New Insights in Anti-Angiogenesis in Multiple Myeloma



Abstract
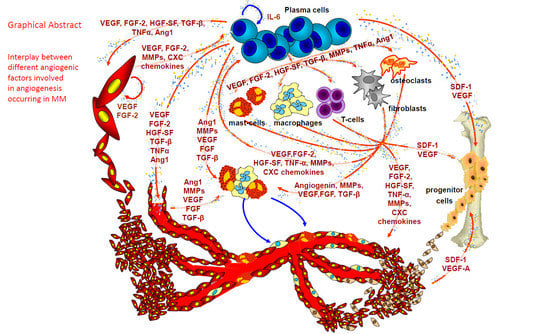
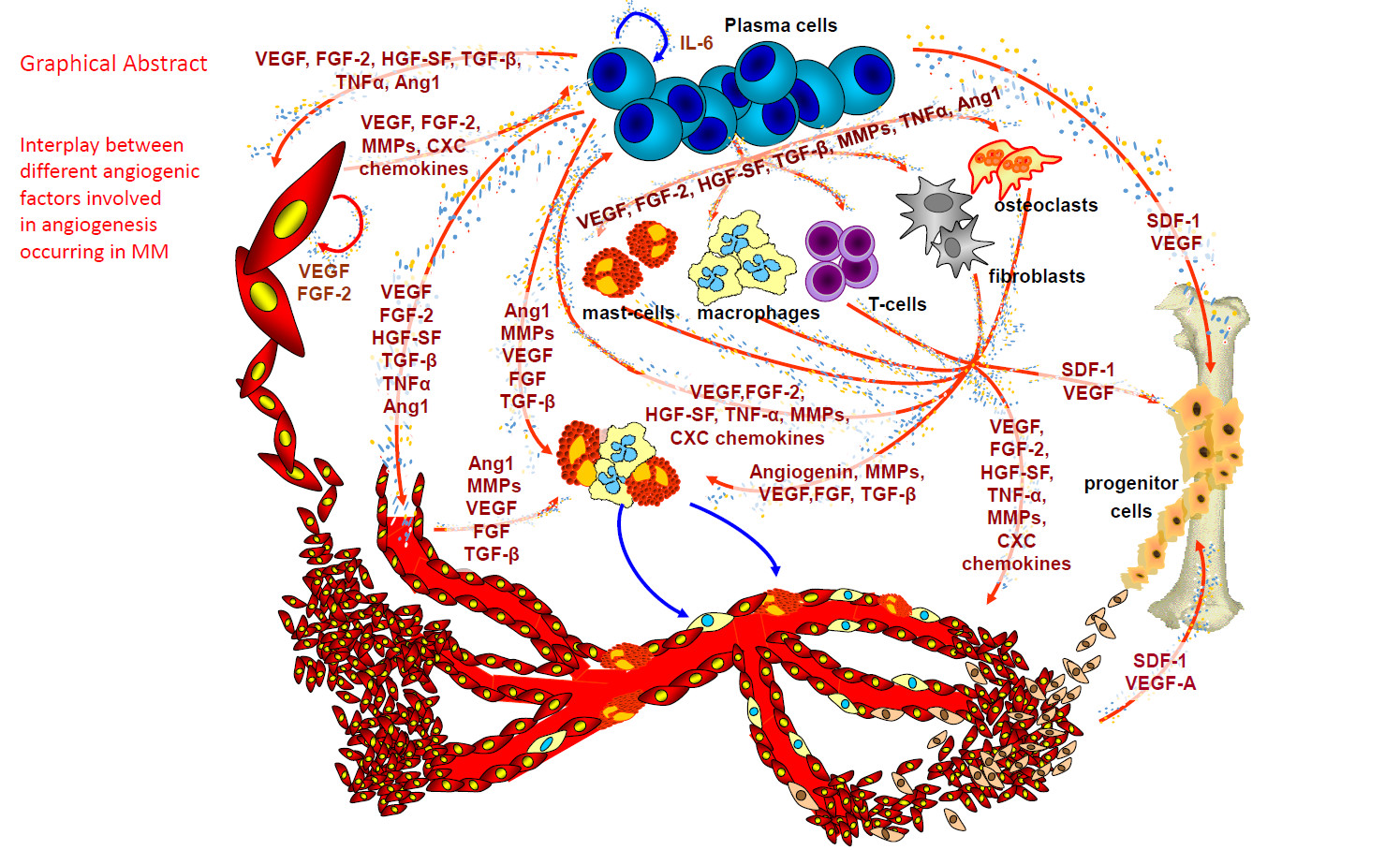
1. Angiogenesis in Multiple Myeloma (MM)
2. Anti-Angiogenesis in Multiple Myeloma
2.1. Thalidomide
2.2. Immunomodulatory Drugs
2.3. Bortezomib and Second-Generation Proteasome Inhibitors
2.4. Bishosphonates
3. New Insights
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MM | multiple myeloma |
| BM | bone marrow |
| PFS | progression free survival |
| OS | overall survival |
| HSC | hematopoietic stem cell |
| BMSCs | bone marrow stromal cells |
| EPCs | endothelial precursor cells |
| VEGF/R | vascular endothelial growth factor/receptor |
| FGF-2/R | fibroblast growth factor-2/ receptor |
| TNF-α | tumor necrosis factor alpha |
| HGF | hepatocyte growth factor |
| IL-6 and IL-8 | interleukin-6 and -8 |
| OPN | osteopontin |
| Ang-1 | angiopoietin-1 |
| Tie-2/Tek | angiopoietin-1 receptor |
| SDF1-α or CXCL12 | stromal cell-derived factor 1α |
| MMPCs | multiple myeloma plasma cells |
| MMECs | multiple myeloma ECs |
| HGF | hepatocyte growth factor |
| c-Met | hepatocyte growth factor receptor |
| Ang 1 | angiopoietin-1 |
| Ang-2 | angiopoietin-2 |
| IGF-1 | insulin-like growth factor-1 |
| OPN | osteopontin |
| MMP-2/9 | matrix metalloproteinases-2/9 |
| PDGF-BB | platelet-derived growth factor-BB |
| ADM | adrenomedullin |
| TGF-1 | transforming growth factor-1 |
| CXCL11/I-TAC | CXCL11-interferon-inducible T-cell chemoattractant |
| MSCs | mesenchymal stem cells |
| BNIP3 | BCL2 Interacting Protein 3 |
| Bcl-2 | B-cell lymphoma 2 |
| IER3 | immediate early response 3 |
| FAS | fas cell surface death receptor |
| rHuEpo | recombinant human erythropoietin |
| IMiDs | immounomodulatory drugs |
| RRMM | refractory multiple myeloma |
| ORR | higher overall response rate |
| TTP | longer time to progression |
| PFS | progression-free survival |
| FVIIIRA | factor VIII-related antigen |
| ERK | Extracellular signal–regulated kinases |
| NFKB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| PDGF | platelet derived growth factor |
| PDGFRβ | BB/PDGF receptor beta |
| AMD3100 | CXCR4 Antagonist |
| SU11274 | cMET inhibitor |
| DC101 | anti-murine VEGFR-2 antibody |
| DARPin | multi-domain designed ankyrin repeat protein |
| mTOR | mammalian target of rapamycin |
| mTORC | mTOR complex |
| CAM | chorioallantoic membrane |
References
- Ribatti, D.; Nico, B.; Crivellato, E.; Roccaro, A.M.; Vacca, A. The history of the angiogenic switch concept. Leukemia 2007, 21, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Ribatti, D.; Roncali, L.; Ranieri, G.; Serio, G.; Silvestris, F.; Dammacco, F. Bone marrow angiogenesis and progression in multiple myeloma. Br. J. Haematol. 1994, 87, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. The discovery of endothelial progenitor cells. Leuk. Res. 2007, 31, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.G.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J.C. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Holash, J. Vessel cooption regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Djonov, V. Intussusceptive microvascular growth in tumors. Cancer Lett. 2012, 316, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Ribatti, D. Bone marrow angiogenesis in multiple myeloma. Leukemia 2005, 20, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Ribatti, D.; Presta, M.; Minischetti, M.; Iurlaro, M.; Ria, R.; Albini, A.; Bussolino, F.; Dammacco, F. Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma. Blood 1999, 93, 3064–3073. [Google Scholar] [PubMed]
