Inhibition of BET Proteins Reduces Right Ventricle Hypertrophy and Pulmonary Hypertension Resulting from Combined Hypoxia and Pulmonary Inflammation

 ,
,  ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
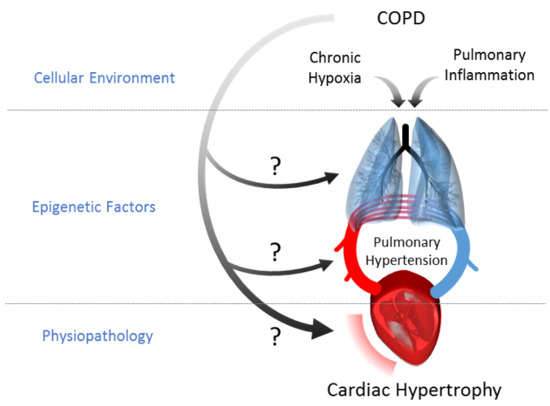
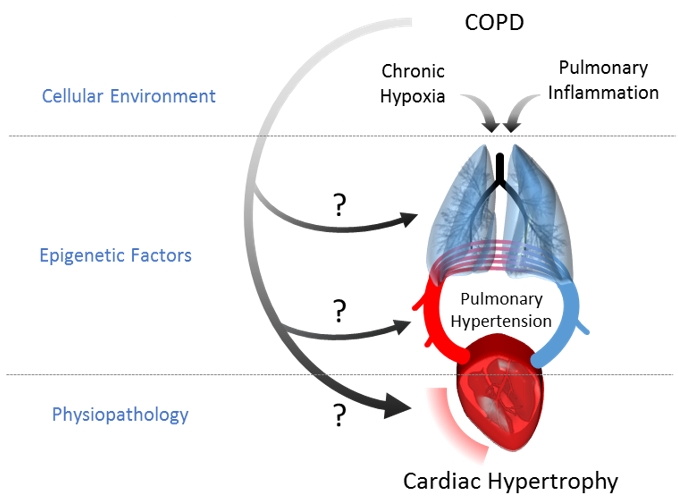
1. Introduction
2. Results
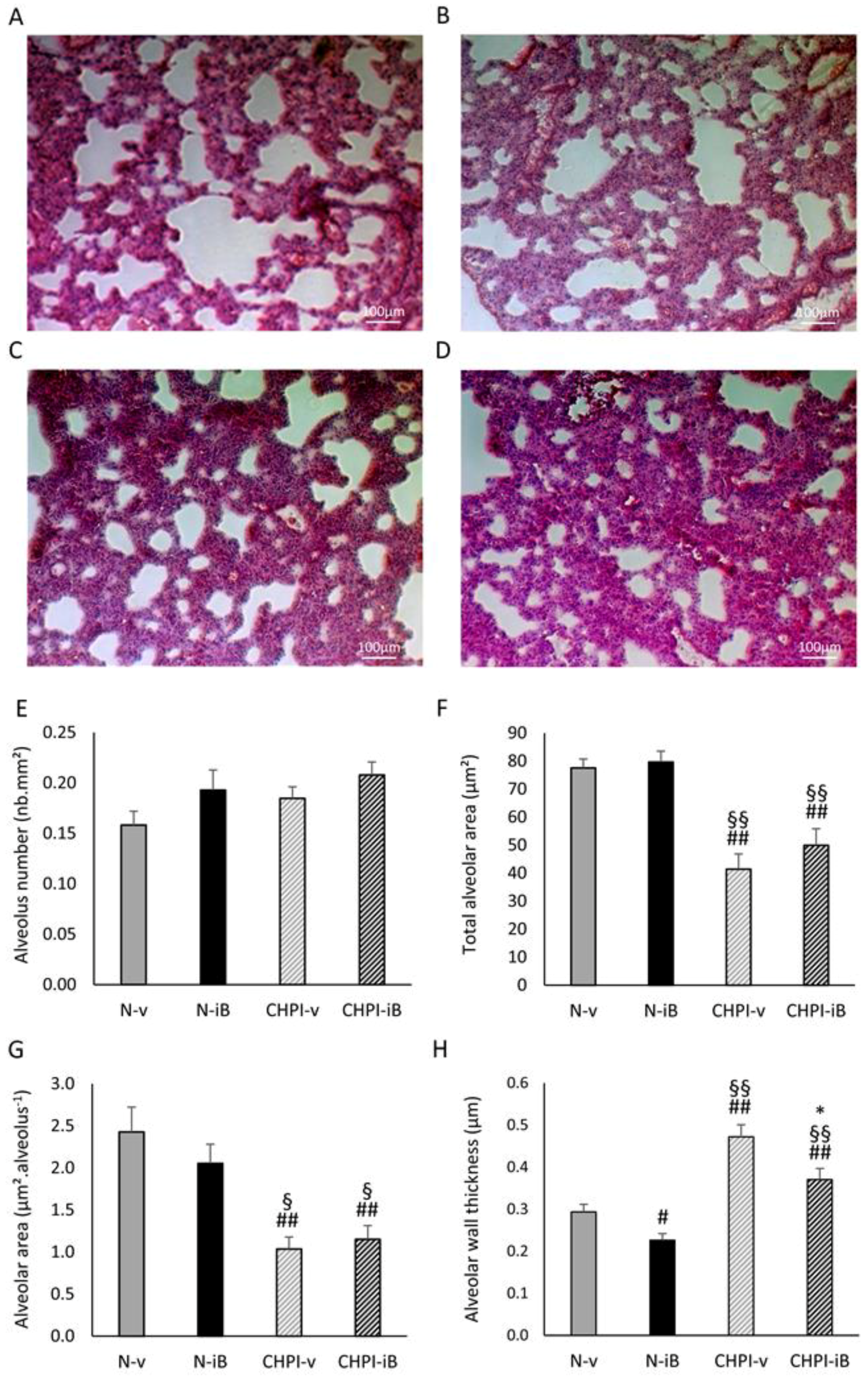
2.1. Lipopolysaccharides (LPS) Instillations Are Associated with Signs of Lung Alterations
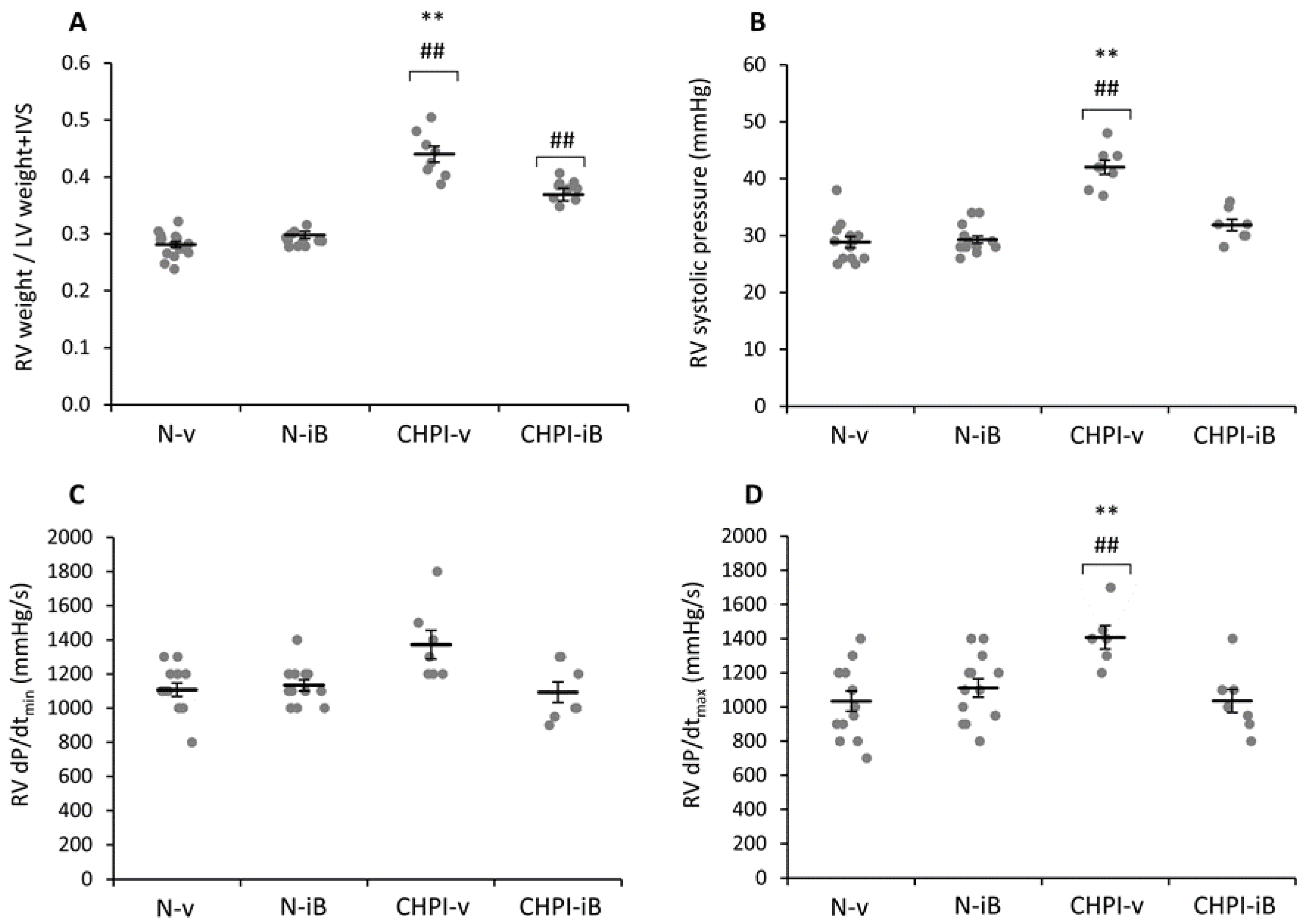
2.2. Inhibition of BET Proteins Reverses Hypertension of the Right Ventricle
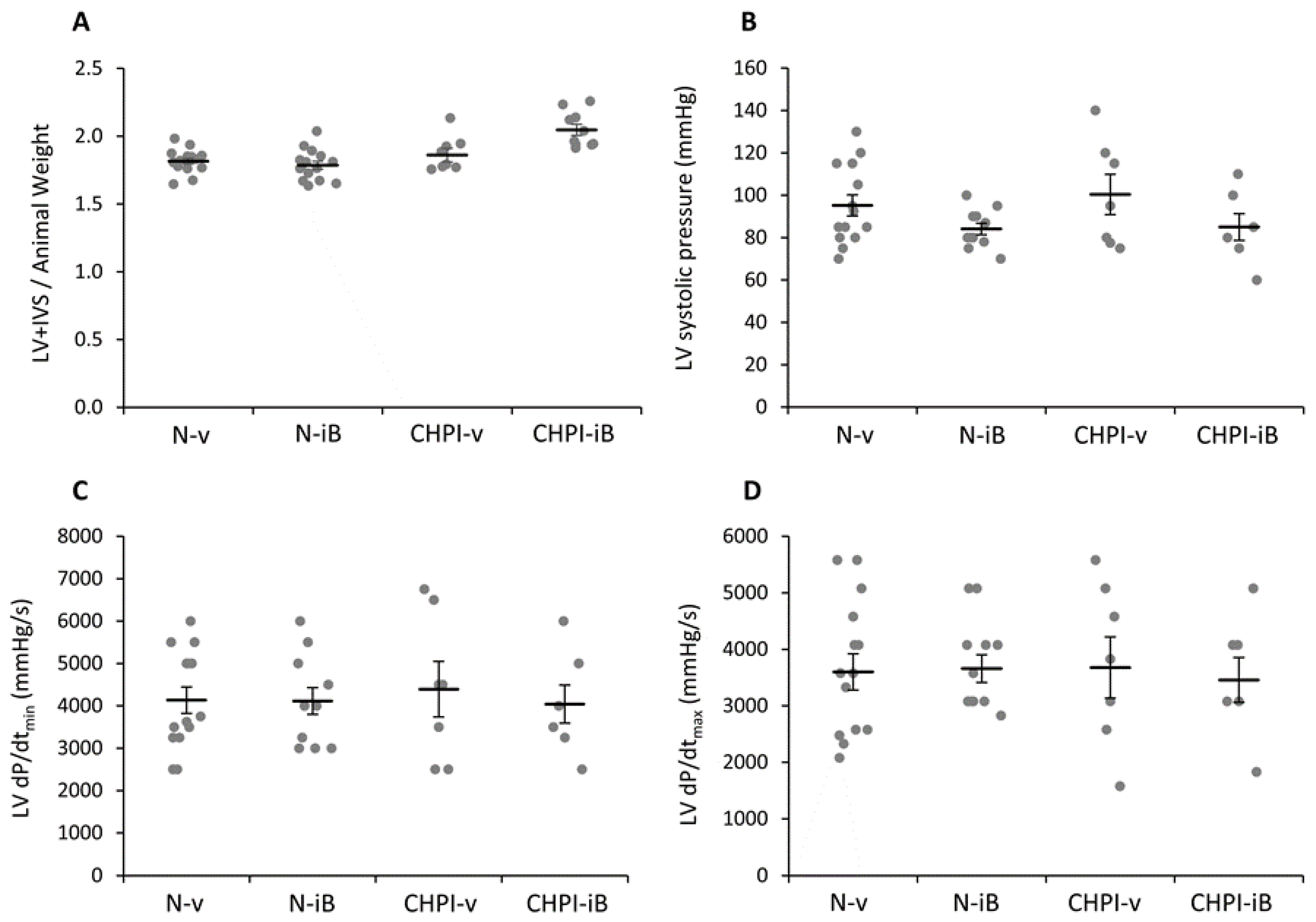
2.3. Inhibition of BET Proteins Has No Effect on Left Ventricle
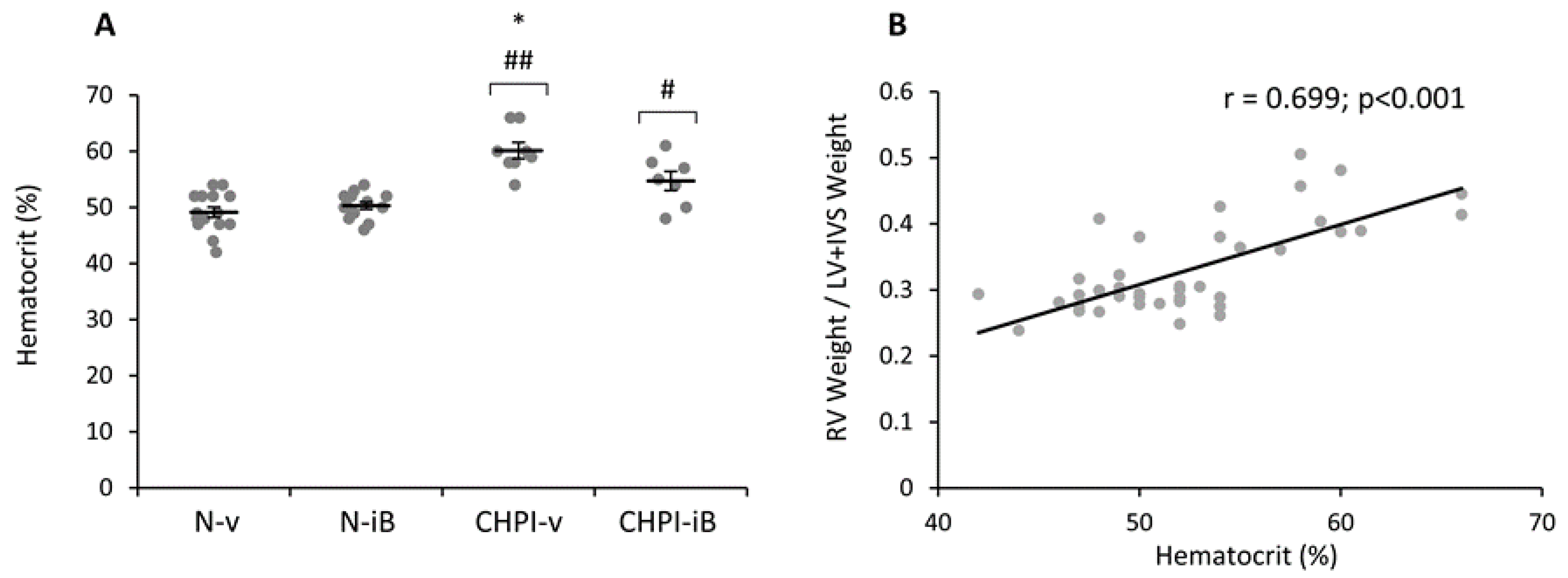
2.4. Inhibition of BET Proteins Blunts Hematocrit Increase Induced by Chronic Hypoxia and Inflammation
3. Discussion
4. Material and Methods
4.1. Animals
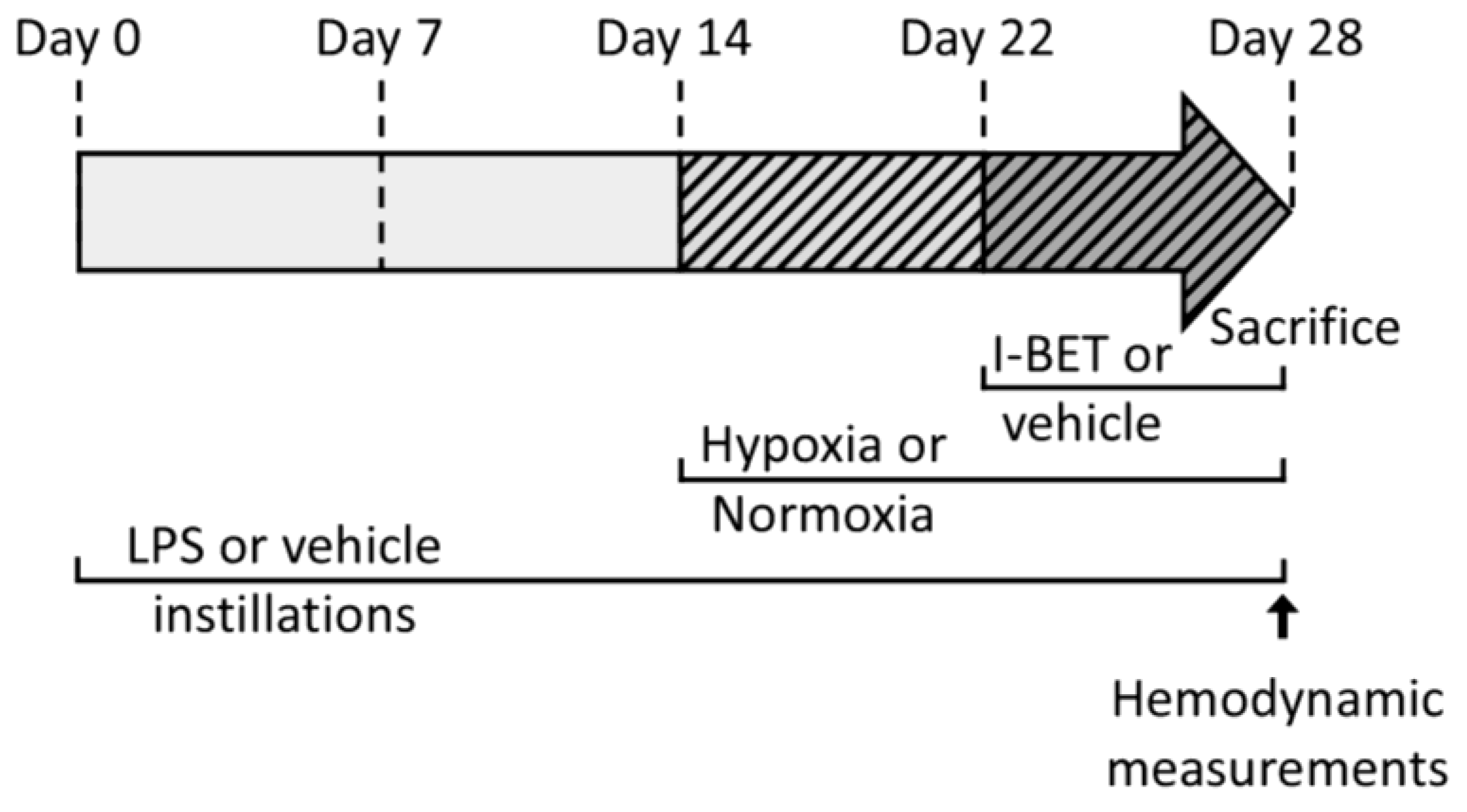
4.2. Right Ventricle Hypertrophy Induction by Pulmonary Inflammation (PI) and Chronic Hypoxia (CH)
4.3. Bromodomain and Extra-C-Terminal Domain Inhibitor (I-BET151) Administration
4.4. Lung Morphometry Analysis
4.5. Hemodynamic Measurements
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoeper, M.M.; McLaughlin, V.V.; Dalaan, A.M.; Satoh, T.; Galie, N. Treatment of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 323–336. [Google Scholar] [CrossRef]
