The Nefarious Nexus of Noncoding RNAs in Cancer



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
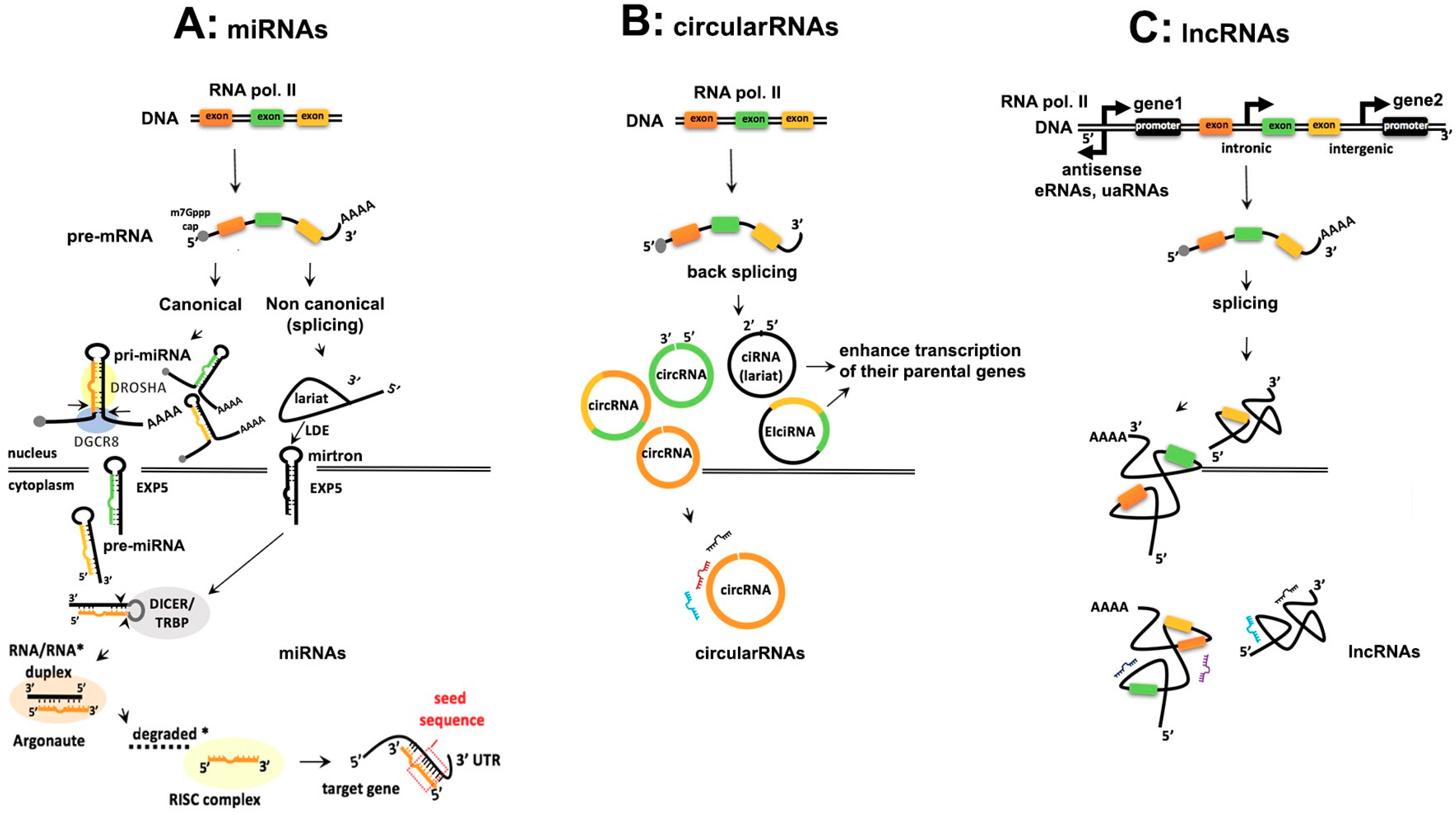
2. Comparison of the Biogenesis of Noncoding RNAs
2.1. Similarities and Differences in Biogenesis of ncRNAs
2.1.1. Transcription, Capping and Adenylation
2.1.2. Processing of ncRNAs
3. Functions of ncRNAs in Cancer
3.1. Deregulation of miRNA Expression in Cancer
3.1.1. miRNA Signatures in Cancer
3.1.2. Cellular miRNAs Interact with Viral Proteins
3.1.3. Defective Biogenesis of miRNAs
3.2. circRNAs in Cancer
3.3. lncRNA in Cancer
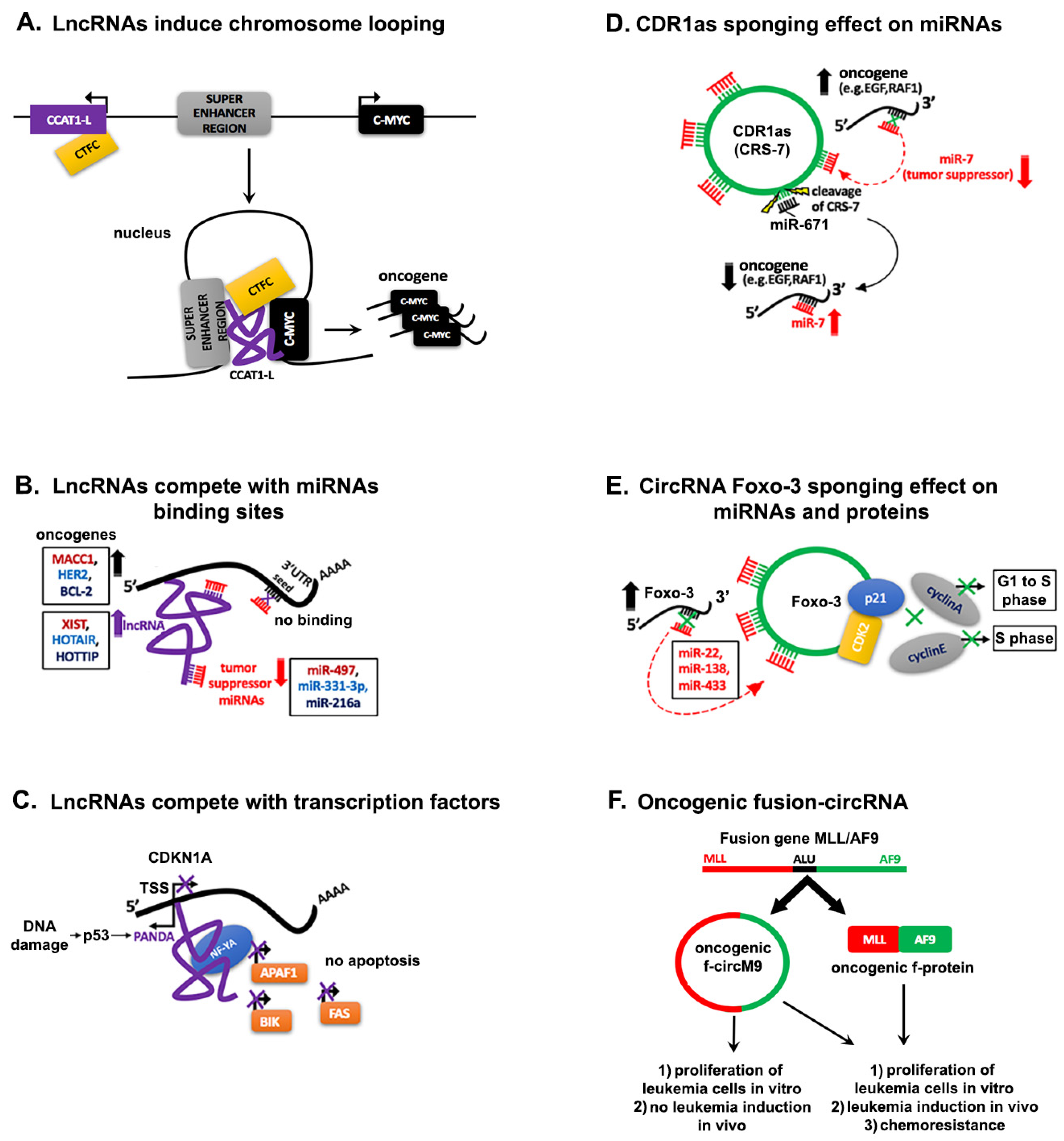
4. Interaction Networks of ncRNAs in Cancer
4.1. Regulation through Competitive Interactions
4.1.1. Competing lncRNAs
4.1.2. Pseudogenes Derived-lncRNAs
4.1.3. Decoying Role of circRNAs
4.1.4. Progenitor miRNA
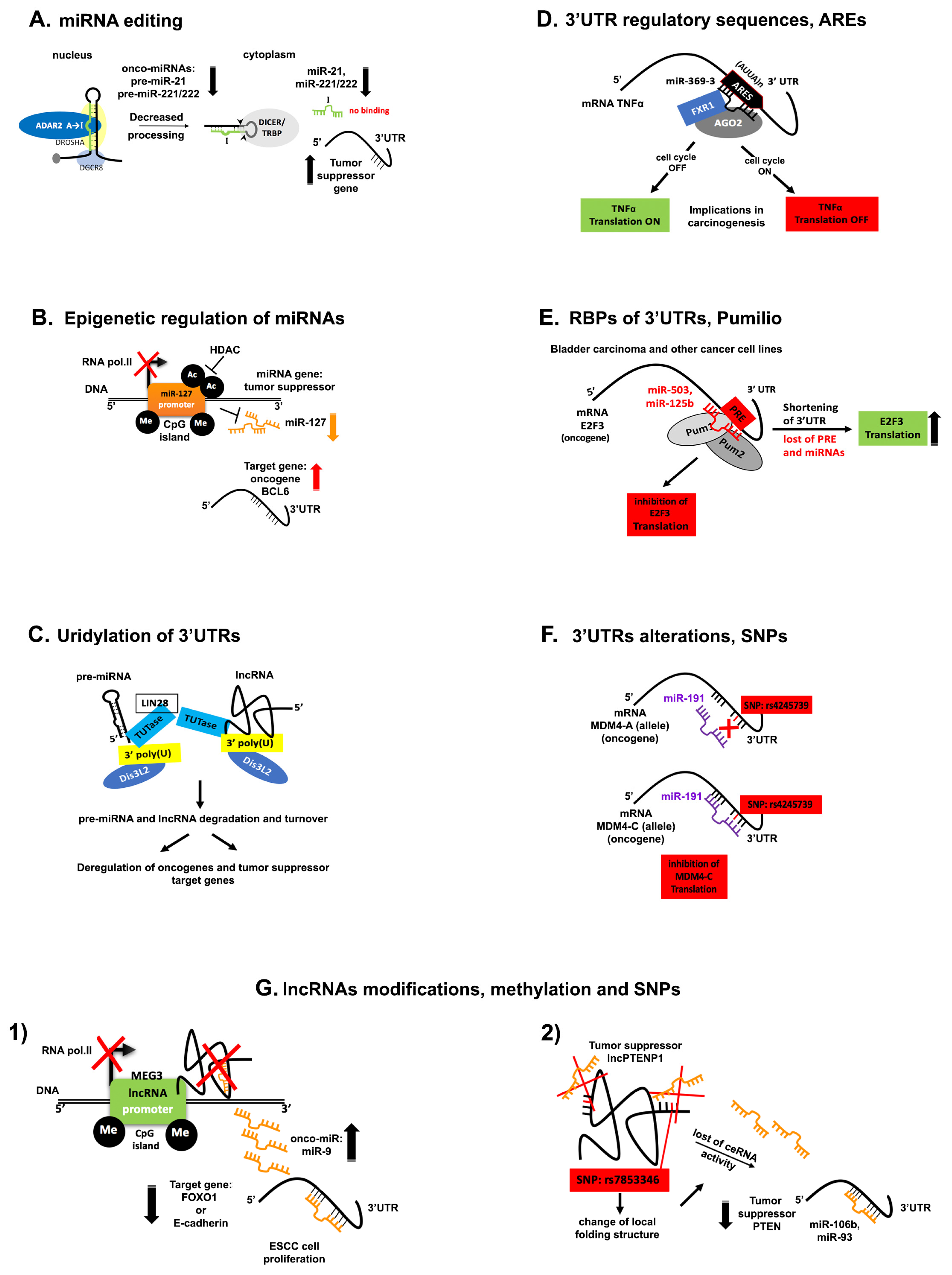
5. Disruption of ncRNA Networks in Cancer
5.1. miRNA Editing
5.2. Epigenetic Regulation of miRNAs
5.3. Uridylation of the 3′-End of ncRNAs
5.4. Phosphorylation of the 5′ End of miRNAs
5.5. Alterations and Regulatory Sequences within 3′UTRs
5.6. Genetic Alterations
5.7. lncRNA Modifications in Cancer
6. Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Eddy, S.R. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001, 2, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W. Origin of life: The RNA world. Nature 1986, 319, 618. [Google Scholar] [CrossRef]
- Altman, S. A view of RNase P. Mol. BioSyst. 2007, 3, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R. Nobel lecture. Self-splicing and enzymatic activity of an intervening sequence RNA from Tetrahymena. Biosci. Rep. 1990, 10, 239–261. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, J.M. Revisiting lncRNAs: How Do You Know Yours Is Not an eRNA? Mol. Cell 2016, 62, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Berget, S.M.; Moore, C.; Sharp, P.A. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. USA 1977, 74, 3171–3175. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.T.; Gelinas, R.E.; Broker, T.R.; Roberts, R.J. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 1977, 12, 1–8. [Google Scholar] [CrossRef]
- Sanger, H.L.; Klotz, G.; Riesner, D.; Gross, H.J.; Kleinschmidt, A.K. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc. Natl. Acad. Sci. USA 1976, 73, 3852–3856. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.T.; Coca-Prados, M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature 1979, 280, 339–340. [Google Scholar] [CrossRef] [PubMed]
- Nigro, J.M.; Cho, K.R.; Fearon, E.R.; Kern, S.E.; Ruppert, J.M.; Oliner, J.D.; Kinzler, K.W.; Vogelstein, B. Scrambled exons. Cell 1991, 64, 607–613. [Google Scholar] [CrossRef]
- Capel, B.; Swain, A.; Nicolis, S.; Hacker, A.; Walter, M.; Koopman, P.; Goodfellow, P.; Lovell-Badge, R. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 1993, 73, 1019–1030. [Google Scholar] [CrossRef]
- Cocquerelle, C.; Mascrez, B.; Hetuin, D.; Bailleul, B. Mis-splicing yields circular RNA molecules. FASEB J. 1993, 7, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Zaphiropoulos, P.G. Exon skipping and circular RNA formation in transcripts of the human cytochrome P-450 2C18 gene in epidermis and of the rat androgen binding protein gene in testis. Mol. Cell. Biol. 1997, 17, 2985–2993. [Google Scholar] [CrossRef] [PubMed]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Memczak, S.; Wyler, E.; Torti, F.; Porath, H.T.; Orejuela, M.R.; Piechotta, M.; Levanon, E.Y.; Landthaler, M.; Dieterich, C.; et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015, 10, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Rybak-Wolf, A.; Stottmeister, C.; Glazar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Pei, B.; Sisu, C.; Frankish, A.; Howald, C.; Habegger, L.; Mu, X.J.; Harte, R.; Balasubramanian, S.; Tanzer, A.; Diekhans, M.; et al. The GENCODE pseudogene resource. Genome Biol. 2012, 13, R51. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Anastasiadou, E.; Esteller, M.; He, L.; Slack, F.J. The Inescapable Influence of Noncoding RNAs in Cancer. Cancer Res. 2015, 75, 5206–5210. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Stroopinsky, D.; Alimperti, S.; Jiao, A.L.; Pyzer, A.R.; Cippitelli, C.; Pepe, G.; Severa, M.; Rosenblatt, J.; Etna, M.P.; et al. Epstein-Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Bonasio, R.; Shiekhattar, R. Regulation of transcription by long noncoding RNAs. Annu. Rev. Genet. 2014, 48, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.K.; Griffiths-Jones, S.; Enright, A.J. Genomic analysis of human microRNA transcripts. Proc. Natl. Acad. Sci. USA 2007, 104, 17719–17724. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.; Lee, R.C.; Lavanway, A.; Williams, P.T.; Jewell, D. MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr. Biol. 2003, 13, 807–818. [Google Scholar] [CrossRef]