- Hose, D.; Moreaux, J.; Meissner, T.; Seckinger, A.; Goldschmidt, H.; Benner, A.; Mahtouk, K.; Hillengass, J.; Reme, T.; De Vos, J.; et al. Induction of angiogenesis by normal and malignant plasma cells. Blood 2009, 114, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Mesa, R.A.; Fonseca, R.; Schroeder, G.; Plevak, M.F.; Dispenzieri, A.; Lacy, M.Q.; Lust, J.A.; Witzig, T.E.; Gertz, M.A.; et al. Bone marrow angiogenesis in 400 patients with monoclonal gammopathy of undetermined significance, multiple myeloma, and primary amyloidosis. Clin. Cancer Res. 2002, 8, 2210–2216. [Google Scholar] [PubMed]
- Jakob, C.; Sterz, J.; Zavrski, I.; Heider, U.; Kleeberg, L.; Fleissner, C.; Kaiser, M.; Sezer, O. Angiogenesis in multiple myeloma. Eur. J. Cancer 2006, 42, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Bone marrow angiogenic ability and expression of angiogenic cytokines in myeloma: Evidence favoring loss of marrow angiogenesis inhibitory activity with disease progression. Blood 2004, 104, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Vacca, A. Importance of the bone marrow microenvironment in inducing the angiogenic response in multiple myeloma. Oncogene 2006, 25, 4257–4266. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A. The role of monocytes-macrophages in vasculogenesis in multiple myeloma. Leukemia 2009, 23, 1535–1536. [Google Scholar] [CrossRef] [PubMed]
- Frassanito, M.A.; Rao, L.; Moschetta, M.; Ria, R.; Di Marzo, L.; De Luisi, A.; Racanelli, V.; Catacchio, I.; Berardi, S.; Basile, A.; et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: In vitro and in vivo studies. Leukemia 2013, 28, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Nico, B.; Mangieri, D.; Crivellato, E.; Vacca, A.; Ribatti, D. Mast cells contribute to vasculogenic mimicry in multiple myeloma. Stem Cells Dev. 2008, 17, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Scavelli, C.; Nico, B.; Cirulli, T.; Ria, R.; Di Pietro, G.; Mangieri, D.; Bacigalupo, A.; Mangialardi, G.; Coluccia, A.M.L.; Caravita, T.; et al. Vasculogenic mimicry by bone marrow macrophages in patients with multiple myeloma. Oncogene 2007, 27, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A. Multiple myeloma bone marrow angiogenesis. In Multiple Myeloma. A New Era of Treatment Strategies; Podar, K., Anderson, K.C., Eds.; Bentham Science Publishers: Sharjah, United Arab Emirates, 2012; pp. 138–148. [Google Scholar]
- Zheng, Y.; Cai, Z.; Wang, S.; Zhang, X.; Qian, J.; Hong, S.; Li, H.; Wang, M.; Yang, J.; Yi, Q. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood 2009, 114, 3625–3628. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A.; Nico, B.; Quondamatteo, F.; Ria, R.; Minischetti, M.; Marzullo, A.; Herken, R.; Roncali, L.; Dammacco, F. Bone marrow angiogenesis and mast cell density increase simultaneously with progression of human multiple myeloma. Br. J. Cancer 1999, 79, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Ria, R.; Piccoli, C.; Cirulli, T.; Falzetti, F.; Mangialardi, G.; Guidolin, D.; Tabilio, A.; Di Renzo, N.; Guarini, A.; Ribatti, D.; et al. Endothelial differentiation of hematopoietic stem and progenitor cells from patients with multiple myeloma. Clin. Cancer Res. 2008, 14, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Vacca, A. Multiple myeloma as a model for the role of bone marrow niches in the control of angiogenesis. Int. Rev. Cell Mol. Biol. 2015, 314, 259–282. [Google Scholar] [PubMed]