- Chaouat, A.; Naeije, R.; Weitzenblum, E. Pulmonary hypertension in COPD. Eur. Respir. J. 2008, 32, 1371–1385. [Google Scholar] [CrossRef] [PubMed]
- Dal Negro, R.W.; Bonadiman, L.; Turco, P. Prevalence of different comorbidities in COPD patients by gender and GOLD stage. Multidiscip. Respir. Med. 2015, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Pak, O.; Aldashev, A.; Welsh, D.; Peacock, A. The effects of hypoxia on the cells of the pulmonary vasculature. Eur. Respir. J. 2007, 30, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Mecham, R.P. Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu. Rev. Phys. 1997, 59, 89–144. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rio, F.; Miravitlles, M.; Soriano, J.B.; Munoz, L.; Duran-Tauleria, E.; Sanchez, G.; Sobradillo, V.; Ancochea, J. Systemic inflammation in chronic obstructive pulmonary disease: A population-based study. Respir. Res. 2010, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, S.C.; Poth, J.M.; Fini, M.A.; Olschewski, A.; El Kasmi, K.C.; Stenmark, K.R. The role of inflammation in hypoxic pulmonary hypertension: From cellular mechanisms to clinical phenotypes. Am. J. Physiol. Lung. Cell Mol. Physiol. 2015, 308, L229–L252. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Dahl, M.J.; Albertine, K.H.; Ramchandran, R.; Sun, M.; Raj, J.U. Role of histone deacetylases in regulation of phenotype of ovine newborn pulmonary arterial smooth muscle cells. Cell Prolif. 2013, 46, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.K.; Eom, G.H.; Kee, H.J.; Kim, H.S.; Choi, W.Y.; Nam, K.I.; Ma, J.S.; Kook, H. Sodium valproate, a histone deacetylase inhibitor, but not captopril, prevents right ventricular hypertrophy in rats. Circ. J. Off. J. Jpn. Circ. Soc. 2010, 74, 760–770. [Google Scholar] [CrossRef]
- Cavasin, M.A.; Demos-Davies, K.; Horn, T.R.; Walker, L.A.; Lemon, D.D.; Birdsey, N.; Weiser-Evans, M.C.; Harral, J.; Irwin, D.C.; Anwar, A.; et al. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ. Res. 2012, 110, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chen, C.N.; Hajji, N.; Oliver, E.; Cotroneo, E.; Wharton, J.; Wang, D.; Li, M.; McKinsey, T.A.; Stenmark, K.R.; et al. Histone deacetylation inhibition in pulmonary hypertension: Therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 2012, 126, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.S.; McKinsey, T.A. Acetyl-lysine erasers and readers in the control of pulmonary hypertension and right ventricular hypertrophy. Biochem. Cell Biol. 2015, 93, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Meloche, J.; Potus, F.; Vaillancourt, M.; Bourgeois, A.; Johnson, I.; Deschamps, L.; Chabot, S.; Ruffenach, G.; Henry, S.; Breuils-Bonnet, S.; et al. Bromodomain-Containing Protein 4: The Epigenetic Origin of Pulmonary Arterial Hypertension. Circ. Res. 2015, 117, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Ciuclan, L.; Bonneau, O.; Hussey, M.; Duggan, N.; Holmes, A.M.; Good, R.; Stringer, R.; Jones, P.; Morrell, N.W.; Jarai, G.; et al. A novel murine model of severe pulmonary arterial hypertension. Am. J. Physiol. Lung. Cell Mol. Physiol. 2011, 184, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Perez-Perri, J.I.; Acevedo, J.M.; Wappner, P. Epigenetics: New questions on the response to hypoxia. Int. J. Mol. Sci. 2011, 12, 4705–4721. [Google Scholar] [CrossRef] [PubMed]
- Bayarsaihan, D. Epigenetic mechanisms in inflammation. J. Dent. Res. 2011, 90, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Puig-Vilanova, E.; Martinez-Llorens, J.; Ausin, P.; Roca, J.; Gea, J.; Barreiro, E. Quadriceps muscle weakness and atrophy are associated with a differential epigenetic profile in advanced COPD. Clin. Sci. 2015, 128, 905–921. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Charron, C.E.; Adcock, I.M. Impact of protein acetylation in inflammatory lung diseases. Pharmacol. Ther. 2007, 116, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Bogaard, H.J.; Mizuno, S.; Hussaini, A.A.; Toldo, S.; Abbate, A.; Kraskauskas, D.; Kasper, M.; Natarajan, R.; Voelkel, N.F. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am. J. Physiol. Lung. Cell Mol. Physiol. 2011, 183, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Koulmann, N.; Novel-Chate, V.; Peinnequin, A.; Chapot, R.; Serrurier, B.; Simler, N.; Richard, H.; Ventura-Clapier, R.; Bigard, X. Cyclosporin A inhibits hypoxia-induced pulmonary hypertension and right ventricle hypertrophy. Am. J. Physiol. Lung. Cell Mol. Physiol. 2006, 174, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Vitali, S.H.; Hansmann, G.; Rose, C.; Fernandez-Gonzalez, A.; Scheid, A.; Mitsialis, S.A.; Kourembanas, S. The Sugen 5416/hypoxia mouse model of pulmonary hypertension revisited: Long-term follow-up. Pulm. Circ. 2014, 4, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung. Cell Mol. Physiol. 2009, 297, L1013–L1032. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.L.; Mandel, J.; Yuan, J.X. Idiopathic pulmonary arterial hypertension. Dis. Models Mech. 2010, 3, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Atochina, E.N.; Morris, K.G.; Danilov, S.M.; Stenmark, K.R. Angiotensin converting enzyme expression is increased in small pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. J. Clin. Investig. 