Melatonin in Prevention of the Sequence from Reflux Esophagitis to Barrett’s Esophagus and Esophageal Adenocarcinoma: Experimental and Clinical Perspectives



 ,
, 
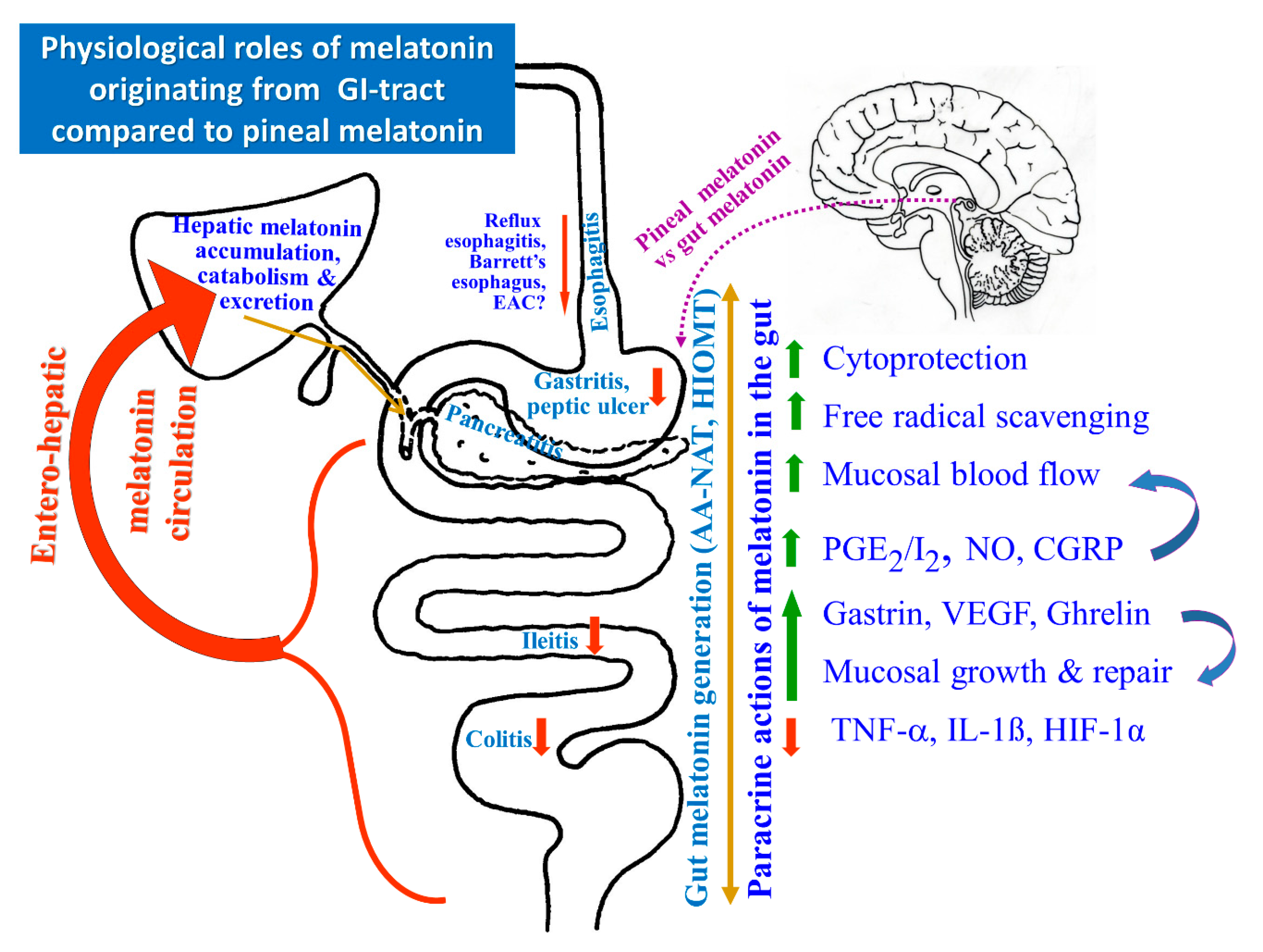{kind=link}
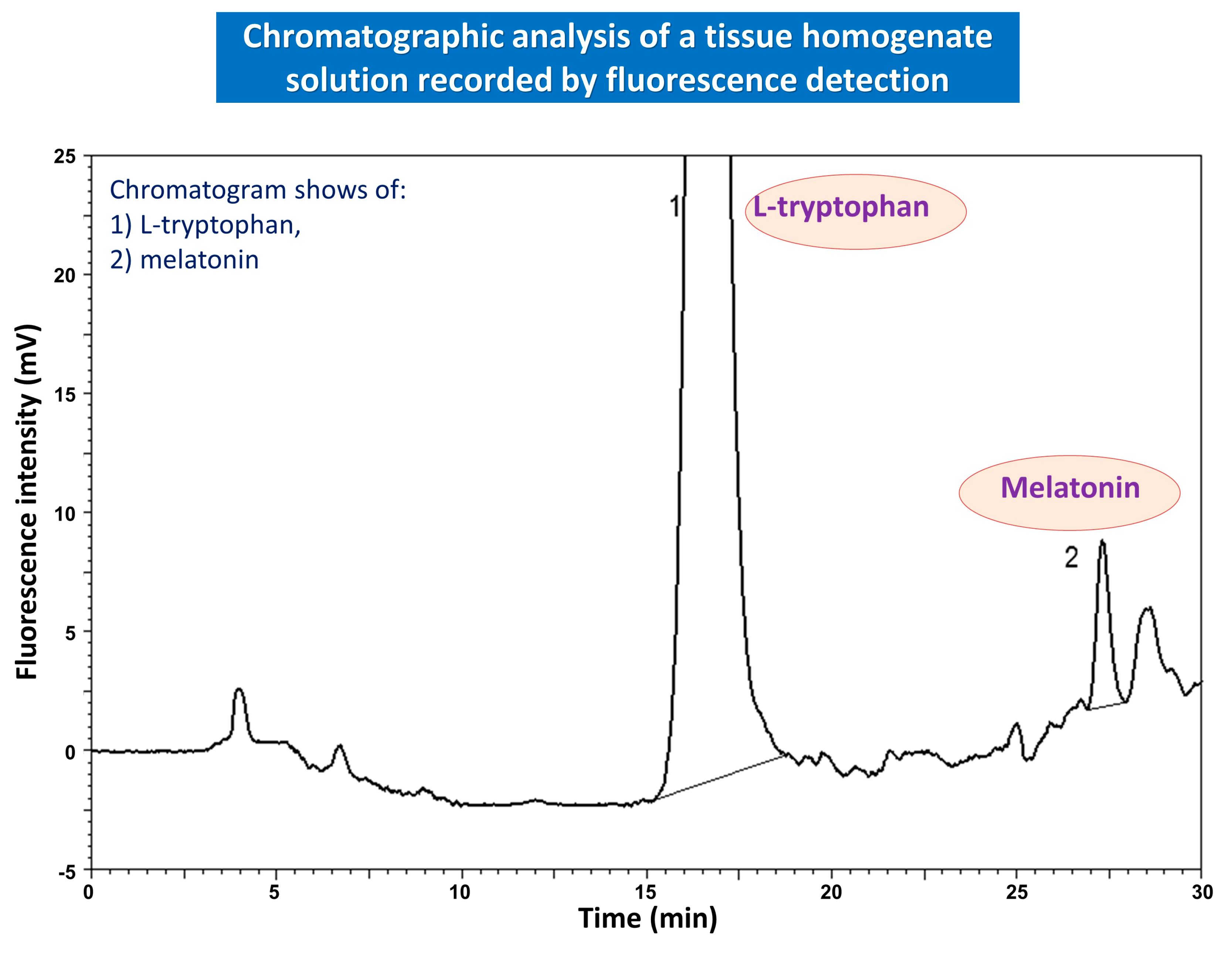{kind=link}
{kind=link}
Abstract
1. Anti-Reflux Esophageal Barrier and Pathogenesis of Esophageal Damage Induced by Gastroduodenal Reflux
2. Reflux Esophagitis in Animal Models and the Process of Progression of Mucosal Changes Caused by Reflux Esophagitis into Barrett’s Esophagus and Esophageal Adenocarcinoma
3. Role of Melatonin in Esophagoprotection and Prevention of Chronic Esophageal Disorders
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dent, J.; El-Serag, H.B.; Wallander, M.A.; Johansson, S. Epidemiology of gastroesophageal reflux disease: A systemic review. Gut 2005, 54, 710–717. [Google Scholar] [CrossRef] [PubMed]
- El-Searag, H.B. Time trends of gastroesophageal reflux disease: A systemic review. Clin. Gastroenterol. Hepatol. 2007, 5, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Vakil, N. Disease definition, clinical manifestations, epidemiology and natural history of GERD. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Boeckxstaens, G.E. Review article: The pathophysiology of gastro-esophageal reflux disease. Aliment. Pharmacol. Ther. 2007, 26, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Farre, R. Pathophysiology of gastro-esophageal reflux disease: A role for mucosa integrity? Neurogastroenterol. Motil. 2013, 25, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Orlando, R.C. The integrity of the esophageal mucosa. Balance between offensive and defensive mechanisms. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, K.; Yamoto, M.; Nishio, H.; Takeuchi, K. Essential role of pepsin in pathogenesis of acid reflux esophagitis in rats. Dig. Dis. Sci. 2006, 51, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Nagahama, K. Animal model of acid-reflux esophagitis: Pathogenic roles of acid/pepsin, prostaglandins, and amino acids. BioMed Res. Int. 2014, 2014, 532594. [Google Scholar] [CrossRef] [PubMed]
- Zayachkivska, O.; Pshyk-Titko, I.; Hrycevych, N.; Savytska, M. New insight into oseophageal injury and protection in physiologically relevant animal models. J. Physiol. Pharmacol. 2014, 65, 295–307. [Google Scholar] [PubMed]
- Naito, Y.; Uchiyama, K.; Kuroda, M.; Takagi, T.; Kokura, S.; Yoshida, N.; Ichikawa, H.; Yoshikawa, T. Role of pancreatic trypsin in chronic esophagitis induced by gastroduodenal reflux in rats. J. Gastroenterol. 2006, 41, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Kikendall, J.W.; Friedman, A.C.; Oyewole, M.A.; Fleischer, D.; Johnson, L.F. Pill-induced esophageal injury: Case reports and review of medical literature. Dig. Dis. Sci. 1983, 28, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Kahrilas, P.J.; Gupta, R.R. The effect of cigarette smoking on salivation and esophageal acid clearance in clinical esophagitis. J. Lab. Clin. Med. 1989, 114, 431–438. [Google Scholar] [PubMed]
- Safaie-Shirazi, S.; Zike, W.L.; Brubacher, M.; Den Bensten, L. Effect of aspirin, alcohol, and pepsin on mucosal permeability of esophageal mucosa. Surg. Forum 1974, 25, 335–337. [Google Scholar] [PubMed]