- Ria, R.; Todoerti, K.; Berardi, S.; Coluccia, A.M.L.; De Luisi, A.; Mattioli, M.; Ronchetti, D.; Morabito, F.; Guarini, A.; Petrucci, M.T.; et al. Gene expression profiling of bone marrow endothelial cells in patients with multiple myeloma. Clin. Cancer Res. 2009, 15, 5369–5378. [Google Scholar] [CrossRef] [PubMed]
- De Luisi, A.; Binetti, L.; Ria, R.; Ruggieri, S.; Berardi, S.; Catacchio, I.; Racanelli, V.; Pavone, V.; Rossini, B.; Vacca, A.; et al. Erythropoietin is involved in the angiogenic potential of bone marrow macrophages in multiple myeloma. Angiogenesis 2013, 16, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Lamanuzzi, A.; Saltarella, I.; Ferrucci, A.; Ria, R.; Ruggieri, S.; Racanelli, V.; Rao, L.; Annese, T.; Nico, B.; Vacca, A.; et al. Role of erythropoietin in the angiogenic activity of bone marrow endothelial cells of MGUS and multiple myeloma patients. Oncotarget 2016, 7, 14510–14521. [Google Scholar] [CrossRef] [PubMed]
- Mangieri, D.; Nico, B.; Benagiano, V.; De Giorgis, M.; Vacca, A.; Ribatti, D. Angiogenic activity of multiple myeloma endothelial cells in vivo in the chick embryo chorioallantoic membrane assay is associated to a down-regulation in the expression of endogenous endostatin. J. Cell Mol. Med. 2008, 12, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A. Therapeutic renaissance of thalidomide in the treatment of haematological malignancies. Leukemia 2005, 19, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Scavelli, C.; Montefusco, V.; Di Pietro, G.; Neri, A.; Mattioli, M.; Bicciato, S.; Nico, B.; Ribatti, D.; Dammacco, F.; et al. Thalidomide downregulates angiogenic genes in bone marrow endothelial cells of patients with active multiple myeloma. J. Clin. Oncol. 2005, 23, 5334–5346. [Google Scholar] [CrossRef] [PubMed]
- Davies, F.E. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood 2001, 98, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N. Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: Therapeutic implications. Blood 2002, 99, 4525–4530. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor activity of thalidomide in refractory multiple myeloma. N. Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Payvandi, F.; Wu, L.; Zhang, L.-H.; Hariri, R.J.; Man, H.-W.; Chen, R.S.; Muller, G.W.; Hughes, C.C.W.; Stirling, D.I.; et al. The anti-cancer drug lenalidomide inhibits angiogenesis and metastasis via multiple inhibitory effects on endothelial cell function in normoxic and hypoxic conditions. Microvasc. Res. 2009, 77, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.H. Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide: Therapeutic implications. Blood 2006, 108, 618–621. [Google Scholar] [CrossRef] [PubMed]
- Dredge, K.; Horsfall, R.; Robinson, S.P.; Zhang, L.-H.; Lu, L.; Tang, Y.; Shirley, M.A.; Muller, G.; Schafer, P.; Stirling, D.; et al. Orally administered lenalidomide (CC-5013) is anti-angiogenic in vivo and inhibits endothelial cell migration and Akt phosphorylation in vitro. Microvasc. Res. 2005, 69, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Gorgun, G.; Calabrese, E.; Soydan, E.; Hideshima, T.; Perrone, G.; Bandi, M.; Cirstea, D.; Santo, L.; Hu, Y.; Tai, Y.T.; et al. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood 2010, 116, 3227–3237. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nature Reviews Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Thalidomide derivative CC-4047 inhibits osteoclast formation by down-regulation of PU.1. Blood 2006, 107, 3098–3105. [Google Scholar] [CrossRef] [PubMed]