1995, 96, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Zhao, X.; Andersson, R.; Song, Z.; Yang, D. Potential effects of peroxisome proliferator-activated receptor activator on LPS-induced lung injury in rats. Pulmon. Pharmacol. Ther. 2009, 22, 318–325. [Google Scholar] [CrossRef]
- Vernooy, J.H.; Dentener, M.A.; van Suylen, R.J.; Buurman, W.A.; Wouters, E.F. Long-term intratracheal lipopolysaccharide exposure in mice results in chronic lung inflammation and persistent pathology. Am. J. Respir. Cell Mol. Biol. 2002, 26, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Chabert, C.; Khochbin, S.; Rousseaux, S.; Furze, R.; Smithers, N.; Prinjha, R.; Schlattner, U.; Pison, C.; Dubouchaud, H. Muscle hypertrophy in hypoxia with inflammation is controlled by bromodomain and extra-terminal domain proteins. Sci. Rep. 2017, 7, 12133. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 2009, 24, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Nagati, J.S.; Xie, J.; Li, J.; Walters, H.; Moon, Y.A.; Gerard, R.D.; Huang, C.L.; Comerford, S.A.; Hammer, R.E.; et al. An acetate switch regulates stress erythropoiesis. Nat. Med. 2014, 20, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Cong, X.; Yun, Z. Differential