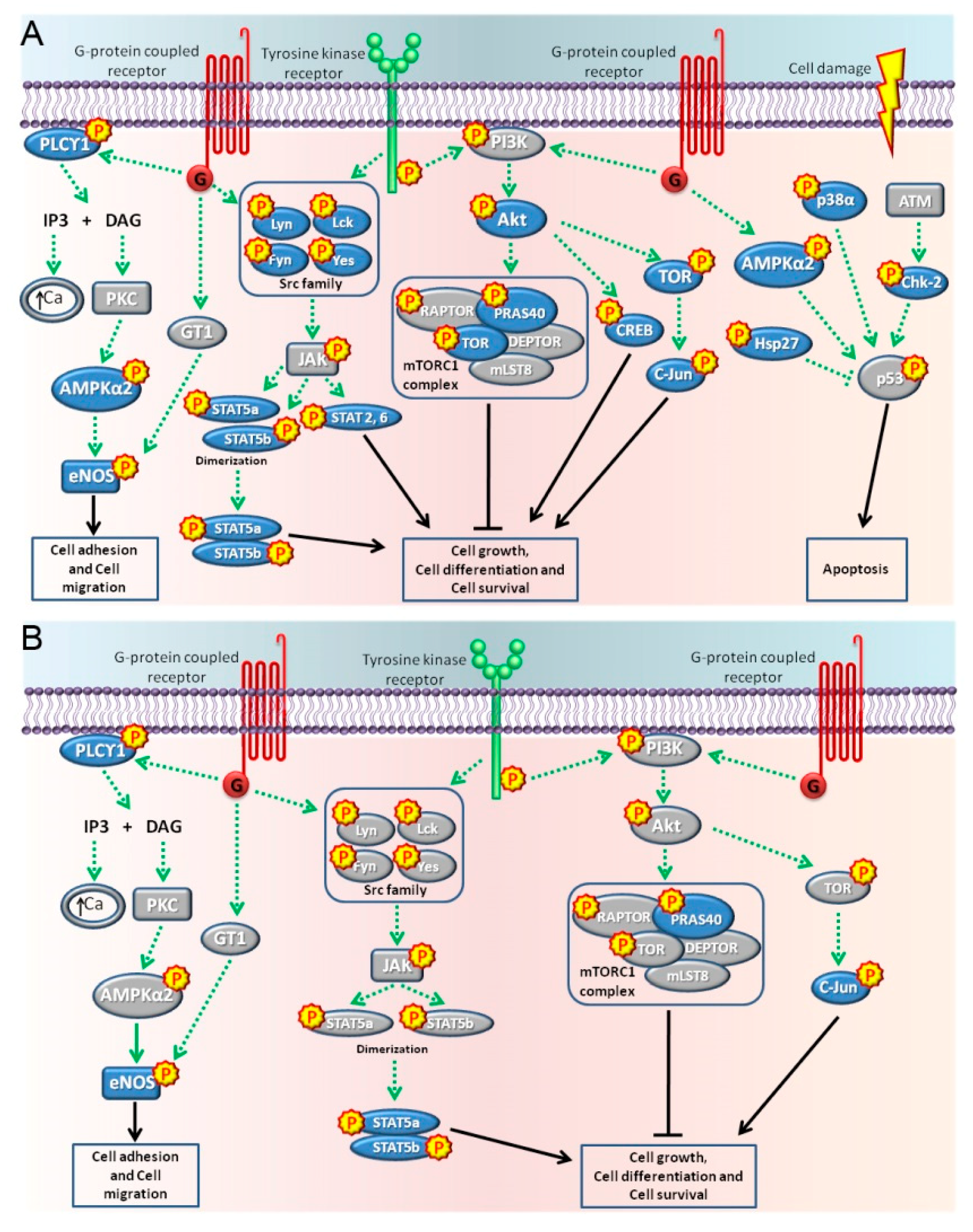
2.2. Pathways Activated by BPA and NP
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Cell Transfection and Generation of a Stable Cell Line
4.3. Alkaline Phosphatase Activity Measurement
4.4. Protein Extraction and Western Blotting
4.5. RNA Extraction and RT-PCR
4.6. Sub-G1 Population Analysis by Flow Cytometry
4.7. Annexin-V Assay
4.8. [Ca2+]i Measurements of LNCaP Cells in Suspension
4.9. Human Phospho-Kinase Array
4.10. Immunohistochemistry and Immunofluorescence
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lagos-Cabre, R.; Moreno, R.D. Contribution of environmental pollutants to male infertily: A working model of germ cell apoptosis induced by plasticizers. Biol. Res. 2012, 45, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, E1–E150. [Google Scholar] [CrossRef] [PubMed]
- Bunay, J.; Larriba, E.; Moreno, R.D.; Del Mazo, J. Chronic low-dose exposure to a mixture of environmental endocrine disruptors induces microRNAs/isomiRs deregulation in mouse concomitant with intratesticular estradiol reduction. Sci. Rep. 2017, 7, 3373. [Google Scholar] [CrossRef] [PubMed]
- Bunay, J.; Larriba, E.; Patino-Garcia, D.; Cruz-Fernandes, L.; Castaneda-Zegarra, S.; Rodriguez-Fernandez, M.; Del Mazo, J.; Moreno, R.D. Differential Effects of Exposure to Single versus a Mixture of Endocrine-Disrupting Chemicals on Steroidogenesis Pathway in Mouse Testes. Toxicol. Sci. 2017. [Google Scholar] [CrossRef]
- Prins, G.S.; Hu, W.Y.; Shi, G.B.; Hu, D.P.; Majumdar, S.; Li, G.; Huang, K.; Nelles, J.L.; Ho, S.M.; Walker, C.L.; et al. Bisphenol A promotes human prostate stem-progenitor cell self-renewal and increases in vivo carcinogenesis in human prostate epithelium. Endocrinology 2014, 155, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Sushil, K. Environmental chemicals targeting estrogen signaling pathways. In Endocrine Disruptors in the Environment; Jon Wiley & Sons: Hoboke, NJ, USA, 2014. [Google Scholar]
- U.S. Environmental Protection Agency. Bisphenol A Action Plan. Available online: https://www.epa.gov/sites/production/files/2015-09/documents/bpa_action_plan.pdf (accessed on 20 October 2017).
- Wetherill, Y.B.; Akingbemi, B.T.; Kanno, J.; McLachlan, J.A.; Nadal, A.; Sonnenschein, C.; Watson, C.S.; Zoeller, R.T.; Belcher, S.M. In vitro molecular mechanisms of bisphenol A action. Reprod. Toxicol. 2007, 24, 178–198. [Google Scholar] [CrossRef] [PubMed]
- Patino-Garcia, D.; Cruz-Fernandes, L.; Bunay, J.; Palomino, J.; Moreno, R.D. Reproductive Alterations in Chronically Exposed Female Mice to Environmentally Relevant Doses of a Mixture of Phthalates and Alkylphenols. Endocrinology 2018, 159, 1050–1061. [Google Scholar] [CrossRef] [PubMed]