- Breitkreutz, I.; Raab, M.S.; Vallet, S.; Hideshima, T.; Raje, N.; Mitsiades, C.; Chauhan, D.; Okawa, Y.; Munshi, N.C.; Richardson, P.G.; et al. Lenalidomide inhibits osteoclastogenesis, survival factors and bone-remodeling markers in multiple myeloma. Leukemia 2008, 22, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- De Luisi, A.; Ferrucci, A.; Coluccia, A.M.L.; Ria, R.; Moschetta, M.; de Luca, E.; Pieroni, L.; Maffia, M.; Urbani, A.; Di Pietro, G.; et al. Lenalidomide restrains motility and overangiogenic potential of bone marrow endothelial cells in patients with active multiple myeloma. Clin. Cancer Res. 2011, 17, 1935–1946. [Google Scholar] [CrossRef] [PubMed]
- Cibeira, M.T.; Rozman, M.; Segarra, M.; Lozano, E.; Rosiñol, L.; Cid, M.C.; Filella, X.; Bladé, J. Bone marrow angiogenesis and angiogenic factors in multiple myeloma treated with novel agents. Cytokine 2008, 41, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Blood, E.; Mitsiades, C.S.; Jagannath, S.; Zeldenrust, S.R.; Alsina, M.; Schlossman, R.L.; Rajkumar, S.V.; Desikan, K.R.; Hideshima, T.; et al. A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood 2006, 108, 3458–3464. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood 2005, 106, 4050–4053. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Dimopoulos, M.A.; Chen, C.; Cibeira, M.T.; Attal, M.; Spencer, A.; Rajkumar, S.V.; Yu, Z.; Olesnyckyj, M.; Zeldis, J.B.; et al. Lenalidomide plus dexamethasone is more effective than dexamethasone alone in patients with relapsed or refractory multiple myeloma regardless of prior thalidomide exposure. Blood 2008, 112, 4445–4451. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, E.; Palumbo, A.; Dimopoulos, M.A. Treatment of relapsed/refractory multiple myeloma. Semin. Hematol. 2009, 46, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Kastritis, E.; Christoulas, D.; Migkou, M.; Gavriatopoulou, M.; Gkotzamanidou, M.; Iakovaki, M.; Matsouka, C.; Mparmparoussi, D.; Roussou, M.; et al. Treatment of patients with relapsed/refractory multiple myeloma with lenalidomide and dexamethasone with or without bortezomib: Prospective evaluation of the impact of cytogenetic abnormalities and of previous therapies. Leukemia 2010, 24, 1769–1778. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G.; Merlini, G.; Billio, A.; Boccadoro, M.; Corradini, P.; Marchetti, M.; Massaia, M.; Tosi, P.; Palumbo, A.; Cavo, M.; et al. SIE, SIES, GITMO evidence-based guidelines on novel agents (thalidomide, bortezomib, and lenalidomide) in the treatment of multiple myeloma. Ann. Hematol. 2012, 91, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Chanan-Khan, A.A.; Swaika, A.; Paulus, A.; Kumar, S.K.; Mikhael, J.R.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q. Pomalidomide: The new immunomodulatory agent for the treatment of multiple myeloma. Blood Cancer J. 2013, 3, e143–e143. [Google Scholar] [CrossRef] [PubMed]
- Schey, S.A.; Fields, P.; Bartlett, J.B.; Clarke, I.A.; Ashan, G.; Knight, R.D.; Streetly, M.; Dalgleish, A.G. Phase I study of an immunomodulatory thalidomide analog, CC-4047, in relapsed or refractory multiple myeloma. J. Clin. Oncol. 2004, 22, 3269–3276. [Google Scholar] [CrossRef] [PubMed]