- Heller, S.R.; Fellows, I.W.; Ogilvie, A.L.; Atkinson, M. Non-steroidal anti-inflammatory drugs and benign oesophageal stricture. Br. Med. J. 1982, 285, 167–168. [Google Scholar] [CrossRef]
- Lanas, A.; Hirschowitz, B.I. Significant role of aspirin use in patients with esophagitis. J. Clin. Gastroenterol. 1991, 13, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, M.; Pajdo, R.; Kwiecien, S.; Ptak-Belowska, A.; Sliwowski, Z.; Mazurkiewicz-Janik, M.; Konturek, S.J.; Pawlik, W.W.; Brzozowski, T. Nitric oxide (NO)-releasing aspirin exhibits a potent esophagoprotection in experimental model of acute reflux esophagitis. Role of nitric oxide and proinflammatory cytokines. J. Physiol. Pharmacol. 2011, 62, 75–86. [Google Scholar] [PubMed]
- Tobey, N.A.; Hossein, S.S.; Argote, C.M.; Dobrucali, A.M.; Awayda, M.S.; Orlando, R.C. Dilated intercellular spaces and shunt permeability in non-erosive acid-damaged esophageal epithelium. Am. J. Gastroenterol. 2004, 9, 13–22. [Google Scholar] [CrossRef]
- Abdulnour-Nakhoul, S.; Nakhoul, N.L.; Wheeler, S.A.; Wang, P.; Swenson, E.R.; Orlando, R.C. HCO3− secretion in the esophageal submucosal glands. Am. J. Physiol. 2006, 288, G736–G744. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Higuchi, K.; Takashima, T.; Hamaguchi, M.; Hayakawa, T.; Tominaga, K.; Watanabe, T.; Oshitani, N.; Shimada, Y.; Arakawa, T. Roles of epidermal growth factor and Na+/H+ exchanger-1 in esophageal epithelial defense against acid-induced injury. Am. J. Physiol. 2006, 290, G665–G673. [Google Scholar] [CrossRef] [PubMed]
- Kaunitz, J.D.; Tanaka, S.; Akiba, Y.D. Gastroduodenal microcirculatory response to luminal acid. In Organ Microcirculation. A Gateway to Diagnostic and Therapeutic Interventions. Series: Keio University International Symposia for Life Sciences and Medicine; Ishii, H., Suematsu, M., Tanishita, K., Suzuki, H., Eds.; Springer: Berlin, Germany, 2005; Volume 13, pp. 79–88. [Google Scholar]
- Tarnawski, A.S.; Chai, J.; Jones, M.K. Esophageal and gastrointestinal microcirculation: Essential for mucosal protection, a target for injury, and critical component of injury and ulcer healing. In Organ Microcirculation. A Gateway to Diagnostic and Therapeutic Interventions. Series: Keito University International Symposia for Life Sciences and Medicine; Ishii, H., Suemastsu, M., Tanishita, K., Suzuki, K., Eds.; Springer: Berlin, Germany, 2005; Volume 13, pp. 49–61. [Google Scholar]
- Hollwarth, M.E.; Smith, M.E.; Kvietys, P.R.; Granger, D.N. Esophageal blood flow in the cat. Normal distribution and effects of acid perfusion. Gastroenterology 1986, 90, 622–628. [Google Scholar] [CrossRef]
- Bass, B.L.; Schweitzer, E.J.; Harmon, J.W.; Kraimer, J. H+ back diffusion interferes with intrinsic reactive regulation of esophageal mucosal blood flow. Surgery 1984, 96, 404–413. [Google Scholar] [PubMed]