- Nagao, T.; Wada, K.; Marumo, H.; Yoshimura, S.; Ono, H. Reproductive effects of nonylphenol in rats after gavage administration: A two-generation study. Reprod. Toxicol. 2001, 15, 293–315. [Google Scholar] [CrossRef]
- McClusky, L.M.; de Jager, C.; Bornman, M.S. Stage-related increase in the proportion of apoptotic germ cells and altered frequencies of stages in the spermatogenic cycle following gestational, lactational, and direct exposure of male rats to p-nonylphenol. Toxicol. Sci. 2007, 95, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Hossaini, A.; Dalgaard, M.; Vinggaard, A.M.; Frandsen, H.; Larsen, J.J. In utero reproductive study in rats exposed to nonylphenol. Reprod. Toxicol. 2001, 15, 537–543. [Google Scholar] [CrossRef]
- Soto, A.M.; Maffini, M.V.; Sonnenschein, C. Neoplasia as development gone awry: The role of endocrine disruptors. Int. J. Androl. 2008, 31, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Fenton, S.E.; Reed, C.; Newbold, R.R. Perinatal environmental exposures affect mammary development, function, and cancer risk in adulthood. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 455–479. [Google Scholar] [CrossRef] [PubMed]
- Tarapore, P.; Ying, J.; Ouyang, B.; Burke, B.; Bracken, B.; Ho, S.M. Exposure to bisphenol A correlates with early-onset prostate cancer and promotes centrosome amplification and anchorage-independent growth in vitro. PLoS ONE 2014, 9, e90332. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, R.; Parnell, P.G.; Villanueva, H.; Chapman, L.M.; Gimenez, T.; Gray, S.L.; Baldwin, W.S. The contribution of hepatic steroid metabolism to serum estradiol and estriol concentrations in nonylphenol treated MMTVneu mice and its potential effects on breast cancer incidence and latency. J. Appl. Toxicol. 2005, 25, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.M.; Tang, W.Y.; Belmonte de Frausto, J.; Prins, G.S. Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res. 2006, 66, 5624–5632. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Hwang, K.A.; Hyun, S.H.; Nam, K.H.; Lee, C.K.; Choi, K.C. Bisphenol A and nonylphenol have the potential to stimulate the migration of ovarian cancer cells by inducing epithelial-mesenchymal transition via an estrogen receptor dependent pathway. Chem. Res. Toxicol. 2015, 28, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Park, M.A.; Choi, K.C. Effects of 4-nonylphenol and bisphenol A on stimulation of cell growth via disruption of the transforming growth factor-beta signaling pathway in ovarian cancer models. Chem. Res. Toxicol. 