- Streetly, M.J.; Gyertson, K.; Daniel, Y.; Zeldis, J.B.; Kazmi, M.; Schey, S.A. Alternate day pomalidomide retains anti-myeloma effect with reduced adverse events and evidence of in vivo immunomodulation. Br. J. Haematol. 2008, 141, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Lacy, M.Q.; Allred, J.B.; Gertz, M.A.; Hayman, S.R.; Short, K.D.; Buadi, F.; Dispenzieri, A.; Kumar, S.; Greipp, P.R.; Lust, J.A.; et al. Pomalidomide plus low-dose dexamethasone in myeloma refractory to both bortezomib and lenalidomide: Comparison of 2 dosing strategies in dual-refractory disease. Blood 2011, 118, 2970–2975. [Google Scholar] [CrossRef] [PubMed]
- Lacy, M.Q.; Hayman, S.R.; Gertz, M.A.; Dispenzieri, A.; Buadi, F.; Kumar, S.; Greipp, P.R.; Lust, J.A.; Russell, S.J.; Dingli, D.; et al. Pomalidomide (CC4047) Plus Low-Dose Dexamethasone As Therapy for Relapsed Multiple Myeloma. J. Clin. Oncol. 2009, 27, 5008–5014. [Google Scholar] [CrossRef] [PubMed]
- Lacy, M.Q.; Hayman, S.R.; Gertz, M.A.; Short, K.D.; Dispenzieri, A.; Kumar, S.; Greipp, P.R.; Lust, J.A.; Russell, S.J.; Dingli, D.; et al. Pomalidomide (CC4047) plus low dose dexamethasone (Pom/dex) is active and well tolerated in lenalidomide refractory multiple myeloma (MM). Leukemia 2010, 24, 1934–1939. [Google Scholar] [CrossRef] [PubMed]
- Miguel, J.S.; Weisel, K.; Moreau, P.; Lacy, M.; Song, K.; Delforge, M.; Karlin, L.; Goldschmidt, H.; Banos, A.; Oriol, A.; et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013, 14, 1055–1066. [Google Scholar] [CrossRef]
- Williams, S.; Pettaway, C.; Song, R.; Papandreou, C.; Logothetis, C.; McConkey, D.J. Differential effects of the proteasome inhibitor bortezomib on apoptosis and angiogenesis in human prostate tumor xenografts. Mol. Cancer Ther. 2003, 2, 835–843. [Google Scholar] [PubMed]
- Hideshima, T.; Chauhan, D.; Hayashi, T.; Akiyama, M.; Mitsiades, N.; Mitsiades, C.; Podar, K.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Proteasome inhibitor PS-341 abrogates IL-6 triggered signaling cascades via caspase-dependent downregulation of gp130 in multiple myeloma. Oncogene 2003, 22, 8386–8393. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Hideshima, T.; Raje, N.; Kumar, S.; Ishitsuka, K.; Yasui, H.; Shiraishi, N.; Ribatti, D.; Nico, B.; Vacca, A.; et al. Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006, 66, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Field-Smith, A.; Morgan, G.J.; Davies, F.E. Bortezomib (Velcadetrade mark) in the Treatment of Multiple Myeloma. Ther. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Ziogas, D.C.; Terpos, E.; Kastritis, E.; Dimopoulos, M.A. An overview of the role of carfilzomib in the treatment of multiple myeloma. Expert Opin. Pharmacother. 2017, 18, 1883–1897. [Google Scholar] [CrossRef] [PubMed]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef] [PubMed]
- Scavelli, C.; Di Pietro, G.; Cirulli, T.; Coluccia, M.; Boccarelli, A.; Giannini, T.; Mangialardi, G.; Bertieri, R.; Coluccia, A.M.L.; Ribatti, D.; et al. Zoledronic acid affects over-angiogenic phenotype of endothelial cells in patients with multiple myeloma. Mol. Cancer Ther. 2007, 6, 3256–3262. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; Di Pietro, G.; Ria, R.; Gnoni, A.; Mangialardi, G.; Guarini, A.; Ditonno, P.; Musto, P.; D’Auria, F.; Ricciardi, M.R.; et al. Bortezomib and zoledronic acid on angiogenic and vasculogenic activities of bone marrow macrophages in patients with multiple myeloma. Eur. J. Cancer 2010, 46, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Denu, R.A.; Dollar, B.A.; Escalante, L.E.; Kuether, J.P.; Callander, N.S.; Asimakopoulos, F.; Hematti, P. Macrophages and mesenchymal stromal cells support survival and proliferation of multiple myeloma cells. Br. J. Haematol. 