hypoxic regulation of hypoxia-inducible factors 1α and 2α. Mol. Cancer Res. 2011, 9, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Weisse, A.B.; Moschos, C.B.; Frank, M.J.; Levinson, G.E.; Cannilla, J.E.; Regan, T.J. Hemodynamic effects of staged hematocrit reduction in patients with stable cor pulmonale and severely elevated hematocrit levels. Am. J. Med. 1975, 58, 92–98. [Google Scholar] [CrossRef]
- Borst, M.M.; Leschke, M.; Konig, U.; Worth, H. Repetitive hemodilution in chronic obstructive pulmonary disease and pulmonary hypertension: Effects on pulmonary hemodynamics, gas exchange, and exercise capacity. Respir. Int. Rev. Thorac. Dis. 1999, 66, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P. Mabley, J.G. Liaudet, L. Evgenov, O.V. Marton, A. Haskó, G. Kollai, M. Szabó, C. Left ventricular pressure-volume relationship in a rat model of advanced aging-associated heart failure. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2132–H2137. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chabert, C.; Khochbin, S.; Rousseaux, S.; Veyrenc, S.; Furze, R.; Smithers, N.; Prinjha, R.K.; Schlattner, U.; Pison, C.; Dubouchaud, H. Inhibition of BET Proteins Reduces Right Ventricle Hypertrophy and Pulmonary Hypertension Resulting from Combined Hypoxia and Pulmonary Inflammation. Int. J. Mol. Sci. 2018, 19, 2224. https://doi.org/10.3390/ijms19082224