- Olive, V.; Li, Q.; He, L. mir-17-92: A polycistronic oncomir with pleiotropic functions. Immunol. Rev. 2013, 253, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.; Baird, A.M.; Brady, L.; Lim, M.; Gray, S.G.; McDermott, R.; Finn, S.P. Circular RNAs: Biogenesis, Function and Role in Human Diseases. Front. Mol. Biosci. 2017, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Lasda, E.; Parker, R. Circular RNAs: Diversity of form and function. RNA 2014, 20, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- Brugiolo, M.; Herzel, L.; Neugebauer, K.M. Counting on co-transcriptional splicing. F1000Prime Rep. 2013, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Domdey, H.; Wiebauer, K. Inverse splicing of a discontinuous pre-mRNA intron generates a circular exon in a HeLa cell nuclear extract. Nucleic Acids Res. 1996, 24, 4152–4157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Sigova, A.A.; Mullen, A.C.; Molinie, B.; Gupta, S.; Orlando, D.A.; Guenther, M.G.; Almada, A.E.; Lin, C.; Sharp, P.A.; Giallourakis, C.C.; et al. Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 2876–2881. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.; Do, B.T.; Webster, D.E.; Khavari, P.A.; Chang, H.Y. Dicer-microRNA-Myc circuit promotes transcription of hundreds of long noncoding RNAs. Nat. Struct. Mol. Biol. 2014, 21, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Alcid, E.A.; Tsukiyama, T. ATP-dependent chromatin remodeling shapes the long noncoding RNA landscape. Genes Dev. 2014, 28, 2348–2360. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, P.; Palanichamy, J.K.; Singh, A.; Das, P.; Bhagat, M.; Kassab, M.A.; Sinha, S.; Chattopadhyay, P. Biogenesis of intronic miRNAs located in clusters by independent transcription and alternative splicing. RNA 2014, 20, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 2006, 13, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.R.; Seow, Y.; Saayman, S.; Dijkstra, K.K.; El Andaloussi, S.; Weinberg, M.S.; Wood, M.J. The biogenesis and characterization of mammalian microRNAs of mirtron origin. Nucleic Acids Res. 2012, 40, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Mourelatos, Z.; Dostie, J.; Paushkin, S.; Sharma, A.; Charroux, B.; Abel, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. miRNPs: A novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev. 2002, 16, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.Y.; Rana, T.M. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol. 2006, 4, e210. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.Y.; Yan, Z.; Xu, Y.; Hu, H.; Menzel, C.; Zhou, Y.H.; Chen, W.; Khaitovich, P. Sequence features associated with microRNA strand selection in humans and flies. BMC Genom. 2009, 10, 413. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K. Anatomy of RISC: How do small RNAs and chaperones activate Argonaute proteins? Wiley Interdiscip. Rev. RNA 2016, 7, 637–660. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Diederichs, S. Argonaute-3 activates the let-7a passenger strand microRNA. RNA Biol. 2013, 10, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Bottini, S.; Hamouda-Tekaya, N.; Mategot, R.; Zaragosi, L.E.; Audebert, S.; Pisano, S.; Grandjean, V.; Mauduit, C.; Benahmed, M.; Barbry, P.; et al. Post-transcriptional gene silencing mediated by microRNAs is controlled by nucleoplasmic Sfpq. Nat. Commun. 2017, 8, 1189. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, K.T.; Li, L.; Chu, Y.; Janowski, B.A.; Corey, D.R. RNAi factors are present and active in human cell nuclei. Cell Rep. 2014, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Kim, B.; Kim, V.N. Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1881–E1889. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Maurin, T.; Robine, N.; Rasmussen, K.D.; Jeffrey, K.L.; Chandwani, R.; Papapetrou, E.P.; Sadelain, M.; O’Carroll, D.; Lai, E.C. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 15163–15168. [Google Scholar] [CrossRef] [PubMed]
- Janas, M.M.; Wang, B.; Harris, A.S.; Aguiar, M.; Shaffer, J.M.; Subrahmanyam, Y.V.; Behlke, M.A.; Wucherpfennig, K.W.; Gygi, S.P.; Gagnon, E.; et al. Alternative RISC assembly: Binding and repression of microRNA-mRNA duplexes by human Ago proteins. RNA 2012, 18, 2041–2055. [Google Scholar] [CrossRef] [PubMed]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Bottini, S.; Hamouda-Tekaya, N.; Tanasa, B.; Zaragosi, L.E.; Grandjean, V.; Repetto, E.; Trabucchi, M. From benchmarking HITS-CLIP peak detection programs to a new method for identification of miRNA-binding sites from Ago2-CLIP data. Nucleic Acids Res. 2017, 45, e71. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Hasler, J.; Samuelsson, T.; Strub, K. Useful ‘junk’: Alu RNAs in the human transcriptome. Cell. Mol. Life Sci. 2007, 64, 1793–1800. [Google Scholar] [CrossRef] [PubMed]
- Pasman, Z.; Been, M.D.; Garcia-Blanco, M.A. Exon circularization in mammalian nuclear extracts. RNA 1996, 2, 603–610. [Google Scholar] [PubMed]
- Zhang, Y.; Yang, L.; Chen, L.L. Characterization of Circular RNAs. Methods Mol. Biol. 2016, 1402, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E. Long noncoding RNAs: Re-writing dogmas of RNA processing and stability. Biochim. Biophys. Acta 2016, 1859, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Dhir, A.; Dhir, S.; Proudfoot, N.J.; Jopling, C.L. Microprocessor mediates transcriptional termination of long noncoding RNA transcripts hosting microRNAs. Nat. Struct. Mol. Biol. 2015, 22, 319–327. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Contreras, A.; Ruggero, D. Small RNAs with big implications: New insights into H/ACA snoRNA function and their role in human disease. Wiley Interdiscip. Rev. RNA 2015, 6, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.F.; Yang, L.; Zhang, Y.; Xiang, J.F.; Wu, Y.W.; Carmichael, G.G.; Chen, L.L. Long noncoding RNAs with snoRNA ends. Mol. Cell 2012, 48, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Ferracin, M.; Cimmino, A.; Di Leva, G.; Shimizu, M.; Wojcik, S.E.; Iorio, M.V.; Visone, R.; Sever, N.I.; Fabbri, M.; et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 353, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Ramkissoon, S.H.; Mainwaring, L.A.; Ogasawara, Y.; Keyvanfar, K.; McCoy, J.P., Jr.; Sloand, E.M.; Kajigaya, S.; Young, N.S. Hematopoietic-specific microRNA expression in human cells. Leuk. Res. 2006, 30, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Metzler, M.; Wilda, M.; Busch, K.; Viehmann, S.; Borkhardt, A. High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma. Genes Chromosomes Cancer 2004, 39, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.Z.; SM, O.C.; van Holst Pellekaan, N.G.; Young, G.P.; James, R.J. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol. Cancer Res. 2003, 1, 882–891. [Google Scholar] [PubMed]