- Szentpali, K.; Eros, G.; Kaszaki, J.; Tiszlavicz, L.; Lazar, G.; Wolfard, A.; Balogh, A.; Boros, M. Microcirculatory changes in the canine oesophageal mucosa during experimental reflux oesophagitis: Comparison of the effects of acid and bile. Scand. J. Gastroenterol. 2003, 38, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Mine, S.; Iida, T.; Tabata, T.; Okada, Y.; Tanaka, Y. Increased esophageal mucosal/submucosal blood flow in patients with gastroesophageal reflux disease: Normalization by treatmentwith proton pump inhibitor. J. Gastroenterol. Hepatol. 2008, 23, 303–309. [Google Scholar] [CrossRef] [PubMed]
- McKie, L.D.; Dunkin, B.J.; Pennanen, M.F.; Dunlap, K.W.; Harmon, J.W.; Bass, B.L. Esophageal blood flow: A central role for calcitonin gene related peptide. Surgery 1994, 116, 409–437. [Google Scholar] [PubMed]
- Holzer, P. Role of visceral afferent neurons in mucosal inflammation. Curr. Opin. Pharmacol. 2007, 7, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Bass, B.L.; Trad, K.S.; Harmon, J.W.; Hakki, F.Z. Capsaicin sensitive nerves mediate esophageal mucosal protection. Surgery 1991, 110, 419–425. [Google Scholar] [PubMed]
- Konturek, P.C.; Brzozowska, I.; Targosz, A.; Pawlik, M.; Kania, J.; Hess, T.; Kwiecien, S.; Konturek, S.J.; Reiter, R.J.; Brzozowski, T. Esophagoprotection mediated by exogenous and endogenous melatonin in an experimental model of reflux esophagitis. J. Pineal Res. 2013, 55, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H.; Fuse, Y.; Fujino, K.H.; Kodama, T. Healing process of experimental esophageal ulcers induced by acetic acid in rats. Scand. J. Gastroenterol. 1989, 24, 6–10. [Google Scholar] [CrossRef]
- Tsukimi, Y.; Okabe, S. Validity of kissing gastric ulcers induced in rats for screening of antiulcer drugs. J. Gastroenterol. Hepatol. 1994, 9, S60–S65. [Google Scholar] [CrossRef] [PubMed]
- Omura, N.; Kashiwagi, H.; Chen, G.; Suzuki, Y.; Yano, F.; Aoki, T. Establishment of surgically induced chronic acid reflux esophagitis in rats. Scand. J. Gastroenterol. 1999, 10, 948–953. [Google Scholar]
- Nishijima, K.; Miwa, K.; Miyashita, T.; Kinami, S.; Ninomiya, I.; Fushida, S.; Fujimura, T.; Hattori, T. Impact of the biliary diversion procedure on carcinogenesis in Barrett’s esophagus surgically induced by duodenoesophageal reflux in rats. Ann. Surg. 2004, 240, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.D.; Jung, K.H.; Ahn, W.S.; Bae, S.H.; Jang, T.J. Expression of inducible nitric oxide synthase is increased in rat Barrett’s esophagus induced by duodenal contents reflux. J. Korean Med. Sci. 2005, 20, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wo, J.M.; Su, R.R.; Ray, M.B.; Jones, W.; Martin, R.C.G. Esophageal injury with external esophageal perfusion. J. Surg. Res. 2005, 129, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Blot, W.J.; McLaughlin, J.K. The changing epidemiology of esophageal cancer. Semin. Oncol. 1999, 26, 2–8. [Google Scholar] [PubMed]
- Paull, A.; Trier, J.S.; Dalton, M.D.; Camp, R.C.; Loeb, P.; Goyal, R.K. The histologic spectrum of Barrett’s esophagus. N. Engl. J. Med. 1976, 295, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, N.; Ransohoff, D.F. Gastroesophageal reflux, Barrett esophagus and esophageal cancer: Scientific review. JAMA 2002, 287, 1972–1981. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.R.; Smith, R.R. The relationship between columnar epithelial dysplasia and invasive adenocarcinoma arising in Barrett’s esophagus. Am. J. Clin. Pathol. 