Therapeutic Opportunities of Targeting Histone Deacetylase Isoforms to Eradicate Cancer Stem Cells


{kind=link}
{kind=link}
Abstract
1. An Overview of Anti-Cancer Stem Cell (CSC) Strategies
1.1. Targeting Pathways that Regulate the Expression of EMT—Inducing Transcription Factors
1.2. Targeting the CXCL12–CXCR4 Signaling Axis
1.3. Targeting CSC Surface Markers
1.4. Targeting Histone Deacetylases
2. Multifaceted Molecular Mechanisms by Which HDAC Inhibitors Eradicate CSCs Independent of Histone Modifications
2.1. Regulation of the Protein Stability and/or Activity of Target Proteins Via Direct Acetylation
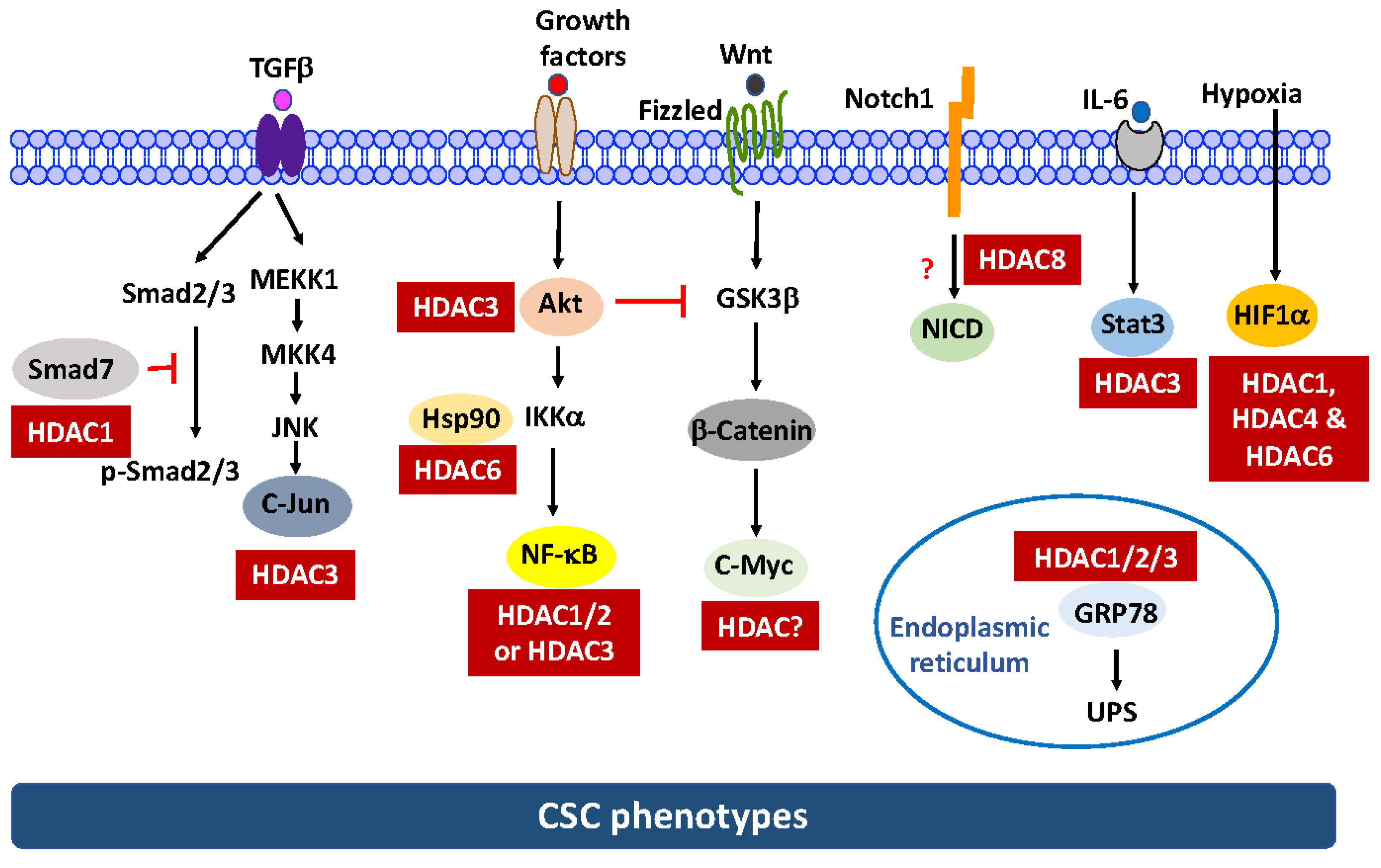
2.2. Targeting Key Signaling Pathways Governing the CSC Population
- Notch1. Data from this and other laboratories indicated that AR-42, SAHA, and the class I HDAC inhibitor depsipeptide were able to suppress the CSC population in TNBC [73] and/or pancreatic cancer cells [34], in part by suppressing Notch1 expression. We obtained evidence that this Notch1 suppression in TNBC was attributable to the ability of HDAC inhibitors to facilitate the proteasomal degradation of Notch1 [73]. Pursuant to this finding, we interrogated the mechanistic link between individual class I isoforms (HDAC1-3 and 8) and this drug-induced Notch1 degradation via genetic knockdown and ectopic expression, which revealed HDAC8 to be the primary mediator for Notch1 degradation [73]. Interestingly, co-immunoprecipitation analysis indicated that HDAC8 did not form complexes with Notch1 and HDAC inhibition had no effect on Notch1 acetylation, suggesting that Notch1 was not a substrate for HDAC8 [73]. However, the signaling mechanism by which HDAC8 regulates the stability of Notch1 remains to be elucidated.