2012, 158, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Tonon, G.; Sattler, M.; Tai, Y.T.; LeGouill, S.; Yasui, H.; Ishitsuka, K.; Kumar, S.; Kumar, R.; Pandite, L.N.; et al. The small-molecule VEGF receptor inhibitor pazopanib (GW786034B) targets both tumor and endothelial cells in multiple myeloma. Proc. Natl. Acad. Sci. USA 2006, 103, 19478–19483. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Hönemann, D.; Spencer, A.; Rizzieri, D.A.; Stadtmauer, E.A.; Roberts, A.W.; Bahlis, N.; Tricot, G.; Bell, B.; DeMarini, D.J.; et al. Vascular endothelial growth factor inhibition is not an effective therapeutic strategy for relapsed or refractory multiple myeloma: A phase 2 study of pazopanib (GW786034): Table 1. Blood 2009, 113, 4819–4820. [Google Scholar] [CrossRef] [PubMed]
- Airoldi, I.; Cocco, C.; Giuliani, N.; Ferrarini, M.; Colla, S.; Ognio, E.; Taverniti, G.; Di Carlo, E.; Cutrona, G.; Perfetti, V.; et al. Constitutive expression of IL-12R beta 2 on human multiple myeloma cells delineates a novel therapeutic target. Blood 2008, 112, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Coluccia, A.M.L.; Cirulli, T.; Neri, P.; Mangieri, D.; Colanardi, M.C.; Gnoni, A.; Di Renzo, N.; Dammacco, F.; Tassone, P.; Ribatti, D.; et al. Validation of PDGFR and c-Src tyrosine kinases as tumor/vessel targets in patients with multiple myeloma: Preclinical efficacy of the novel, orally available inhibitor dasatinib. Blood 2008, 112, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Thompson, B.; Leleu, X.; Azab, A.K.; Azab, F.; Runnels, J.; Jia, X.; Ngo, H.T.; Melhem, M.R.; et al. MicroRNAs 15a and 16 regulate tumor proliferation in multiple myeloma. Blood 2009, 113, 6669–6680. [Google Scholar] [CrossRef] [PubMed]
- Azab, A.K.; Roccaro, A.M.; Runnels, J.; Sacco, A.; Burwick, N.; Azab, F.; Pitsillides, C.; Ngo, H.T.; Jia, X.; Melhem, M.R.; et al. B622 Rho-A and Rac-1 GTPases in multiple myeloma. Clin. Lymphoma Myeloma 2009, 9, S159. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Mishima, Y.; Sacco, A.; Moschetta, M.; Tai, Y.-T.; Shi, J.; Zhang, Y.; Reagan, M.R.; Huynh, D.; Kawano, Y.; et al. CXCR4 regulates extra-medullary myeloma through epithelial-mesenchymal-transition-like transcriptional activation. Cell Rep. 2015, 12, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, V.; Timm, M.; Haug, J.L.; Kimlinger, T.K.; Wellik, L.E.; Witzig, T.E.; Rajkumar, S.V.; Adjei, A.A.; Kumar, S. Sorafenib, a dual Raf kinase/vascular endothelial growth factor receptor inhibitor has significant anti-myeloma activity and synergizes with common anti-myeloma drugs. Oncogene 2009, 29, 1190–1202. [Google Scholar] [CrossRef] [PubMed]
- Yordanova, A.; Hose, D.; Neben, K.; Witzens-Harig, M.; Gutgemann, I.; Raab, M.S.; Moehler, T.; Goldschmidt, H.; Schmidt-Wolf, I.G. Sorafenib in patients with refractory or recurrent multiple myeloma. Hematol. Oncol. 2013, 31, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Zimmerhackl, A.; Fulciniti, M.; Tonon, G.; Hainz, U.; Tai, Y.-T.; Vallet, S.; Halama, N.; Jäger, D.; Olson, D.L.; et al. The selective adhesion molecule inhibitor Natalizumab decreases multiple myeloma cell growth in the bone marrow microenvironment: Therapeutic implications. Br. J. Haematol. 