2014, 27, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Li, H.; Lu, G.; Chen, Z.; Sun, W.; Shi, Y.; Fu, Z.; Huang, B.; Zhu, X.; Lu, W.; et al. Low Dose of Bisphenol A Modulates Ovarian Cancer Gene Expression Profile and Promotes Epithelial to Mesenchymal Transition via Canonical Wnt Pathway. Toxicol. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Urriola-Munoz, P.; Lizama, C.; Lagos-Cabre, R.; Reyes, J.G.; Moreno, R.D. Differential expression and localization of ADAM10 and ADAM17 during rat spermatogenesis suggest a role in germ cell differentiation. Biol. Res. 2014, 47, 31. [Google Scholar] [CrossRef] [PubMed]
- Urriola-Munoz, P.; Li, X.; Maretzky, T.; McIlwain, D.R.; Mak, T.W.; Reyes, J.G.; Blobel, C.P.; Moreno, R.D. The xenoestrogens biphenol-A and nonylphenol differentially regulate metalloprotease-mediated shedding of EGFR ligands. J. Cell. Physiol. 2018, 233, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Urriola-Munoz, P.; Lagos-Cabre, R.; Moreno, R.D. A mechanism of male germ cell apoptosis induced by bisphenol-A and nonylphenol involving ADAM17 and p38 MAPK activation. PLoS ONE 2014, 9, e113793. [Google Scholar] [CrossRef] [PubMed]
- Moreno, R.D.; Urriola-Munoz, P.; Lagos-Cabre, R. The emerging role of matrix metalloproteases of the ADAM family in male germ cell apoptosis. Spermatogenesis 2011, 1, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Quan, C.; Duan, P.; Tang, S.; Chen, W.; Yang, K. Nonylphenol induced apoptosis and autophagy involving the Akt/mTOR pathway in prepubertal Sprague-Dawley male rats in vivo and in vitro. Toxicology 2016, 373, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta 2017, 1864, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Pruessmeyer, J.; Ludwig, A. The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin. Cell Dev. Biol. 2009, 20, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Kudo, C.; Wada, K.; Masuda, T.; Yonemura, T.; Shibuya, A.; Fujimoto, Y.; Nakajima, A.; Niwa, H.; Kamisaki, Y. Nonylphenol induces the death of neural stem cells due to activation of the caspase cascade and regulation of the cell cycle. J. Neurochem. 2004, 88, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Yang, L.; Hu, Y.; Liang, J.; Liang, J.; Hou, Y. Nonylphenol-induced thymocyte apoptosis involved caspase-3 activation and mitochondrial depolarization. Mol. Immunol. 