- Calin, G.A.; Liu, C.G.; Sevignani, C.; Ferracin, M.; Felli, N.; Dumitru, C.D.; Shimizu, M.; Cimmino, A.; Zupo, S.; Dono, M.; et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc. Natl. Acad. Sci. USA 2004, 101, 11755–11760. [Google Scholar] [CrossRef] [PubMed]
- Medina, P.P.; Nolde, M.; Slack, F.J. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 2010, 467, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Srinivasan, L.; Calado, D.P.; Patterson, H.C.; Zhang, B.; Wang, J.; Henderson, J.M.; Kutok, J.L.; Rajewsky, K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 2008, 9, 405–414. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Beachy, S.H.; Onozawa, M.; Chung, Y.J.; Slape, C.; Bilke, S.; Francis, P.; Pineda, M.; Walker, R.L.; Meltzer, P.; Aplan, P.D. Enforced expression of Lin28b leads to impaired T-cell development, release of inflammatory cytokines, and peripheral T-cell lymphoma. Blood 2012, 120, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Asslaber, D.; Pinon, J.D.; Seyfried, I.; Desch, P.; Stocher, M.; Tinhofer, I.; Egle, A.; Merkel, O.; Greil, R. microRNA-34a expression correlates with MDM2 SNP309 polymorphism and treatment-free survival in chronic lymphocytic leukemia. Blood 2010, 115, 4191–4197. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—Micrornas with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Pyzer, A.R.; Stroopinsky, D.; Rosenblatt, J.; Anastasiadou, E.; Rajabi, H.; Washington, A.; Tagde, A.; Chu, J.H.; Coll, M.; Jiao, A.L.; et al. MUC1 inhibition leads to decrease in PD-L1 levels via upregulation of miRNAs. Leukemia 2017, 31, 2780–2790. [Google Scholar] [CrossRef] [PubMed]
- Costinean, S.; Zanesi, N.; Pekarsky, Y.; Tili, E.; Volinia, S.; Heerema, N.; Croce, C.M. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7024–7029. [Google Scholar] [CrossRef] [PubMed]
- Gilles, M.E.; Hao, L.; Huang, L.; Rupaimoole, R.; Lopez-Casas, P.P.; Pulver, E.; Jeong, J.C.; Muthuswamy, S.K.; Hidalgo, M.; Bhatia, S.N.; et al. Personalized RNA Medicine for Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 1734–1747. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.K.; Li, Y.; Hafner, M.; Banerjee, N.S.; Tang, S.; Briskin, D.; Meyers, C.; Chow, L.T.; Xie, X.; et al. microRNAs are biomarkers of oncogenic human papillomavirus infections. Proc. Natl. Acad. Sci. USA 2014, 111, 4262–4267. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Schutz, S.; Sarnow, P. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 2008, 4, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. MicroRNAs as mediators of viral evasion of the immune system. Nat. Immunol. 2013, 14, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, A.; Al-Jadiri, M.F.; Talerico, C.; Duranti, E.; Pilozzi, E.; Trivedi, P.; Anastasiadou, E.; Alsaadawi, A.R.; Al-Darraji, A.F.; Al-Hadad, S.A.; et al. Epstein-Barr virus (EBV) positive classical Hodgkin lymphoma of Iraqi children: An immunophenotypic and molecular characterization of Hodgkin/Reed-Sternberg cells. Pediatr. Blood Cancer 2013, 60, 2068–2072. [Google Scholar] [CrossRef] [PubMed]
- Farina, A.; Peruzzi, G.; Lacconi, V.; Lenna, S.; Quarta, S.; Rosato, E.; Vestri, A.R.; York, M.; Dreyfus, D.H.; Faggioni, A.; et al. Epstein-Barr virus lytic infection promotes activation of Toll-like receptor 8 innate immune response in systemic sclerosis monocytes. Arthritis Res. Ther. 2017, 19, 39. [Google Scholar] [CrossRef] [PubMed]
- Farina, A.; Farina, G.A. Fresh Insights into Disease Etiology and the Role of Microbial Pathogens. Curr. Rheumatol. Rep. 2016, 18, 1. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Boccellato, F.; Cirone, M.; Kis, L.L.; Klein, E.; Frati, L.; Faggioni, A.; Trivedi, P. Epigenetic mechanisms do not control viral latency III in primary effusion lymphoma cells infected with a recombinant Epstein-Barr virus. Leukemia 2005, 19, 1854–1856. [Google Scholar] [CrossRef] [PubMed]
- Rosato, P.; Anastasiadou, E.; Garg, N.; Lenze, D.; Boccellato, F.; Vincenti, S.; Severa, M.; Coccia, E.M.; Bigi, R.; Cirone, M.; et al. Differential regulation of miR-21 and miR-146a by Epstein-Barr virus-encoded EBNA2. Leukemia 2012, 26, 2343–2352. [Google Scholar] [CrossRef] [PubMed]
- Gatto, G.; Rossi, A.; Rossi, D.; Kroening, S.; Bonatti, S.; Mallardo, M. Epstein-Barr virus latent membrane protein 1 trans-activates miR-155 transcription through the NF-κB pathway. Nucleic Acids Res. 2008, 36, 6608–6619. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Boccellato, F.; Vincenti, S.; Rosato, P.; Bozzoni, I.; Frati, L.; Faggioni, A.; Presutti, C.; Trivedi, P. Epstein-Barr virus encoded LMP1 downregulates TCL1 oncogene through miR-29b. Oncogene 2010, 29, 1316–1328. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.E.; Fewell, C.; Yin, Q.; McBride, J.; Wang, X.; Lin, Z.; Flemington, E.K. Epstein-Barr virus growth/latency III program alters cellular microRNA expression. Virology 2008, 382, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Garg, N.; Bigi, R.; Yadav, S.; Campese, A.F.; Lapenta, C.; Spada, M.; Cuomo, L.; Botta, A.; Belardelli, F.; et al. Epstein-Barr virus infection induces miR-21 in terminally differentiated malignant B cells. Int. J. Cancer 2015, 137, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.; Motsch, N.; Zhu, J.Y.; Barth, S.; Okoniewski, M.; Reineke, T.; Tinguely, M.; Faggioni, A.; Trivedi, P.; Meister, G.; et al. microRNA profiling in Epstein-Barr virus-associated B-cell lymphoma. Nucleic Acids Res. 2011, 39, 1880–1893. [Google Scholar] [CrossRef] [PubMed]
