Regulation of CYP2J2 and EET Levels in Cardiac Disease and Diabetes


Abstract
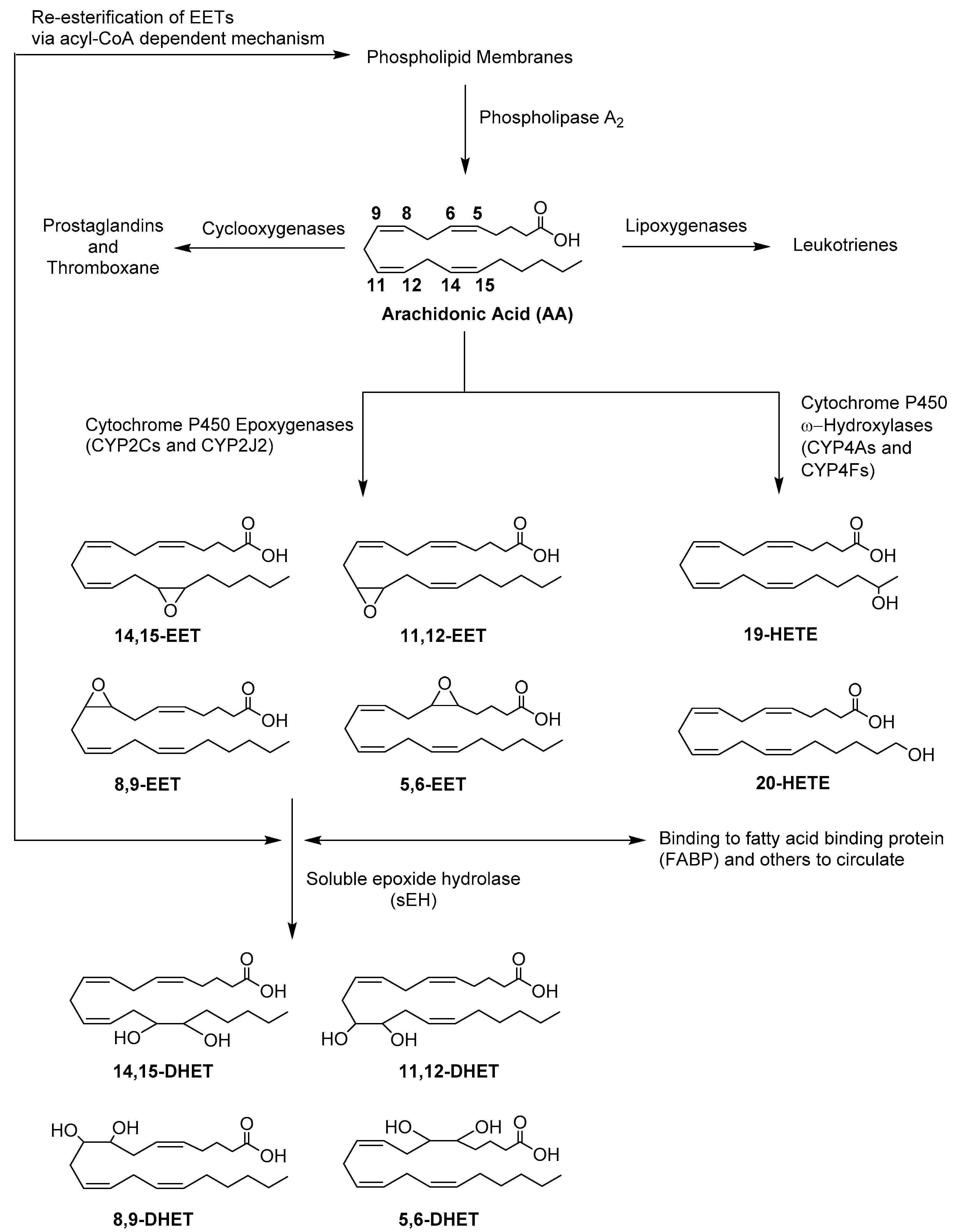
1. Introduction
2. CYP2J2 Expression and Regulation in the Heart
3. CYP2J2 and CVD
3.1. Ischemic Cardiomyopathy
3.1.1. Impact of CYP2J2 on Ischemia-Reperfusion Injury and MI
3.1.2. Coronary Artery Disease (CAD)
3.2. Non-Ischemic Cardiomyopathy
3.2.1. Drug-Induced Cardiotoxicity
3.2.2. Hypertrophy and Arrhythmias
4. Protective Role of CYP2J2 in the Kidney
5. CYP2J2, EETs, and Risk of Diabetes
CYP2J2 in the Pancreas
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- King, L.M.; Ma, J.; Srettabunjong, S.; Graves, J.; Bradbury, J.A.; Li, L.; Spiecker, M.; Liao, J.K.; Mohrenweiser, H.; Zeldin, D.C. Cloning of CYP2J2 gene and identification of functional polymorphisms. Mol. Pharmacol. 2002, 61, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Moomaw, C.R.; Tomer, K.B.; Falck, J.R.; Zeldin, D.C. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J. Biol. Chem. 1996, 271, 3460–3468. [Google Scholar] [CrossRef] [PubMed]
- Dutheil, F.; Dauchy, S.; Diry, M.; Sazdovitch, V.; Cloarec, O.; Mellottee, L.; Bieche, I.; Ingelman-Sundberg, M.; Flinois, J.P.; de Waziers, I.; et al. Xenobiotic-metabolizing enzymes and transporters in the normal human brain: Regional and cellular mapping as a basis for putative roles in cerebral function. Drug Metab. Dispos. Biol. Fate Chem. 2009, 37, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Polonikov, A.V.; Ivanov, V.P.; Solodilova, M.A.; Khoroshaya, I.V.; Kozhuhov, M.A.; Panfilov, V.I. Promoter polymorphism G-50T of a human CYP2J2 epoxygenase gene is associated with common susceptibility to asthma. Chest 2007, 132, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Okayama, A.; Imai, N.; Guengerich, F.P.; Shimizu, M. Inter-individual variation of cytochrome P4502J2 expression and catalytic activities in liver microsomes from Japanese and Caucasian populations. Xenobiotica Fate Foreign Compd. Biol. Syst. 2006, 36, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, P.E.; Ma, J.; Qu, W.; Zeldin, D.C. P450 subfamily CYP2J and their role in the bioactivation of arachidonic acid in extrahepatic tissues. Drug Metab. Rev. 1999, 31, 205–234. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.H.; Mitchell, L.A.; Bradbury, J.A.; Qu, W.; Zeldin, D.C.; Schnellmann, R.G.; Grant, D.F. Analysis of the cytotoxic properties of linoleic acid metabolites produced by renal and hepatic P450s. Toxicol. Appl. Pharmacol. 