1987, 87, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Haggitt, R.C.; Tryzelaar, J.; Ellis, F.H.; Colher, H. Adenocarcinoma complicating columnar epithelium-lined (Barrett’s) esophagus. Am. J. Clin. Pathol. 1978, 70, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lagergren, J.; Bergstrom, R.; Lingren, A.; Nyren, O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N. Engl. Med. 1999, 340, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Orlando, R.C.; Peterson, W.G.; Harnett, K.M.; Ma, J.; Behar, J.; Biancani, P.; Guarino, M.P.L.; Altomare, A.; Cicala, M.; Cao, W. Esophageal disease: Updated information on inflammation. Ann. N. Y. Acad. Sci. 2011, 1232, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Guarino, M.P.; Cocca, S.; Emerenziani, S.; Cicala, M. Gastroesophageal reflux disease: Update on inflammation and symptom perception. World J. Gastroenterol. 2013, 19, 6523–6528. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.F.; Shewmake, K.; Terada, L.S.; Spechler, S.J. Acid exposure activates the mitogen-activated protein kinase pathways in Barrett’s esophagus. Gastroenterology 2002, 122, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Fein, M.; Ireland, A.P.; Ritter, M.P.; Peters, J.H.; Hagen, J.A.; Bremner, C.G.; DeMeester, T.R. Duodenogastric reflux potentiate the injurious effects of gastroesophageal reflux. J. Gastrointest. Surg. 1997, 1, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Evett, G.E.; Xie, W.; Chipman, J.G.; Robertson, D.I.; Simmons, D.L. Prostaglandin G/H synthase isoenzyme 2 expression in fibroblasts: Regulation by dexamethasone, mitogens and oncogenes. Arch. Biochem. Biophys. 1993, 306, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.S.; Triadafilopoulos, G. Acid-and bile-induced PGE2 release and hyperproliferation in Barrett’s esophagus are COX-2 and PKC-epsilon dependent. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G327–G334. [Google Scholar] [CrossRef] [PubMed]
- Gately, S.; Li, W.W. Multiple roles of COX-2 in tumor angiogenesis: A target for antiangiogenetic therapy. Semin. Oncol. 2004, 31, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, H.; Vallbohmer, D.; Uchida, K.; Schneider, S.; Hamoui, N.; Shimizu, D.; Chandrasoma, P.T.; Demeester, T.R.; Danenberg, K.D.; Danenberg, P.V.; et al. Quantitative, tissue-specific analysis of cyclooxygenase gene expression in the pathogenesis of Barrett’s adenocarcinoma. J. Gastrointest. Surg. 2004, 8, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Tsujii, M.; DuBois, R.N. Alternations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin end peroxide synthase 2. Cell 1995, 83, 493–501. [Google Scholar] [CrossRef]
- Zimmermann, K.C.; Sarbia, M.; Weber, A.A.; Borchard, R.F.; Gabbert, H.E.; Schror, K. Cyclooxygenase-2 expression in human esophageal carcinoma. Cancer Res. 1999, 59, 198–204. [Google Scholar] [PubMed]
- Morgan, G. Deleterious effects of prostaglandin E2 in esophageal carcinogenesis. Med. Hypotheses 1997, 48, 177–181. [Google Scholar] [CrossRef]
- Funkhouser, E.M.; Sharp, G.B. Aspirin and reduced risk of esophageal carcinoma. Cancer 1995, 76, 1116–1119. [Google Scholar] [CrossRef]