- Merritt, W.M.; Lin, Y.G.; Han, L.Y.; Kamat, A.A.; Spannuth, W.A.; Schmandt, R.; Urbauer, D.; Pennacchio, L.A.; Cheng, J.F.; Nick, A.M.; et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N. Engl. J. Med. 2008, 359, 2641–2650. [Google Scholar] [CrossRef] [PubMed]
- Vaksman, O.; Hetland, T.E.; Trope, C.G.; Reich, R.; Davidson, B. Argonaute, Dicer, and Drosha are up-regulated along tumor progression in serous ovarian carcinoma. Hum. Pathol. 2012, 43, 2062–2069. [Google Scholar] [CrossRef] [PubMed]
- Chiosea, S.; Jelezcova, E.; Chandran, U.; Acquafondata, M.; McHale, T.; Sobol, R.W.; Dhir, R. Up-regulation of dicer, a component of the MicroRNA machinery, in prostate adenocarcinoma. Am. J. Pathol. 2006, 169, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Chiosea, S.; Jelezcova, E.; Chandran, U.; Luo, J.; Mantha, G.; Sobol, R.W.; Dacic, S. Overexpression of Dicer in precursor lesions of lung adenocarcinoma. Cancer Res. 2007, 67, 2345–2350. [Google Scholar] [CrossRef] [PubMed]
- Muralidhar, B.; Winder, D.; Murray, M.; Palmer, R.; Barbosa-Morais, N.; Saini, H.; Roberts, I.; Pett, M.; Coleman, N. Functional evidence that Drosha overexpression in cervical squamous cell carcinoma affects cell phenotype and microRNA profiles. J. Pathol. 2011, 224, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Rakheja, D.; Chen, K.S.; Liu, Y.; Shukla, A.A.; Schmid, V.; Chang, T.C.; Khokhar, S.; Wickiser, J.E.; Karandikar, N.J.; Malter, J.S.; et al. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat. Commun. 2014, 2, 4802. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, Y.; Wood, C.G.; Yang, H.; Zhao, H.; Ye, Y.; Gu, J.; Lin, J.; Habuchi, T.; Wu, X. Single nucleotide polymorphisms of microRNA machinery genes modify the risk of renal cell carcinoma. Clin. Cancer Res. 2008, 14, 7956–7962. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; An, J.; Lin, J.; Liu, Y.; Bao, L.; Zhang, W.; Zhao, J.J. Single nucleotide polymorphisms of microRNA processing machinery genes and outcome of hepatocellular carcinoma. PLoS ONE 2014, 9, e92791. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.L.; Cui, R.; Zhou, J.; Teng, K.Y.; Hsiao, Y.H.; Nakanishi, K.; Fassan, M.; Luo, Z.; Shi, G.; Tili, E.; et al. ERK Activation Globally Downregulates miRNAs through Phosphorylating Exportin-5. Cancer Cell 2016, 30, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Corrigendum: Global quantification of mammalian gene expression control. Nature 2013, 495, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Harries, L.W. Circular RNAs (circRNAs) in Health and Disease. Genes 2017, 8. [Google Scholar] [CrossRef]
- Ghosal, S.; Das, S.; Sen, R.; Basak, P.; Chakrabarti, J. Circ2Traits: A comprehensive database for circular RNA potentially associated with disease and traits. Front. Genet. 2013, 4, 283. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Han, P.; Zhou, T.; Guo, X.; Song, X.; Li, Y. circRNADb: A comprehensive database for human circular RNAs with protein-coding annotations. Sci. Rep. 2016, 6, 34985. [Google Scholar] [CrossRef] [PubMed]
- Glazar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G.; et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016, 7, 11215. [Google Scholar] [CrossRef] [PubMed]
- Boeckel, J.N.; Jae, N.; Heumuller, A.W.; Chen, W.; Boon, R.A.; Stellos, K.; Zeiher, A.M.; John, D.; Uchida, S.; Dimmeler, S. Identification and Characterization of Hypoxia-Regulated Endothelial Circular RNA. Circ. Res. 2015, 117, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.A.; Chang, H.Y. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell 2014, 14, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Volinia, S.; Papaioannou, D.; Nicolet, D.; Kohlschmidt, J.; Yan, P.S.; Mrozek, K.; Bucci, D.; Carroll, A.J.; Baer, M.R.; et al. Expression and prognostic impact of lncRNAs in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, 18679–18684. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell. 2016, 29, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011, 472, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yan, T.; Chai, Y.; Jiang, Y.; Zhu, X. Long noncoding RNA HOTTIP as an independent prognostic marker in cancer. Clin. Chim. Acta 2017. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.F.; Yin, Q.F.; Chen, T.; Zhang, Y.; Zhang, X.O.; Wu, Z.; Zhang, S.; Wang, H.B.; Ge, J.; Lu, X.; et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014, 24, 513–531. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, C.; Cech, T.R. The recruitment of chromatin modifiers by long noncoding RNAs: Lessons from PRC2. RNA 2015, 21, 2007–2022. [Google Scholar] [CrossRef] [PubMed]