- β-Catenin. We recently reported that genetic knockdown or pharmacological inhibition of HDAC3 was effective in eliminating the CSC population in TNBC cells [74]. We obtained evidence that HDAC3 is mechanistically linked to CSC homeostasis by increasing β-catenin expression through the Akt/GSK3β pathway. This finding is consistent with the report that HDAC3 binds and deacetylates Akt at Lys20, which increases Akt phosphorylation [63]. Consequently, HDAC3 inhibition leads to β-catenin degradation via the inactivation of Akt signaling.
3. HDAC Isoforms as Anti-CSC Targets—Challenges and Opportunities
4. Conclusions
Funding
Conflicts of Interest
References
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, X.; Ren, Y.; Zhang, J.; Chen, J.; Zhou, W.; Guo, W.; Wang, X.; Chen, H.; Li, M.; et al. Cisplatin-enriching cancer stem cells confer multidrug resistance in non-small cell lung cancer via enhancing TRIB1/HDAC activity. Cell Death Dis. 2017, 8, e2746. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhou, K.; Gao, L.; Zhang, B.; Li, W.; Yan, W.; Song, X.; Yu, H.; Wang, S.; Yu, N.; et al. Radiation induces the generation of cancer stem cells: A novel mechanism for cancer radioresistance. Oncol. Lett. 2016, 12, 3059–3065. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Huang, Y.H.; Chen, J.L. Understanding and targeting cancer stem cells: Therapeutic implications and challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Dragu, D.L.; Necula, L.G.; Bleotu, C.; Diaconu, C.C.; Chivu-Economescu, M. Therapies targeting cancer stem cells: Current trends and future challenges. World J. Stem Cells 2015, 7, 1185–1201. [Google Scholar] [PubMed]
- Radpour, R. Tracing and targeting cancer stem cells: New venture for personalized molecular cancer therapy. World J. Stem Cells 2017, 9, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Semba, T.; Saya, H.; Arima, Y. Concise Review: Stem Cells and Epithelial-Mesenchymal Transition in Cancer: Biological Implications and Therapeutic Targets. Stem Cells 2016, 34, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Eaton, E.N.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. Weinberg, The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; Hausen, A.Z.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Bertran, E.; Campbell, J.; Fausto, N.; Fabregat, I. The transforming growth factor-beta (TGF-beta) mediates acquisition of a mesenchymal stem cell-like phenotype in human liver cells. J. Cell. Physiol. 2011, 226, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.; Ding, W.; Emerson, D.; Rountree, C.B. Snail1 induces epithelial-to-mesenchymal transition and tumor initiating stem cell characteristics. BMC Cancer 2011, 11, 396. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Ding, J.; Chen, C.; Sun, W.; Ning, B.F.; Wen, W.; Huang, L.; Han, T.; Yang, W.; Wang, C.; et al. Hepatic transforming growth factor beta gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology 2012, 56, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Rinkenbaugh, A.L.; Baldwin, A.S. The NF-kappaB Pathway and Cancer Stem Cells. Cells 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2016, 35, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Brooks, J.; Willard, M.; Liang, K.; Yoon, Y.; Kang, S.; Shim, H. CXCR4/CXCL12 axis promotes VEGF-mediated tumor angiogenesis through Akt signaling pathway. Biochem. Biophys. Res. Commun. 2007, 359, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Wei, S.; Keller, E.; Liu, R.; Zou, W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am. J. Physiol. Cell Physiol. 2007, 292, C987–C995. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Li, Q.; Liu, S.; Ning, N.; Zhang, X.; Xu, Y.; Chang, A.E.; Wicha, M.S. Concise Review: Targeting Cancer Stem Cells Using Immunologic Approaches. Stem Cells 2015, 33, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Canter, R.J.; Grossenbacher, S.K.; Ames, E.; Murphy, W.J. Immune targeting of cancer stem cells in gastrointestinal oncology. J. Gastrointest. Oncol. 2016, 7, S1–S10. [Google Scholar] [PubMed]