- Sharma, P.; Morales, T.G.; Sampliner, R.E. Short segment Barrett’s esophagus—The need for standardization of the definition and of endoscopic criteria. Am. J. Gastroenterol. 1998, 93, 1033–1036. [Google Scholar] [PubMed]
- Caygill, C.P.; Watson, A.; Reed, P.I.; Hill, M.J.; UK National Barrett’s Oesophagus Registry (UKBOR) and the 27 Participating Centres. Characteristics and regional variations of patients with Barrett’s oesophagus in the UK. Eur. J. Gastroenterol. Hepatol. 2003, 15, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.J.; Lomboy, C.T.; Pera, M.; Carpenter, H.A. Adenocarcinoma of the esophagogastric junction and Barrett’s esophagus. Gastroenterology 1995, 109, 1541–1546. [Google Scholar] [CrossRef]
- Reid, B.J.; Blount, P.L.; Rabinovitch, P.S. Biomarkers in Barrett’s esophagus. Gastrointest. Endosc. Clin. N. Am. 2003, 13, 369–397. [Google Scholar] [CrossRef]
- Migaczewski, M.; Budzynski, A.; Rembiasz, K.; Ryszard, C. Quality of life of patients with gastroesophageal reflux disease after laparoscopic Nissen fundoplication. Videosurg. Miniinvasive Tech. 2008, 3, 119–125. [Google Scholar]
- Migaczewski, M.; Budzynski, A.; Rembiasz, K. Argon plasma coagulation (APC) for treatment of Barrett’s oesophagus. Videosurg. Miniinvasive Tech. 2009, 4, 102–109. [Google Scholar]
- Moore, R.Y. Circadian rhythms: Basic neurobiology and clinical applications. Annu. Rev. Med. 1997, 48, 253. [Google Scholar] [CrossRef] [PubMed]
- De Masi, L.; Castaldo, D.; Pignone, D.; Servillo, L. Facchiano A. Experimental evidence and in silico identification of tryptophan decarboxylase in citrus genus. Molecules 2017, 22, 272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Wang, R.; Liu, D.; Wu, Y.; Sun, J.; Tao, J. Melatonin and expression of tryptophan decarboxylase Gene (TDC) in Herbaceous Peony (Paeonia lactiflora Pall.) Flowers. Molecules 2018, 23, 1164. [Google Scholar] [CrossRef] [PubMed]
- Sudgen, D. Melatonin biosynthesis in the mammalian pineal gland. Experientia 1989, 45, 922–931. [Google Scholar]
- Raikhlin, N.T.; Kvetnoy, I.M. Melatonin and enterochromaffine cells. Acta Histochem. 1976, 55, 19–24. [Google Scholar] [CrossRef]
- Lee, P.P.N.; Pang, S.F. Melatonin and its receptors in the gastrointestinal tract. Biol. Signals 1993, 2, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Bubenik, G.A.; Pang, S.F.; Hacker, R.R.; Smith, P.S. Melatonin concentrations in serum and tissues of porcine gastrointestinal tract and their relationship to the intake and passage of food. J. Pineal Res. 1996, 21, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Bubenik, G.A.; Pang, S.F.; Cockshut, J.R.; Smith, P.S.; Grovum, L.W.; Friendship, R.M.; Hacker, R.R. Circadian variation of portent arterial and venous blood levels of melatonin in pigs and its relationship to food intake and sleep. J. Pineal Res. 2000, 28, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Huether, G.; Poegeller, G.; Reimer, R.; George, A. Effect of tryptophan administration on circulating melatonin levels in chicks and rats: Evidence for stimulation of melatonin synthesis and release in the gastrointestinal tract. Life Sci. 1992, 51, 945–953. [Google Scholar] [CrossRef]