2006, 43, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Kusunoki, T.; Shimoke, K.; Komatsubara, S.; Kishi, S.; Ikeuchi, T. p-Nonylphenol induces endoplasmic reticulum stress-mediated apoptosis in neuronally differentiated PC12 cells. Neurosci. Lett. 2008, 431, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, J.; Wang, Q.; Wu, W.; Huan, F.; Xiao, H. Bisphenol A modulates calcium currents and intracellular calcium concentration in rat dorsal root ganglion neurons. J. Membr. Biol. 2013, 246, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Liu, C.S.; Lin, K.L.; Chou, C.T.; Hsieh, C.H.; Chang, C.H.; Chen, W.C.; Liu, S.I.; Hsu, S.S.; Chang, H.T.; et al. Nonylphenol-induced Ca2+ elevation and Ca2+-independent cell death in human osteosarcoma cells. Toxicol. Lett. 2005, 160, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.Z.; Kirk, C.J.; Michelangeli, F. Alkylphenol endocrine disrupters inhibit IP3-sensitive Ca2+ channels. Biochem. Biophys. Res. Commun. 2003, 310, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Wu, J.; Huang, Y.; Shen, S.; Han, X. Nonylphenol induces apoptosis in rat testicular Sertoli cells via endoplasmic reticulum stress. Toxicol. Lett. 2009, 186, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.; Zanatta, A.P.; Cavalari, F.C.; do Nascimento, M.A.W.; Delalande-Lecapitaine, C.; Bouraima-Lelong, H.; Silva, F. Acute effect of bisphenol A: Signaling pathways on calcium influx in immature rat testes. Reprod. Toxicol. 2018, 77, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, S.M.; Bobe, P.; Reiss, K.; Horiuchi, K.; Niu, X.D.; Lundell, D.; Gibb, D.R.; Conrad, D.; Saftig, P.; Blobel, C.P. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Mol. Biol. Cell 2009, 20, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Gutierrez, D.; Bauer, M.A.; Zimmermann, A.; Aguilera, A.; Austriaco, N.; Ayscough, K.; Balzan, R.; Bar-Nun, S.; Barrientos, A.; Belenky, P.; et al. Guidelines and recommendations on yeast cell death nomenclature. Microb. Cell 2018, 5, 4–31. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.L.G.; Salvesen, G.S. A primer on caspase mechanisms. Semin. Cell Dev. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Rheaume, E.; Cohen, L.Y.; Uhlmann, F.; Lazure, C.; Alam, A.; Hurwitz, J.; Sekaly, R.P.; Denis, F. The large subunit of replication factor C is a substrate for caspase-3 in vitro and is cleaved by a caspase-3-like protease during Fas-mediated apoptosis. EMBO J. 1997, 16, 6346–6354. [Google Scholar] [CrossRef] [PubMed]
- ECACC and E.c.o.A.C. Ovarian Cancer Cell Line A2780 (ECACC Catalogue no. 93112519). Available online: https://www.phe-culturecollections.org.uk/media/113526/a112780-cell-line-profile.pdf (accessed on 23 March 2018).