2011, 155, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Mimura, N.; Hideshima, T.; Shimomura, T.; Suzuki, R.; Ohguchi, H.; Rizq, O.; Kikuchi, S.; Yoshida, Y.; Cottini, F.; Jakubikova, J.; et al. Selective and potent Akt inhibition triggers anti-myeloma activities and enhances fatal endoplasmic reticulum stress induced by proteasome inhibition. Cancer Res. 2014, 74, 4458–4469. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, A.; Moschetta, M.; Frassanito, M.A.; Berardi, S.; Catacchio, I.; Ria, R.; Racanelli, V.; Caivano, A.; Solimando, A.G.; Vergara, D.; et al. A HGF/cMET autocrine loop is operative in multiple myeloma bone marrow endothelial cells and may represent a novel therapeutic target. Clin. Cancer Res. 2014, 20, 5796–5807. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; Basile, A.; Ferrucci, A.; Frassanito, M.A.; Rao, L.; Ria, R.; Solimando, A.G.; Giuliani, N.; Boccarelli, A.; Fumarola, F.; et al. Novel targeting of phospho-cMET overcomes drug resistance and induces antitumor activity in multiple myeloma. Clin. Cancer Res. 2013, 19, 4371–4382. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Imanishi, S.; Azuma, K.; Kobayashi, C.; Yoshizawa, S.; Ohyashiki, K.; Ohyashiki, J.H. Replenishing exosomes from older bone marrow stromal cells with miR-340 inhibits myeloma-related angiogenesis. Blood Adv. 2017, 1, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; Mishima, Y.; Kawano, Y.; Manier, S.; Paiva, B.; Palomera, L.; Aljawai, Y.; Calcinotto, A.; Unitt, C.; Sahin, I.; et al. Targeting vasculogenesis to prevent progression in multiple myeloma. Leukemia 2016, 30, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; De Veirman, K.; Giannico, D.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Solimando, A.G.; Ribatti, D.; Prete, M.; Harstrick, A.; et al. Targeting angiogenesis in multiple myeloma by the VEGF and HGF blocking DARPin ® protein MP0250: A preclinical study. Oncotarget 2018, 9, 13366–13381. [Google Scholar] [CrossRef] [PubMed]
- Lamanuzzi, A.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Leone, P.; Racanelli, V.; Nico, B.; Ribatti, D.; Ditonno, P.; Prete, M.; et al. Inhibition of mTOR complex 2 restrains tumor angiogenesis in multiple myeloma. Oncotarget 2018, 9, 20563–20577. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.C. The 39th David A. Karnofsky Lecture: Bench-to-Bedside Translation of Targeted Therapies in multiple myeloma. J. Clin. Oncol. 2012, 30, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Lonial, S. Integration of novel agents into the care of patients with multiple myeloma. Clin. Cancer Res. 2016, 22, 5443–5452. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Anderson, K.C. Immune therapies in multiple myeloma. Clin. Cancer Res. 2016, 22, 5453–5460. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S. Monoclonal antibodies for the treatment of myeloma. Cancer J. 2016, 22, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Vij, R.; Lendvai, N.; Martin, T.G.; Baz, R.C.; Campana, F.; Mazuir, F.; Charpentier, E.; Benson, D.M. A phase Ib dose escalation trial of isatuximab (SAR650984, anti-CD38 mAb) plus lenalidomide and dexamethasone (Len/Dex) in relapsed/refractory multiple myeloma (RRMM): Interim results from two new dose cohorts. J. Clin. Oncol. 2016, 34 (Suppl. 15), S8009. [Google Scholar]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Angiogenic Factor | Origin | Receptors | Experimental Observation |
|---|---|---|---|