- Kvetnoy, I.M.; Raikhlin, N.T.; Yuzhakov, V.V.; Ingel, I.E. Extrapineal melatonin and its role in neuroendocrine regulation of homeostasis. Bull. Exp. Biol. Med. 1999, 127, 329–334. [Google Scholar] [CrossRef]
- Raikhlin, N.T.; Kvetnoy, I.M.; Kadagidze, Z.G.; Sokolov, A.V. Immunomorphological studies on synthesis of melatonin in enterochromaffine cells. Acta Histochem. Cytochem. 1978, 11, 75–77. [Google Scholar] [CrossRef]
- Messner, M.; Huether, G.; Lorf, T.; Ramadori, G.; Schworer, H. Presence of melatonin in the human hepatobiliary-gastrointestinal tract. Life Sci. 2001, 69, 543–551. [Google Scholar] [CrossRef]
- Ma, X.; Idle, J.R.; Krausz, K.W.; Gonzales, F.J. Metabolism of melatonin by human cytochromes p450. Drug Metab. Dis. 2005, 33, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Pandi-Perumal, S.R.; Srinivasan, V.; Maestroni, G.J.M.; Cardinali, D.P.; Poeggeler, B.; Hardeland, R. Melatonin nature’s most versatile biological signal? FEBS J. 2006, 273, 2813–2838. [Google Scholar] [CrossRef] [PubMed]
- Bubenik, G.A. Therapeutic perspectives of gastrointestinal melatonin. In The Melatonin: From Molecules to Therapy; Pandi-Perumal, S.R., Cardinali, D.P., Eds.; Nova Science Publishers Inc.: Hauppauge, NY, USA, 2006; pp. 1–20. [Google Scholar]
- Konturek, S.J.; Konturek, P.C.; Brzozowska, I.; Pawlik, M.; Sliwowski, Z.; Cześnikiewicz-Guzik, M.; Kwiecień, S.; Brzozowski, T.; Bubenik, G.A.; Pawlik, W.W. Localization and biological activities of melatonin in intact and diseased gastrointestinal tract (GIT). J. Physiol. Pharmacol. 2007, 58, 381–405. [Google Scholar] [PubMed]
- Poon, A.M.; Chow, P.H.; Mak, A.S.; Pang, S.F. Autoradiographic localization of 2[125I]iodomelatonin binding sites in the gastrointestinal tract of mammals including humans and birds. J. Pineal Res. 1997, 23, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Bubenik, G.A.; Hacker, R.R.; Brown, G.M.; Bartos, L. Melatonin concentrations in the luminal fluid, mucosa and muscularis of the bovine and porcine gastrointestinal tract. J. Pineal Res. 1999, 29, 56–63. [Google Scholar] [CrossRef]
- Brzozowska, I.; Strzalka, M.; Drozdowicz, D.; Konturek, S.J.; Brzozowski, T. Mechanisms of esophageal protection, gastroprotection and ulcer healing by melatonin. Implications for the therapeutic use of melatonin in gastroesophageal reflux disease (GERD) and peptic ulcer disease. Curr. Pharm. Des. 2014, 20, 4807–4815. [Google Scholar] [CrossRef] [PubMed]
- De Souza Pereira, R. Regression of gastroesophageal reflux disease symptoms using dietary supplementation with melatonin, vitamins and amino acids: Comparison with omeprazole. J. Pineal Res. 2006, 41, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Patrick, L. Gastroesophageal reflux disease (GERD): A review of conventional and alternative treatments. Altern. Med. Rev. 2011, 16, 116–133. [Google Scholar] [PubMed]
- Konturek, S.J.; Zayachkiwska, O.; Hawryluk, O.; Havryluk, X.O.; Brzozowski, T.; Sliwowski, Z.; Pawlik, M.; Konturek, P.C.; Cześnikiewicz-Guzik, M.; Gzhegotsky, M.R.; et al. Protective influence of melatonin against acute esophageal lesions involves prostaglandins, nitric oxide and sensory nerves. J. Physiol. Pharmacol. 2007, 58, 371–387. [Google Scholar]