- McCudden, C.R.; Hains, M.D.; Kimple, R.J.; Siderovski, D.P.; Willard, F.S. G-protein signaling: Back to the future. Cell. Mol. Life Sci. 2005, 62, 551–577. [Google Scholar] [CrossRef] [PubMed]
- Rozengurt, E. Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell. Physiol. 2007, 213, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Premont, R.T.; Rockey, D.C. G-protein-coupled receptor kinase interactor-1 (GIT1) is a new endothelial nitric-oxide synthase (eNOS) interactor with functional effects on vascular homeostasis. J. Biol. Chem. 2012, 287, 12309–12320. [Google Scholar] [CrossRef] [PubMed]
- Nishino, Y.; Miura, T.; Miki, T.; Sakamoto, J.; Nakamura, Y.; Ikeda, Y.; Kobayashi, H.; Shimamoto, K. Ischemic preconditioning activates AMPK in a PKC-dependent manner and induces GLUT4 up-regulation in the late phase of cardioprotection. Cardiovasc. Res 2004, 61, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.P.; Mitchelhill, K.I.; Michell, B.J.; Stapleton, D.; Rodriguez-Crespo, I.; Witters, L.A.; Power, D.A.; Ortiz de Montellano, P.R.; Kemp, B.E. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999, 443, 285–289. [Google Scholar] [CrossRef]
- Al-Jarallah, A.; Chen, X.; Gonzalez, L.; Trigatti, B.L. High density lipoprotein stimulated migration of macrophages depends on the scavenger receptor class B, type I, PDZK1 and Akt1 and is blocked by sphingosine 1 phosphate receptor antagonists. PLoS ONE 2014, 9, e106487. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Tomura, H.; Mogi, C.; Kuwabara, A.; Damirin, A.; Ishizuka, T.; Sekiguchi, A.; Ishiwara, M.; Im, D.S.; Sato, K.; et al. Role of scavenger receptor class B type I and sphingosine 1-phosphate receptors in high density lipoprotein-induced inhibition of adhesion molecule expression in endothelial cells. J. Biol. Chem. 2006, 281, 37457–37467. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, M.P.; Cheng, L.; Holland, E.C.; Federspiel, M.J.; Donovan, P.J. Dissection of the c-Kit signaling pathway in mouse primordial germ cells by retroviral-mediated gene transfer. Proc. Natl. Acad. Sci. USA 2002, 99, 10458–10463. [Google Scholar] [CrossRef] [PubMed]
- Brizzi, M.F.; Dentelli, P.; Rosso, A.; Yarden, Y.; Pegoraro, L. STAT protein recruitment and activation in c-Kit deletion mutants. J. Biol. Chem. 1999, 274, 16965–16972. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.J.; Huang, H.; McReynolds, L.J.; Shelburne, C.; Hu-Li, J.; Huff, T.F.; Paul, W.E. Stem cell factor activates STAT-5 DNA binding in IL-3-derived bone marrow mast cells. Exp. Hematol. 1997, 25, 357–362. [Google Scholar] [PubMed]
- Matsui, J.; Wakabayashi, T.; Asada, M.; Yoshimatsu, K.; Okada, M. Stem cell factor/c-kit signaling promotes the survival, migration, and capillary tube formation of human umbilical vein endothelial cells. J. Biol. Chem. 2004, 279, 18600–18607. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Montminy, M. CREB is a regulatory target for the protein kinase Akt/PKB. J. Biol. Chem. 1998, 273, 32377–32379. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell. 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C., Jr. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.H.; Sasaki, T.; Chao, M.V. JNK-interacting protein 1 promotes Akt1 activation. J. Biol. Chem. 2003, 278, 29830–29836. [Google Scholar] [CrossRef] [PubMed]