- Hock, H. A complex Polycomb issue: The two faces of EZH2 in cancer. Genes Dev. 2012, 26, 751–755. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Cai, Q.; Sun, F.; Zhong, G.; Wang, P.; Liu, H.; Luo, J.; Yu, H.; Huang, J.; Lin, T. linc-UBC1 physically associates with polycomb repressive complex 2 (PRC2) and acts as a negative prognostic factor for lymph node metastasis and survival in bladder cancer. Biochim. Biophys. Acta 2013, 1832, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long noncoding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.K.; Lee, J.H.; Kim, W.K.; Yun, S.; Youn, Y.H.; Park, C.H.; Choi, Y.Y.; Kim, H.; Lee, S.K. Promoter methylation of PCDH10 by HOTAIR regulates the progression of gastrointestinal stromal tumors. Oncotarget 2016, 7, 75307–75318. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, L.; Zheng, S. Role of the long non-coding RNA HOTAIR in hepatocellular carcinoma. Oncol. Lett. 2017, 14, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T. Lessons from X-chromosome inactivation: Long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009, 23, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Ma, J.; Xue, Y.; Wang, P.; Li, Z.; Liu, J.; Chen, L.; Xi, Z.; Teng, H.; Wang, Z.; et al. Knockdown of long non-coding RNA XIST exerts tumor-suppressive functions in human glioblastoma stem cells by up-regulating miR-152. Cancer Lett. 2015, 359, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhou, Y.; Luo, X.; Gao, H.; Deng, X.; Jiang, Y. Long non-coding RNA XIST promotes cell growth and invasion through regulating miR-497/MACC1 axis in gastric cancer. Oncotarget 2017, 8, 4125–4135. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.H.; Yao, R.W.; Zhang, Y.; Guo, C.J.; Jiang, S.; Xu, G.; Dong, R.; Yang, L.; Chen, L.L. SLERT Regulates DDX21 Rings Associated with Pol I Transcription. Cell 2017, 169, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Nguyen le, X.T.; Raval, A.; Garcia, J.S.; Mitchell, B.S. Regulation of ribosomal gene expression in cancer. J. Cell. Physiol. 2015, 230, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hammerle, M.; Diederichs, S. MALAT1—A paradigm for long noncoding RNA function in cancer. J. Mol. Med. 2013, 91, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Sun, L.; Liu, Q.; Gong, C.; Yao, Y.; Lv, X.; Lin, L.; Yao, H.; Su, F.; Li, D.; et al. A cytoplasmic NF-κB interacting long noncoding RNA blocks IκB phosphorylation and suppresses breast cancer metastasis. Cancer Cell 2015, 27, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Cui, X.; Chen, J.; Feng, Y.; Song, E.; Li, J.; Liu, Y. Long non-coding RNA NKILA inhibits migration and invasion of tongue squamous cell carcinoma cells via suppressing epithelial-mesenchymal transition. Oncotarget 2016, 7, 62520–62532. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Li, Y.; Wang, J.; Che, Y.; Sun, S.; Huang, J.; Chen, Z.; He, J. Long non-coding RNA NKILA inhibits migration and invasion of non-small cell lung cancer via NF-κB/Snail pathway. J. Exp. Clin. Cancer Res. 2017, 36, 54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shields, T.; Crenshaw, T.; Hao, Y.; Moulton, T.; Tycko, B. Imprinting of human H19: Allele-specific CpG methylation, loss of the active allele in Wilms tumor, and potential for somatic allele switching. Am. J. Hum. Genet. 1993, 53, 113–124. [Google Scholar] [PubMed]
- Luo, J.H.; Ren, B.; Keryanov, S.; Tseng, G.C.; Rao, U.N.; Monga, S.P.; Strom, S.; Demetris, A.J.; Nalesnik, M.; Yu, Y.P.; et al. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology 2006, 44, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- De Kok, J.B.; Verhaegh, G.W.; Roelofs, R.W.; Hessels, D.; Kiemeney, L.A.; Aalders, T.W.; Swinkels, D.W.; Schalken, J.A. DD3(PCA3), a very sensitive and specific marker to detect prostate tumors. Cancer Res. 2002, 62, 2695–2698. [Google Scholar] [PubMed]
- Mei, D.; Song, H.; Wang, K.; Lou, Y.; Sun, W.; Liu, Z.; Ding, X.; Guo, J. Up-regulation of SUMO1 pseudogene 3 (SUMO1P3) in gastric cancer and its clinical association. Med. Oncol. 2013, 30, 709. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L. Linking Long Noncoding RNA Localization and Function. Trends Biochem. Sci. 2016, 41, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jin, X.; Deng, X.; Zhang, G.; Zhang, B.; Ma, L. The putative tumor suppressor microRNA-497 modulates gastric cancer cell proliferation and invasion by repressing eIF4E. Biochem. Biophys. Res. Commun. 2014, 449, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, Y.H.; Yao, Y.L.; Li, Z.; Li, Z.Q.; Ma, J.; Xue, Y.X. Long non-coding RNA CASC2 suppresses malignancy in human gliomas by miR-21. Cell. Signal. 2015, 27, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Sun, M.; Nie, F.Q.; Ge, Y.B.; Zhang, E.B.; Yin, D.D.; Kong, R.; Xia, R.; Lu, K.H.; Li, J.H.; et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol. Cancer 2014, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Huang, J.; Zhou, N.; Zhang, Z.; Zhang, A.; Lu, Z.; Wu, F.; Mo, Y.Y. LncRNA loc285194 is a p53-regulated tumor suppressor. Nucleic Acids Res. 2013, 41, 4976–4987. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhao, W.; Lu, Z.; Zhang, W.; Yang, X. Long non-coding RNA GAS5 promotes proliferation, migration and invasion by regulation of miR-301a in esophageal cancer. Oncol. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hu, B.; Wang, Q.; Ye, M.; Qiu, Q.; Zhou, Y.; Zeng, F.; Zhang, X.; Guo, Y.; Guo, L. Long non-coding RNA HOTTIP promotes BCL-2 expression and induces chemoresistance in small cell lung cancer by sponging miR-216a. Cell Death Dis. 2018, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wen, L.; Zhu, H. Unveiling the hidden function of long non-coding RNA by identifying its major partner-protein. Cell Biosci. 2015, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.; Wang, Y.; Lin, M.F.; Koegel, A.K.; Kotake, Y.; Grant, G.D.; Horlings, H.M.; Shah, N.; Umbricht, C.; Wang, P.; et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 2011, 43, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Jacq, C.; Miller, J.R.; Brownlee, G.G. A pseudogene structure in 5S DNA of Xenopus laevis. Cell 1977, 12, 109–120. [Google Scholar] [CrossRef]