- Hamaguchi, M.; Fujiwara, Y.; Takashima, T.; Hayakawa, T.; Sasaki, E.; Shiba, M.; Watanabe, T.; Tominaga, K.; Oshitani, N.; Matsumoto, T.; et al. Increased expression of cytokines and adhesion molecules in rat chronic esophagitis. Digestion 2003, 68, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Klinkenberg-Knol, E.C.; Nelis, F.; Dent, J.; Snel, P.; Mitchell, B.; Prichard, P.; Lloyd, D.; Havu, N.; Frame, M.H.; Romàn, J.; et al. Long-term omeprazole treatment in resistant gastroesophageal reflux disease. Efficacy, safety, and influence on gastric mucosa. Gastroenterology 2000, 118, 661–669. [Google Scholar] [CrossRef]
- Abdalla, S.I.; Lao-Sirieix, P.; Novelli, M.R.; Lovat, L.B.; Sanderson, I.R.; Fitzgerald, R.C. Gastrin induced cyclooxygenase-2 expression in Barrett’s carcinogenesis. Clin. Cancer Res. 2004, 10, 4784–4792. [Google Scholar] [CrossRef] [PubMed]
- Boeckxstaens, G.E. Alterations confined to the gastro-oesophageal junction: The relationship between low LOSP, TLOSRs, hiatus hernia and acid pocket. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Wang, Y.; Xia, Y.; Zhang, N.; Sun, X.; Yu, T.; Lin, L. Melatonin protects the esophageal epithelial barrier by suppressing the transcription, expression and activity of myosin light chain kinase through ERK1/2 signal transduction. Cell. Physiol. Biochem. 2014, 34, 2117–2127. [Google Scholar] [CrossRef] [PubMed]
- Bubenik, G.A.; Konturek, S.J. Melatonin and aging: Prospects for human treatment. J. Physiol. Pharmacol. 2011, 62, 13–19. [Google Scholar] [PubMed]
- Madalinski, M.H. Does a melatonin supplement alter the course of gastro-esophageal reflux disease? World J. Gastrointest. Pharmacol. Ther. 2011, 2, 50–51. [Google Scholar] [CrossRef] [PubMed]
- Klupinska, G.A.; Wisniowska-Jarosinska, M.; Harsiuk, A.; Chojnacki, C.; Stec-Michalska, K.; Błasiak, J.; Reiter, R.J.; Chojnacki, J. Nocturnal secretion of melatonin in patients with upper digestive tract disorders. J. Physiol. Pharmacol. 2006, 57, 41–50. [Google Scholar] [PubMed]
- Lewy, A.J. Melatonin as a marker and phase-resetter of circadian rhythms in humans. Adv. Exp. Med. Biol. 1999, 460, 425–434. [Google Scholar] [PubMed]
- Buscemi, N.; Vandermeer, B.; Pandya, R.; Hooton, N.; Tjosvold, L.; Hartling, L.; Baker, G.; Vohra, S.; Klassen, T. Melatonin for treatment of sleep disorders. Evid. Rep. Technol. Assess. (Summ.) 2004, 108, 1–7. [Google Scholar]
- Middleton, B.A.; Stone, B.M.; Arendt, J. Melatonin and fragmented sleep patterns. Lancet 1996, 348, 551–552. [Google Scholar] [CrossRef]
- Rogers, N.L.; Phan, O.; Kennaway, D.J.; Dawson, D. Effect of day time oral melatonin administration on neurobehavioral performance in humans. J. Pineal Res. 1998, 25, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Calvo, J.R.; Guerrero, J.M.; Osuna, C.; Molinero, P.; Carrillo-Vico, A. Melatonin triggers Crohn’s disease symptoms. J. Pineal Res. 2002, 32, 277–278. [Google Scholar] [PubMed]
- Acuna-Castroviejo, D.; Escames, G.; Venegas, C.; Díaz-Casado, M.E.; Lima-Cabello, E.; López, L.C.; Rosales-Corral, S.; Tan, D.X.; Reiter, R.J. Extrapineal melatonin: Sources, regulation, and potential functions. Cell. Mol. Life Sci. 2014, 71, 2997–3025. [Google Scholar] [CrossRef] [PubMed]