- Krystal, G.W.; DeBerry, C.S.; Linnekin, D.; Litz, J. Lck associates with and is activated by Kit in a small cell lung cancer cell line: Inhibition of SCF-mediated growth by the Src family kinase inhibitor PP1. Cancer Res. 1998, 58, 4660–4666. [Google Scholar] [PubMed]
- New, D.C.; Wong, Y.H. Molecular mechanisms mediating the G protein-coupled receptor regulation of cell cycle progression. J. Mol. Signal. 2007, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.M. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene 2004, 23, 8017–8023. [Google Scholar] [CrossRef] [PubMed]
- Tichy, A.; Zaskodova, D.; Rezacova, M.; Vavrova, J.; Vokurkova, D.; Pejchal, J.; Vilasova, Z.; Cerman, J.; Osterreicher, J. Gamma-radiation-induced ATM-dependent signalling in human T-lymphocyte leukemic cells, MOLT-4. Acta Biochim. Pol. 2007, 54, 281–287. [Google Scholar] [PubMed]
- Dorion, S.; Landry, J. Activation of the mitogen-activated protein kinase pathways by heat shock. Cell Stress Chaperones 2002, 7, 200–206. [Google Scholar] [CrossRef]
- Venkatakrishnan, C.D.; Dunsmore, K.; Wong, H.; Roy, S.; Sen, C.K.; Wani, A.; Zweier, J.L.; Ilangovan, G. HSP27 regulates p53 transcriptional activity in doxorubicin-treated fibroblasts and cardiac H9c2 cells: P21 upregulation and G2/M phase cell cycle arrest. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1736–H1744. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Prieto, R.; Rojas, J.M.; Taya, Y.; Gutkind, J.S. A role for the p38 mitogen-acitvated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res. 2000, 60, 2464–2472. [Google Scholar] [PubMed]
- In, S.J.; Kim, S.H.; Go, R.E.; Hwang, K.A.; Choi, K.C. Benzophenone-1 and nonylphenol stimulated MCF-7 breast cancer growth by regulating cell cycle and metastasis-related genes via an estrogen receptor alpha-dependent pathway. J. Toxicol. Environ. Health Part A 2015, 78, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.H.; Hwang, K.A.; Kim, T.H.; Hyun, S.H.; Jeung, E.B.; Choi, K.C. Induced growth of BG-1 ovarian cancer cells by 17beta-estradiol or various endocrine disrupting chemicals was reversed by resveratrol via downregulation of cell cycle progression. Mol. Med. Rep. 2012, 6, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, F.; Zhang, J.; Hao, L.; Jiang, J.; Dang, L.; Mei, D.; Fan, S.; Yu, Y.; Jiang, L. Bisphenol A and estrogen induce proliferation of human thyroid tumor cells via an estrogen-receptor-dependent pathway. Arch. Biochem. Biophys. 2017, 633, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhou, Z.; Qing, D.; He, Y.; Wu, T.; Miao, M.; Wang, J.; Weng, X.; Ferber, J.R.; Herrinton, L.J.; et al. Occupational exposure to bisphenol-A (BPA) and the risk of self-reported male sexual dysfunction. Hum. Reprod. 2010, 25, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Kim, B.K.; Shim, J.H.; Gil, J.E.; Yoon, Y.D.; Kim, J.H. Nonylphenol and octylphenol-induced apoptosis in human embryonic stem cells is related to Fas-Fas ligand pathway. Toxicol. Sci. 2006, 94, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Sauer, S.J.; Tarpley, M.; Shah, I.; Save, A.V.; Lyerly, H.K.; Patierno, S.R.; Williams, K.P.; Devi, G.R. Bisphenol A activates EGFR and ERK promoting proliferation, tumor spheroid formation and resistance to EGFR pathway inhibition in estrogen receptor-negative inflammatory breast cancer cells. Carcinogenesis 2017, 38, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Matthews, A.L.; Noy, P.J.; Reyat, J.S.; Tomlinson, M.G. Regulation of A disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: The emerging role of tetraspanins and rhomboids. Platelets 2017, 28, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Bilancio, A.; Bontempo, P.; Di Donato, M.; Conte, M.; Giovannelli, P.; Altucci, L.; Migliaccio, A.; Castoria, G. Bisphenol A induces cell cycle arrest in primary and prostate cancer cells through EGFR/ERK/p53 signaling pathway activation. Oncotarget 2017, 8, 115620–115631. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Steinle, J.J. IGFBP-3 inhibits TNF-alpha production and TNFR-2 signaling to protect against retinal endothelial cell apoptosis. Microvasc. Res. 2014, 95, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Piprode, V.; Mhaske, S.T.; Barhanpurkar-Naik, A.; Wani, M.R. IL-3 Differentially Regulates Membrane and Soluble RANKL in Osteoblasts through Metalloproteases and the JAK2/STAT5 Pathway and Improves the RANKL/OPG Ratio in Adult Mice. J. Immunol. 2018, 200, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Xu, D.; Ai, Y.; Zhao, S.; Zhang, L.; Ming, G.; Liu, Z. Angiotensin-(1-7)/Mas Signaling Inhibits Lipopolysaccharide-Induced ADAM17 Shedding Activity and Apoptosis in Alveolar Epithelial Cells. Pharmacology 2016, 97, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Michaela, P.; Maria, K.; Silvia, H.; L’Ubica, L. Bisphenol A differently inhibits CaV3.1, Ca V3.2 and Ca V3.3 calcium channels. Naunyn Schmiedebergs Arch. Pharmacol. 2014, 387, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Liu, S.; Guo, F.; Liu, S.; Yu, X.; Hu, H.; Sun, X.; Hao, L.; Zhu, T. Nonylphenol affects myocardial contractility and L-type Ca(2+) channel currents in a non-monotonic manner via G protein-coupled receptor 30. Toxicology 2015, 334, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.M.; Weskamp, G.; Chesneau, V.; Sahin, U.; Vortkamp, A.; Horiuchi, K.; Chiusaroli, R.; Hahn, R.; Wilkes, D.; Fisher, P.; et al. Essential role for ADAM19 in cardiovascular morphogenesis. Mol. Cell. Biol 2004, 24, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [PubMed]
- Sokal, R.R. Biometry: The Principles and Practice of Statistic in Biological Research; W. H. Freeman: New York, NY, USA, 1995; p. 887. [Google Scholar]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urriola-Muñoz, P.; Lagos-Cabré, R.; Patiño-García, D.; Reyes, J.G.; Moreno, R.D. Bisphenol-A and Nonylphenol Induce Apoptosis in Reproductive Tract Cancer Cell Lines by the Activation of ADAM17. Int. J. Mol. Sci. 2018, 19, 2238. https://doi.org/10.3390/ijms19082238
Urriola-Muñoz P, Lagos-Cabré R, Patiño-García D, Reyes JG, Moreno RD. Bisphenol-A and Nonylphenol Induce Apoptosis in Reproductive Tract Cancer Cell Lines by the Activation of ADAM17. International Journal of Molecular Sciences. 2018; 19(8):2238. https://doi.org/10.3390/ijms19082238
Chicago/Turabian StyleUrriola-Muñoz, Paulina, Raúl Lagos-Cabré, Daniel Patiño-García, Juan G. Reyes, and Ricardo D. Moreno. 2018. "Bisphenol-A and Nonylphenol Induce Apoptosis in Reproductive Tract Cancer Cell Lines by the Activation of ADAM17" International Journal of Molecular Sciences 19, no. 8: 2238. https://doi.org/10.3390/ijms19082238
APA StyleUrriola-Muñoz, P., Lagos-Cabré, R., Patiño-García, D., Reyes, J. G., & Moreno, R. D. (2018). Bisphenol-A and Nonylphenol Induce Apoptosis in Reproductive Tract Cancer Cell Lines by the Activation of ADAM17. International Journal of Molecular Sciences, 19(8), 2238. https://doi.org/10.3390/ijms19082238