- Milligan, M.J.; Lipovich, L. Pseudogene-derived lncRNAs: Emerging regulators of gene expression. Front. Genet. 2014, 5, 476. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Ackley, A.; Vidarsdottir, L.; Lui, W.O.; Corcoran, M.; Grander, D.; Morris, K.V. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat. Struct. Mol. Biol. 2013, 20, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.W.; Xie, M.; Sun, M.; Chen, T.Y.; Jin, R.R.; Ma, T.S.; Chen, Q.N.; Zhang, E.B.; He, X.Z.; De, W.; et al. The pseudogene derived long noncoding RNA DUXAP8 promotes gastric cancer cell proliferation and migration via epigenetically silencing PLEKHO1 expression. Oncotarget 2017, 8, 52211–52224. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.; Xu, Y.; Xiao, C.; Xia, R.; Gong, H.; Yang, P.; Chen, T.; Wu, D.; Cai, Z.; Zhang, J.; et al. The pseudogene derived from long non-coding RNA DUXAP10 promotes colorectal cancer cell growth through epigenetically silencing of p21 and PTEN. Sci. Rep. 2017, 7, 7312. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Wiklund, E.D.; Bramsen, J.B.; Villadsen, S.B.; Statham, A.L.; Clark, S.J.; Kjems, J. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011, 30, 4414–4422. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Kjems, J.; Damgaard, C.K. Circular RNA and miR-7 in cancer. Cancer Res. 2013, 73, 5609–5612. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Agarwal, V.; Guo, H.; Bartel, D.P. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014, 15, 409. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, L.; Li, W.; Deng, J.; Zheng, J.; An, M.; Lu, J.; Zhou, Y. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/beta-catenin pathway. Oncotarget 2015, 6, 6001–6013. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ma, K.; Sun, M.; Shi, S. Identification of the tumor-suppressive function of circular RNA ITCH in glioma cells through sponging miR-214 and promoting linear ITCH expression. Am. J. Transl. Res. 2018, 10, 1373–1386. [Google Scholar] [PubMed]
- Du, W.W.; Yang, W.; Liu, E.; Yang, Z.; Dhaliwal, P.; Yang, B.B. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016, 44, 2846–2858. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Du, W.W.; Li, X.; Yee, A.J.; Yang, B.B. Foxo3 activity promoted by non-coding effects of circular RNA and Foxo3 pseudogene in the inhibition of tumor growth and angiogenesis. Oncogene 2016, 35, 3919–3931. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.Y. Roles of the circular RNA circ-Foxo3 in breast cancer progression. Cell Cycle 2017, 16, 589–590. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, Q.; Bao, C.; Li, S.; Guo, W.; Zhao, J.; Chen, D.; Gu, J.; He, X.; Huang, S. Circular RNA is enriched and stable in exosomes: A promising biomarker for cancer diagnosis. Cell Res. 2015, 25, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Wang, L.; Sliz, P.; Gregory, R.I. A Biogenesis Step Upstream of Microprocessor Controls miR-17 approximately 92 Expression. Cell 2015, 162, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Olive, V.; Sabio, E.; Bennett, M.J.; De Jong, C.S.; Biton, A.; McGann, J.C.; Greaney, S.K.; Sodir, N.M.; Zhou, A.Y.; Balakrishnan, A.; et al. A component of the mir-17-92 polycistronic oncomir promotes oncogene-dependent apoptosis. Elife 2013, 2, e00822. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, D.; Gacquer, D.; Rothe, F.; Lefort, A.; Libert, F.; Brown, D.; Kheddoumi, N.; Shlien, A.; Konopka, T.; Salgado, R.; et al. Principles Governing A-to-I RNA Editing in the Breast Cancer Transcriptome. Cell Rep. 2015, 13, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.D.; Kim, T.T.; Walsh, T.; Kobayashi, Y.; Matise, T.C.; Buyske, S.; Gabriel, A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004, 14, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.; Lagergren, J.; Ohman, M. RNA editing of non-coding RNA and its role in gene regulation. Biochimie 2015, 117, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Veroni, C.; Marnetto, F.; Granieri, L.; Bertolotto, A.; Ballerini, C.; Repice, A.M.; Schirru, L.; Coghe, G.; Cocco, E.; Anastasiadou, E.; et al. Immune and Epstein-Barr virus gene expression in cerebrospinal fluid and peripheral blood mononuclear cells from patients with relapsing-remitting multiple sclerosis. J. Neuroinflamm. 2015, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, S.; Galeano, F.; Alon, S.; Raho, S.; Galardi, S.; Polito, V.A.; Presutti, C.; Vincenti, S.; Eisenberg, E.; Locatelli, F.; et al. Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol. 2015, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Nemlich, Y.; Greenberg, E.; Ortenberg, R.; Besser, M.J.; Barshack, I.; Jacob-Hirsch, J.; Jacoby, E.; Eyal, E.; Rivkin, L.; Prieto, V.G.; et al. MicroRNA-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. J. Clin. Investig. 2013, 123, 2703–2718. [Google Scholar] [CrossRef] [PubMed]