- Triadafilopoulos, G. Proton pomp inhibitors for Barrett’s oesophagus. Gut 2000, 46, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Haigh, C.R.; Attwood, S.E.; Thompson, D.G.; Jankowski, J.A.; Kirton, C.M.; Pritchard, D.M.; Varro, A.; Dimaline, R. Gastrin induces proliferation in Barrett’s metaplasia through activation of the CCK2 receptor. Gastroenterology 2003, 124, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Shinomura, Y.; Tsutsui, S.; Zushi, S.; Higashimoto, Y.; Kanayama, S.; Higashiyama, S.; Taniguchi, N.; Matsuzawa, Y. Gastrin induces heparin-binding epidermal growth factor-like growth factor in rat gastric epithelial cells transfected with gastrin receptor. Gastroenterology 1999, 116, 78–89. [Google Scholar] [CrossRef]
- Sawaoka, H.; Tsuji, S.; Tsuji, M.; Gunawan, E.S.; Nakama, A.; Takei, Y.; Nagano, K.; Matsui, H.; Kawano, S.; Hori, M. Expression of the cyclooxygenase-2 gene in gastric epithelium. J. Clin. Gastroenterol. 1997, 25, S105–S110. [Google Scholar] [CrossRef] [PubMed]
- Sawaoka, H.; Tsuji, S.; Tsuji, M.; Gunawan, E.S.; Kawai, N.; Sasaki, Y.; Hori, M.; Kawano, S. Involvement of cyclooxygenase-2 in proliferation and morphogenesis induced by transforming growth factor alpha in gastric epithelial cells. Prostaglandins Leukot. Essent. Fatty Acids 1999, 61, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Rybak, A.; Pesce, M.; Thapar, N.; Borrelli, O. Gastro-esophageal reflux in children. Int. J. Mol. Sci. 2017, 18, 1671. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majka, J.; Wierdak, M.; Brzozowska, I.; Magierowski, M.; Szlachcic, A.; Wojcik, D.; Kwiecien, S.; Magierowska, K.; Zagajewski, J.; Brzozowski, T. Melatonin in Prevention of the Sequence from Reflux Esophagitis to Barrett’s Esophagus and Esophageal Adenocarcinoma: Experimental and Clinical Perspectives. Int. J. Mol. Sci. 2018, 19, 2033. https://doi.org/10.3390/ijms19072033
Majka J, Wierdak M, Brzozowska I, Magierowski M, Szlachcic A, Wojcik D, Kwiecien S, Magierowska K, Zagajewski J, Brzozowski T. Melatonin in Prevention of the Sequence from Reflux Esophagitis to Barrett’s Esophagus and Esophageal Adenocarcinoma: Experimental and Clinical Perspectives. International Journal of Molecular Sciences. 2018; 19(7):2033. https://doi.org/10.3390/ijms19072033
Chicago/Turabian StyleMajka, Jolanta, Mateusz Wierdak, Iwona Brzozowska, Marcin Magierowski, Aleksandra Szlachcic, Dagmara Wojcik, Slawomir Kwiecien, Katarzyna Magierowska, Jacek Zagajewski, and Tomasz Brzozowski. 2018. "Melatonin in Prevention of the Sequence from Reflux Esophagitis to Barrett’s Esophagus and Esophageal Adenocarcinoma: Experimental and Clinical Perspectives" International Journal of Molecular Sciences 19, no. 7: 2033. https://doi.org/10.3390/ijms19072033
APA StyleMajka, J., Wierdak, M., Brzozowska, I., Magierowski, M., Szlachcic, A., Wojcik, D., Kwiecien, S., Magierowska, K., Zagajewski, J., & Brzozowski, T. (2018). Melatonin in Prevention of the Sequence from Reflux Esophagitis to Barrett’s Esophagus and Esophageal Adenocarcinoma: Experimental and Clinical Perspectives. International Journal of Molecular Sciences, 19(7), 2033. https://doi.org/10.3390/ijms19072033