Chabert C, Khochbin S, Rousseaux S, Veyrenc S, Furze R, Smithers N, Prinjha RK, Schlattner U, Pison C, Dubouchaud H. Inhibition of BET Proteins Reduces Right Ventricle Hypertrophy and Pulmonary Hypertension Resulting from Combined Hypoxia and Pulmonary Inflammation. International Journal of Molecular Sciences. 2018; 19(8):2224. https://doi.org/10.3390/ijms19082224
Chicago/Turabian StyleChabert, Clovis, Saadi Khochbin, Sophie Rousseaux, Sylvie Veyrenc, Rebecca Furze, Nicholas Smithers, Rab K Prinjha, Uwe Schlattner, Christophe Pison, and Hervé Dubouchaud. 2018. "Inhibition of BET Proteins Reduces Right Ventricle Hypertrophy and Pulmonary Hypertension Resulting from Combined Hypoxia and Pulmonary Inflammation" International Journal of Molecular Sciences 19, no. 8: 2224. https://doi.org/10.3390/ijms19082224
APA StyleChabert, C., Khochbin, S., Rousseaux, S., Veyrenc, S., Furze, R., Smithers, N., Prinjha, R. K., Schlattner, U., Pison, C., & Dubouchaud, H. (2018). Inhibition of BET Proteins Reduces Right Ventricle Hypertrophy and Pulmonary Hypertension Resulting from Combined Hypoxia and Pulmonary Inflammation. International Journal of Molecular Sciences, 19(8), 2224. https://doi.org/10.3390/ijms19082224