- Shoshan, E.; Mobley, A.K.; Braeuer, R.R.; Kamiya, T.; Huang, L.; Vasquez, M.E.; Salameh, A.; Lee, H.J.; Kim, S.J.; Ivan, C.; et al. Reduced adenosine-to-inosine miR-455-5p editing promotes melanoma growth and metastasis. Nat. Cell Biol. 2015, 17, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Kuang, L.; Lv, G.; Wang, B.; Li, L.; Dai, Y.; Li, Y. Overexpression of adenosine deaminase acting on RNA 1 in chordoma tissues is associated with chordoma pathogenesis by reducing miR125a and miR10a expression. Mol. Med. Rep. 2015, 12, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Kikuchi, J.; Furukawa, Y. Histone deacetylase 1 enhances microRNA processing via deacetylation of DGCR8. EMBO Rep. 2012, 13, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Phan, R.T.; Dalla-Favera, R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 2004, 432, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Calin, G.A.; Villanueva, A.; Ropero, S.; Sánchez-Céspedes, M.; Blanco, D.; Montuenga, L.M.; Rossi, S.; Nicoloso, M.S.; Faller, W.J.; et al. A microRNA DNA methylation signature for human cancer metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 13556–13561. [Google Scholar] [CrossRef] [PubMed]
- He, D.X.; Gu, F.; Gao, F.; Hao, J.J.; Gong, D.; Gu, X.T.; Mao, A.Q.; Jin, J.; Fu, L.; Ma, X. Genome-wide profiles of methylation, microRNAs, and gene expression in chemoresistant breast cancer. Sci. Rep. 2016, 6, 24706. [Google Scholar] [CrossRef] [PubMed]
- Ameres, S.L.; Horwich, M.D.; Hung, J.H.; Xu, J.; Ghildiyal, M.; Weng, Z.; Zamore, P.D. Target RNA-directed trimming and tailing of small silencing RNAs. Science 2010, 328, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Mullen, T.E.; Marzluff, W.F. Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5′ to 3′ and 3′ to 5′. Genes Dev. 2008, 22, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Dahlberg, J.E. Cyclic 2′,3′-phosphates and nontemplated nucleotides at the 3′-end of spliceosomal U6 small nuclear RNA’s. Science 1992, 255, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Norbury, C.J. 3′ Uridylation and the regulation of RNA function in the cytoplasm. Biochem. Soc. Trans. 2010, 38, 1150–1153. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Patena, W.; Leavitt, A.D.; McManus, M.T. Widespread RNA 3′-end oligouridylation in mammals. RNA 2012, 18, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.A.; Mani, V.; Hammond, S.M. Deep sequencing of microRNA precursors reveals extensive 3′-end modification. RNA 2011, 17, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Heo, I.; Joo, C.; Kim, Y.K.; Ha, M.; Yoon, M.J.; Cho, J.; Yeom, K.H.; Han, J.; Kim, V.N. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 2009, 138, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Ustianenko, D.; Hrossova, D.; Potesil, D.; Chalupnikova, K.; Hrazdilova, K.; Pachernik, J.; Cetkovska, K.; Uldrijan, S.; Zdrahal, Z.; Vanacova, S. Mammalian DIS3L2 exoribonuclease targets the uridylated precursors of let-7 miRNAs. RNA 2013, 19, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Greimann, J.C.; Lima, C.D. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell 2006, 127, 1223–1237. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.M.; Triboulet, R.; Thornton, J.E.; Gregory, R.I. A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature 2013, 497, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Pirouz, M.; Du, P.; Munafò, M.; Gregory, R.I. Dis3l2-Mediated Decay Is a Quality Control Pathway for Noncoding RNAs. Cell Rep. 2016, 16, 1861–1873. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.E.; Gregory, R.I. How does Lin28 let-7 control development and disease? Trends Cell. Biol. 2012, 22, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Salzman, D.W.; Nakamura, K.; Nallur, S.; Dookwah, M.T.; Metheetrairut, C.; Slack, F.J.; Weidhaas, J.B. miR-34 activity is modulated through 5′-end phosphorylation in response to DNA damage. Nat. Commun. 2016, 7, 10954. [Google Scholar] [CrossRef] [PubMed]
- Welch, C.; Chen, Y.; Stallings, R.L. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 2007, 26, 5017–5022. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, N.J. Ending the message: Poly(A) signals then and now. Genes Dev. 2011, 25, 1770–1782. [Google Scholar] [CrossRef] [PubMed]
- Majoros, W.H.; Ohler, U. Spatial preferences of microRNA targets in 3′ untranslated regions. BMC Genom. 2007, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Bartel, D.P. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009, 138, 673. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, C.J.; Wormington, M.; Peltz, S.W. The cap-to-tail guide to mRNA turnover. Nat. Rev. Mol. Cell Biol. 2001, 2, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: MicroRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Liu, L.Z.; Addison, J.B.; Wonderlin, W.F.; Ivanov, A.V.; Ruppert, J.M. A KLF4-miRNA-206 autoregulatory feedback loop can promote or inhibit protein translation depending upon cell context. Mol. Cell. Biol. 2011, 31, 2513–2527. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J. From birth to death: The complex lives of eukaryotic mRNAs. Science 2005, 309, 1514–1518. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kogan, N.; Slack, F.J. Cis-acting elements in its 3′UTR mediate post-transcriptional regulation of KRAS. Oncotarget 2016, 7, 11770–11784. [Google Scholar] [CrossRef] [PubMed]
- Barrett, L.W.; Fletcher, S.; Wilton, S.D. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell. Mol. Life Sci. 2012, 69, 3613–3634. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.J.; Furneaux, H. Localization of the human HuR gene to chromosome 19p13.2. Hum. Genet. 1997, 99, 32–33. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.C.; Steitz, J.A. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. 1998, 17, 3448–3460. [Google Scholar] [CrossRef] [PubMed]