- Qiu, L.; Li, H.; Fu, S.; Chen, X.; Lu, L. Surface markers of liver cancer stem cells and innovative targeted-therapy strategies for HCC. Oncol. Lett. 2018, 15, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, C.; Liu, G.; Liu, H.; Zhou, C.; Lu, Y.; Zhou, C.; Yuan, L.; Li, X. A novel mouse CD133 binding-peptide screened by phage display inhibits cancer cell motility in vitro. Clin. Exp. Metast. 2012, 29, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Lee, K.J.; Lee, S.J.; Yoon, M.Y. Screening of peptides bound to breast cancer stem cell specific surface marker CD44 by phage display. Mol. Biotechnol. 2012, 51, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Botrugno, O.A.; Santoro, F.; Minucci, S. Histone deacetylase inhibitors as a new weapon in the arsenal of differentiation therapies of cancer. Cancer Lett. 2009, 280, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Dvorakova, M.; Vanek, T. Histone deacetylase inhibitors for the treatment of cancer stem cells. MedChemComm 2016, 7, 2217–2231. [Google Scholar] [CrossRef]
- Liu, N.; Li, S.; Wu, N.; Cho, K.S. Acetylation and deacetylation in cancer stem-like cells. Oncotarget 2017, 8, 89315–89325. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Ceccacci, E.; Minucci, S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.L.; Yang, N.; Sharma, K.K.; Balys, M.; Corbett, C.A.; Jordan, C.T.; Becker, M.W.; Steidl, U.; Abdel-Wahab, O.; Levine, R.L.; et al. Selective activity of the histone deacetylase inhibitor AR-42 against leukemia stem cells: A novel potential strategy in acute myelogenous leukemia. Mol. Cancer Ther. 2014, 13, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Nalls, D.; Tang, S.N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Yadav, A.; Lang, J.C.; Teknos, T.N.; Kumar, P. Suberoylanilide hydroxamic acid (SAHA) reverses chemoresistance in head and neck cancer cells by targeting cancer stem cells via the downregulation of nanog. Genes Cancer 2015, 6, 169–181. [Google Scholar] [PubMed]
- Salvador, M.A.; Wicinski, J.; Cabaud, O.; Toiron, Y.; Finetti, P.; Josselin, E.; Lelievre, H.; Kraus-Berthier, L.; Depil, S.; Bertucci, F.; et al. The histone deacetylase inhibitor abexinostat induces cancer stem cells differentiation in breast cancer with low Xist expression. Clin. Cancer Res. 2013, 19, 6520–6531. [Google Scholar] [CrossRef] [PubMed]
- Di Pompo, G.; Salerno, M.; Rotili, D.; Valente, S.; Zwergel, C.; Avnet, S.; Lattanzi, G.; Baldini, N.; Mai, A. Novel histone deacetylase inhibitors induce growth arrest, apoptosis, and differentiation in sarcoma cancer stem cells. J. Med. Chem. 2015, 58, 4073–4079. [Google Scholar] [CrossRef] [PubMed]
- Pathania, R.; Ramachandran, S.; Mariappan, G.; Thakur, P.; Shi, H.; Choi, J.H.; Manicassamy, S.; Kolhe, R.; Prasad, P.D.; Sharma, S.; et al. Combined Inhibition of DNMT and HDAC Blocks the Tumorigenicity of Cancer Stem-like Cells and Attenuates Mammary Tumor Growth. Cancer Res. 2016, 76, 3224–3235. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- MDoherty, R.; Smigiel, J.M.; Junk, D.J.; Jackson, M.W. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, C.; Vassilopoulos, A. Sirtuins at the crossroads of stemness, aging, and cancer. Aging Cell 2017, 16, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Palmirotta, R.; Cives, M.; Della-Morte, D.; Capuani, B.; Lauro, D.; Guadagni, F.; Silvestris, F. Sirtuins and Cancer: Role in the Epithelial-Mesenchymal Transition. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, P.M.; Cole, K.E.; Dowling, D.P.; Christianson, D.W. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 2011, 21, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Liu, Y. Hypoxia-inducible factors in cancer stem cells and inflammation. Trends Pharmacol. Sci. 2015, 36, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Malek, S.N.; Zheng, P.; Liu, Y. Targeting HIF1alpha eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 2011, 8, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Jeong, J.W.; Park, J.A.; Lee, J.W.; Seo, J.H.; Jung, B.K.; Bae, M.K.; Kim, K.W. Regulation of the HIF-1alpha stability by histone deacetylases. Oncol. Rep. 2007, 17, 647–651. [Google Scholar] [PubMed]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Geng, H.; Liu, Q.; Xue, C.; David, L.L.; Beer, T.M.; Thomas, G.V.; Dai, M.S.; Qian, D.Z. HIF1alpha protein stability is increased by acetylation at lysine 709. J. Biol. Chem. 2012, 287, 35496–35505. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.Z.; Kachhap, S.K.; Collis, S.J.; Verheul, H.M.; Carducci, M.A.; Atadja, P.; Pili, R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006, 66, 8814–8821. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Han, J.J.; Stenson, M.; Wellik, L.; Witzig, T.E. Regulation of STAT3 by histone deacetylase-3 in diffuse large B-cell lymphoma: Implications for therapy. Leukemia 2012, 26, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Carafa, V.; Conte, M.; Tambaro, F.P.; Abbondanza, C.; Martens, J.; Nees, M.; Benedetti, R.; Pallavicini, I.; Minucci, S.; et al. c-Myc Modulation and Acetylation Is a Key HDAC Inhibitor Target in Cancer. Clin. Cancer Res. 2017, 23, 2542–2555. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Hermann, P.C.; Liebau, S.; Weidgang, C.; Seufferlein, T.; Kleger, A.; Perkhofer, L. The role of pluripotency factors to drive stemness in gastrointestinal cancer. Stem Cell Res. 2016, 16, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Qin, S.; Schulte, B.A.; Ethier, S.P.; Tew, K.D.; Wang, G.Y. MYC Inhibition Depletes Cancer Stem-like Cells in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 6641–6650. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, H.; Yoon, S.O.; Xu, X.; Hottiger, M.O.; Svaren, J.; Nave, K.A.; Kim, H.A.; Olson, E.N.; Lu, Q.R. HDAC-mediated deacetylation of NF-kappaB is critical for Schwann cell myelination. Nat. Neurosci. 2011, 14, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, R.; Bres, V.; Ng, R.W.; Coudart, M.P.; el Messaoudi, S.; Sardet, C.; Jin, D.Y.; Emiliani, S.; Benkirane, M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 2003, 278, 2758–2766. [Google Scholar] [CrossRef] [PubMed]
- Greene, W.C.; Chen, L.F. Regulation of NF-kappaB action by reversible acetylation. Novartis Found. Symp. 2004, 259, 208–217; discussion 218–225. [Google Scholar] [PubMed]
- Long, J.; Fang, W.Y.; Chang, L.; Gao, W.H.; Shen, Y.; Jia, M.Y.; Zhang, Y.X.; Wang, Y.; Dou, H.B.; Zhang, W.J.; et al. Targeting HDAC3, a new partner protein of AKT in the reversal of chemoresistance in acute myeloid leukemia via DNA damage response. Leukemia 2017, 31, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Kaoud, T.S.; Edupuganti, R.; Zhang, T.; Kogawa, T.; Zhao, Y.; Chauhan, G.B.; Giannoukos, D.N.; Qi, Y.; Tripathy, D.; et al. c-Jun N-terminal kinase promotes stem cell phenotype in triple-negative breast cancer through upregulation of Notch1 via activation of c-Jun. Oncogene 2017, 36, 2599–2608. [Google Scholar] [CrossRef] [PubMed]
- Weiss, C.; Schneider, S.; Wagner, E.F.; Zhang, X.; Seto, E.; Bohmann, D. JNK phosphorylation relieves HDAC3-dependent suppression of the transcriptional activity of c-Jun. EMBO J. 2003, 22, 3686–3695. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; Dijke, P.T. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gong, W.; Ma, X.; Sun, X.; Jiang, H.; Chen, T. Smad7 maintains epithelial phenotype of ovarian cancer stem-like cells and supports tumor colonization by mesenchymal-epithelial transition. Mol. Med. Rep. 2015, 11, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Simonsson, M.; Heldin, C.H.; Ericsson, J.; Gronroos, E. The balance between acetylation and deacetylation controls Smad7 stability. J. Biol. Chem. 2005, 280, 21797–21803. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.C.; Lee, L.Y.; Li, Y.C.; Chen, Y.J.; Lu, Y.C.; Li, Y.L.; Wang, H.M.; Chang, J.T.; Cheng, A.J. Grp78 as a therapeutic target for refractory head-neck cancer with CD24(−)CD44(+) stemness phenotype. Cancer Gene Ther. 2013, 20, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Kahali, S.; Sarcar, B.; Prabhu, A.; Seto, E.; Chinnaiyan, P. Class I histone deacetylases localize to the endoplasmic reticulum and modulate the unfolded protein response. FASEB J. 2012, 26, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cheung, H.H.; Tu, J.; Miu, K.K.; Chan, W.Y. New insights into the unfolded protein response in stem cells. Oncotarget 2016, 7, 54010–54027. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.W.; Chu, P.C.; Chuang, H.C.; Shen, F.H.; Chou, C.C.; Hsu, E.C.; Himmel, L.E.; Huang, H.L.; Tu, H.J.; Kulp, S.K.; et al. Non-epigenetic function of HDAC8 in regulating breast cancer stem cells by maintaining Notch1 protein stability. Oncotarget 2016, 7, 1796–1807. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.Y.; Chuang, H.C.; Shen, F.H.; Detroja, K.; Hsin, L.W.; Chen, C.S. Targeting breast cancer stem cells by novel HDAC3-selective inhibitors. Eur. J. Med. Chem. 2017, 140, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Witt, A.E.; Lee, C.W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 2017, 36, 1707–1720. [Google Scholar] [CrossRef] [PubMed]
- Milde, T.; Oehme, I.; Korshunov, A.; Kopp-Schneider, A.; Remke, M.; Northcott, P.; Deubzer, H.E.; Lodrini, M.; Taylor, M.D.; von Deimling, A.; et al. HDAC5 and HDAC9 in medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clin. Cancer Res. 2010, 16, 3240–3252. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, M.; Crompot, E.; Meuleman, N.; Mineur, P.; Bron, D.; Lagneaux, L.; Stamatopoulos, B. HDAC isoenzyme expression is deregulated in chronic lymphocytic leukemia B-cells and has a complex prognostic significance. Epigenetics 2012, 7, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Clocchiatti, A.; di Giorgio, E.; Ingrao, S.; Meyer-Almes, F.J.; Tripodo, C.; Brancolini, C. Class IIa HDACs repressive activities on MEF2-depedent transcription are associated with poor prognosis of ER(+) breast tumors. FASEB J. 2013, 27, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Li, L.; Zhou, J.; Cui, X.; Tian, Q.; Jin, Y.; Zhu, Y. MiR-2861 Behaves as a Biomarker of Lung Cancer Stem Cells and Regulates the HDAC5-ERK System Genes. Cell Reprogram. 2018, 20, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Gil, V.S.; Bhagat, G.; Howell, L.; Zhang, J.; Kim, C.H.; Stengel, S.; Vega, F.; Zelent, A.; Petrie, K. Deregulated expression of HDAC9 in B cells promotes development of lymphoproliferative disease and lymphoma in mice. Dis. Model. Mech. 2016, 9, 1483–1495. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, P.-C.; Hsieh, H.-Y.; Chu, P.-C.; Chen, C.S. Therapeutic Opportunities of Targeting Histone Deacetylase Isoforms to Eradicate Cancer Stem Cells. Int. J. Mol. Sci. 2018, 19, 1939. https://doi.org/10.3390/ijms19071939
Lin P-C, Hsieh H-Y, Chu P-C, Chen CS. Therapeutic Opportunities of Targeting Histone Deacetylase Isoforms to Eradicate Cancer Stem Cells. International Journal of Molecular Sciences. 2018; 19(7):1939. https://doi.org/10.3390/ijms19071939
Chicago/Turabian StyleLin, Peng-Chan, Hao-Yu Hsieh, Po-Chen Chu, and Ching S. Chen. 2018. "Therapeutic Opportunities of Targeting Histone Deacetylase Isoforms to Eradicate Cancer Stem Cells" International Journal of Molecular Sciences 19, no. 7: 1939. https://doi.org/10.3390/ijms19071939
APA StyleLin, P.-C., Hsieh, H.-Y., Chu, P.-C., & Chen, C. S. (2018). Therapeutic Opportunities of Targeting Histone Deacetylase Isoforms to Eradicate Cancer Stem Cells. International Journal of Molecular Sciences, 19(7), 1939. https://doi.org/10.3390/ijms19071939