| VEGF (Vascular endothelial growth factor) | Multiple myeloma (MM) Plasma cells (MMPCs) Multiple myeloma ECs (MMECs) | 1. VEGFR-2 on endothelial cells (ECs) 2. VEGFR-1 on bone marrow stromal cells (BMSCs) 3. VEGFR-3 on MMPCs | Promotes MMECs and BMSCs proliferation, growth and chemotaxis Promotes IL-6 and VEGF secretion by BMSCs Up-regulated in active MM Its expression correlates with bone marrow microvascular density |
| FGF-2 (Fibroblast growth factor-2) | MMPCs | FGF-2R-2 | Promotes MMECs and BMSCs proliferation, growth and chemotaxis Promotes IL-6 and VEGF secretion by BMSCs Up-regulated in active MMIts expression correlates with bone marrow microvascular density |
| HGF (Hepatocyte growth factor) | MMPCs | c-Met | Identified in MM cell lines Up-regulated in active MM |
| Ang-1 (Angiopoietin-1) | MMPCs | Tie-2/Tek | Up-regulated in active MM Its expression correlates with bone marrow microvascular density Stabilizes nascent vessels by tightening endo-periendothelial cell interactions |
| Ang-2 (Angiopoietin-2) | MMPCs | Tie-2/Tek | Angiogenic in the presence of VEGF via loosening of periendothelial cells |
| IGF-1 (Insulin-like growth factor-1) | MMPCs (BMSCs) MMECs | IGF-1R | Stimulates MMPCs to secrete VEGF Up-regulated in active MM Promotes MMECs and BMSCs proliferation, growth and chemotaxis |
| IL-8 (Interleukin-8) | BMSCs MMECs | IL-8R | Up-regulated in active MM |
| OPN (Osteopontin) | MMPCs BMSCs MMECs | CD44 | Its expression correlates with bone marrow microvascular density |
| MMP-2/9 (Matrix metalloproteinases-2/9) | MMPCs BMSCs MMECs | Collagen isoforms | Up-regulated in active MM MMP-2 expression is stronger |
| PDGF-BB (Platelet-derived growth factor-BB) | MMPCs | PDGFRβ | This pathway was selectively induced by VEGF Recruits smooth muscle cells around nascent endothelial channels |
| ADM (Adrenomedullin) | MMPCs | ADMR | Induces in vitro angiogenesis |
| TNF-α (Tumor necrosis factor-α) | MMPCs BMSCs MMECs | TNF-αR | Stimulation of IL-6 secretion, bone resorption and expression of adhesion molecules |
| TGF-β1 (Transforming growth factor-β1) | MMPCs BMSCs MMECs | TGF-β1R | Stabilizes nascent vessels by stimulating ECM production |
| CXCL8/IL8 (CXC-chemokine CXCl8/IL8) | MMECs | CXCR2 | Takes part in a paracrine loop between MMECs and MMPCs to mediate MMPCs proliferation and chemotaxis |
| CXCL11/I-TAC (CXCL11-interferon-inducible T-cell α chemoattractant) | MMECs | CXCR3 | Takes part in a paracrine loop between MMECs and MMPCs to mediate MMPCs proliferation and chemotaxis |
| CXCL12/SDF-1α (CXCL12/stromal cell-derived factor-1α) | MMECs | CXCR4 | Takes part in a paracrine loop between MMECs and MMPCs to mediate MMPCs proliferation and chemotaxis |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribatti, D.; Vacca, A. New Insights in Anti-Angiogenesis in Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 2031. https://doi.org/10.3390/ijms19072031
Ribatti D, Vacca A. New Insights in Anti-Angiogenesis in Multiple Myeloma. International Journal of Molecular Sciences. 2018; 19(7):2031. https://doi.org/10.3390/ijms19072031
Chicago/Turabian StyleRibatti, Domenico, and Angelo Vacca. 2018. "New Insights in Anti-Angiogenesis in Multiple Myeloma" International Journal of Molecular Sciences 19, no. 7: 2031. https://doi.org/10.3390/ijms19072031
APA StyleRibatti, D., & Vacca, A. (2018). New Insights in Anti-Angiogenesis in Multiple Myeloma. International Journal of Molecular Sciences, 19(7), 2031. https://doi.org/10.3390/ijms19072031