- Quenault, T.; Lithgow, T.; Traven, A. PUF proteins: Repression, activation and mRNA localization. Trends Cell Biol. 2011, 21, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Kedde, M.; van Kouwenhove, M.; Zwart, W.; Oude Vrielink, J.A.; Elkon, R.; Agami, R. A Pumilio-induced RNA structure switch in p27-3′UTR controls miR-221 and miR-222 accessibility. Nat. Cell Biol. 2010, 12, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Miles, W.O.; Tschop, K.; Herr, A.; Ji, J.Y.; Dyson, N.J. Pumilio facilitates miRNA regulation of the E2F3 oncogene. Genes Dev. 2012, 26, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Kedde, M.; Strasser, M.J.; Boldajipour, B.; Oude Vrielink, J.A.; Slanchev, K.; le Sage, C.; Nagel, R.; Voorhoeve, P.M.; van Duijse, J.; Orom, U.A.; et al. RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell 2007, 131, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Dutta, A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007, 21, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Preskill, C.; Weidhaas, J.B. SNPs in microRNA Binding Sites as Prognostic and Predictive Cancer Biomarkers. Crit. Rev. Oncog. 2013, 18, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Li, Z.; Jolicoeur, N.; Zhang, L.; Fortin, Y.; Wang, E.; Wu, M.; Shen, S.H. Aberrant allele frequencies of the SNPs located in microRNA target sites are potentially associated with human cancers. Nucleic Acids Res. 2007, 35, 4535–4541. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.J.; Ratner, E.; Leng, S.; Zhai, R.; Nallur, S.; Babar, I.; Muller, R.U.; Straka, E.; Su, L.; Burki, E.A.; et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008, 68, 8535–8540. [Google Scholar] [CrossRef] [PubMed]
- Wynendaele, J.; Bohnke, A.; Leucci, E.; Nielsen, S.J.; Lambertz, I.; Hammer, S.; Sbrzesny, N.; Kubitza, D.; Wolf, A.; Gradhand, E.; et al. An illegitimate microRNA target site within the 3′UTR of MDM4 affects ovarian cancer progression and chemosensitivity. Cancer Res. 2010, 70, 9641–9649. [Google Scholar] [CrossRef] [PubMed]
- Nicoloso, M.S.; Sun, H.; Spizzo, R.; Kim, H.; Wickramasinghe, P.; Shimizu, M.; Wojcik, S.E.; Ferdin, J.; Kunej, T.; Xiao, L.; et al. Single-nucleotide polymorphisms inside microRNA target sites influence tumor susceptibility. Cancer Res. 2010, 70, 2789–2798. [Google Scholar] [CrossRef] [PubMed]
- Guarnerio, J.; Bezzi, M.; Jeong, J.C.; Paffenholz, S.V.; Berry, K.; Naldini, M.M.; Lo-Coco, F.; Tay, Y.; Beck, A.H.; Pandolfi, P.P. Oncogenic Role of Fusion-circRNAs Derived from Cancer-Associated Chromosomal Translocations. Cell 2016, 166, 1055–1056. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, G.A.; Kats, L.; Pandolfi, P.P. Synergy against PML-RARa: Targeting transcription, proteolysis, differentiation, and self-renewal in acute promyelocytic leukemia. J. Exp. Med. 2013, 210, 2793–2802. [Google Scholar] [CrossRef] [PubMed]
- Martelli, M.P.; Sozzi, G.; Hernandez, L.; Pettirossi, V.; Navarro, A.; Conte, D.; Gasparini, P.; Perrone, F.; Modena, P.; Pastorino, U.; et al. EML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissues. Am. J. Pathol. 2009, 174, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Dong, Z.; Shi, Y.; Liu, S.; Liang, J.; Guo, Y.; Guo, X.; Shen, S.; Shan, B. Aberrant methylation-mediated downregulation of long noncoding RNA LOC100130476 correlates with malignant progression of esophageal squamous cell carcinoma. Dig. Liver Dis. 2016, 48, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Liu, S.; Dong, Z.; Guo, Y.; Ding, C.; Shen, S.; Liang, J.; Shan, B. Aberrant methylation-mediated silencing of lncRNA CTC-276P9.1 is associated with malignant progression of esophageal squamous cell carcinoma. Clin. Exp. Metastasis 2018, 35, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Zhang, A.; Liu, S.; Lu, F.; Guo, Y.; Zhang, G.; Xu, F.; Shi, Y.; Shen, S.; Liang, J.; et al. Aberrant Methylation-Mediated Silencing of lncRNA MEG3 Functions as a ceRNA in Esophageal Cancer. Mol. Cancer Res. 2017, 15, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y. Polymorphisms in lncRNA PTENP1 and the Risk of Gastric Cancer in a Chinese Population. Dis. Markers 2017, 2017, 6807452. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Tang, R.; Ma, X.; Wang, Y.; Luo, D.; Xu, Z.; Zhu, Y.; Yang, L. Tag SNPs in long non-coding RNA H19 contribute to susceptibility to gastric cancer in the Chinese Han population. Oncotarget 2015, 6, 15311–15320. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anastasiadou, E.; Faggioni, A.; Trivedi, P.; Slack, F.J. The Nefarious Nexus of Noncoding RNAs in Cancer. Int. J. Mol. Sci. 2018, 19, 2072. https://doi.org/10.3390/ijms19072072
Anastasiadou E, Faggioni A, Trivedi P, Slack FJ. The Nefarious Nexus of Noncoding RNAs in Cancer. International Journal of Molecular Sciences. 2018; 19(7):2072. https://doi.org/10.3390/ijms19072072
Chicago/Turabian StyleAnastasiadou, Eleni, Alberto Faggioni, Pankaj Trivedi, and Frank J. Slack. 2018. "The Nefarious Nexus of Noncoding RNAs in Cancer" International Journal of Molecular Sciences 19, no. 7: 2072. https://doi.org/10.3390/ijms19072072
APA StyleAnastasiadou, E., Faggioni, A., Trivedi, P., & Slack, F. J. (2018). The Nefarious Nexus of Noncoding RNAs in Cancer. International Journal of Molecular Sciences, 19(7), 2072. https://doi.org/10.3390/ijms19072072