2000, 168, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Hirama, T.; Matsubara, T.; Nagata, K.; Yamazoe, Y. Involvement of CYP2J2 on the intestinal first-pass metabolism of antihistamine drug, astemizole. Drug Metab. Dispos. Biol. Fate Chem. 2002, 30, 1240–1245. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.A.; Neul, D.; Clouser-Roche, A.; Dalvie, D.; Wester, M.R.; Jiang, Y.; Jones, J.P., 3rd; Freiwald, S.; Zientek, M.; Totah, R.A. Identification of novel substrates for human cytochrome P450 2J2. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.A.; Jones, J.P., 3rd; Katayama, J.; Kaspera, R.; Jiang, Y.; Freiwald, S.; Smith, E.; Walker, G.S.; Totah, R.A. Identifying a selective substrate and inhibitor pair for the evaluation of CYP2J2 activity. Drug Metab. Dispos. Biol. Fate Chem. 2012, 40, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Narjoz, C.; Favre, A.; McMullen, J.; Kiehl, P.; Montemurro, M.; Figg, W.D.; Beaune, P.; de Waziers, I.; Rochat, B. Important role of CYP2J2 in protein kinase inhibitor degradation: A possible role in intratumor drug disposition and resistance. PLoS ONE 2014, 9, e95532. [Google Scholar] [CrossRef] [PubMed]
- Kaspera, R.; Kirby, B.J.; Sahele, T.; Collier, A.C.; Kharasch, E.D.; Unadkat, J.D.; Totah, R.A. Investigating the contribution of CYP2J2 to ritonavir metabolism in vitro and in vivo. Biochem. Pharmacol. 2014, 91, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Aliwarga, T.; Raccor, B.S.; Lemaitre, R.N.; Sotoodehnia, N.; Gharib, S.A.; Xu, L.; Totah, R.A. Enzymatic and free radical formation of cis- and trans-epoxyeicosatrienoic acids in vitro and in vivo. Free Radic. Biol. Med. 2017, 112, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.Y.; Li, Y.H.; Chao, T.H.; Wu, H.L.; Lin, L.J.; Tsai, L.M.; Chen, J.H. Synergistic effect of cytochrome P450 epoxygenase CYP2J2*7 polymorphism with smoking on the onset of premature myocardial infarction. Atherosclerosis 2007, 195, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Borgel, J.; Bulut, D.; Hanefeld, C.; Neubauer, H.; Mugge, A.; Epplen, J.T.; Holland-Letz, T.; Spiecker, M. The CYP2J2 G-50T polymorphism and myocardial infarction in patients with cardiovascular risk profile. BMC Cardiovasc. Disord. 2008, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Marciante, K.D.; Totah, R.A.; Heckbert, S.R.; Smith, N.L.; Lemaitre, R.N.; Lumley, T.; Rice, K.M.; Hindorff, L.A.; Bis, J.C.; Hartman, B.; et al. Common variation in cytochrome P450 epoxygenase genes and the risk of incident nonfatal myocardial infarction and ischemic stroke. Pharmacogenet. Genom. 2008, 18, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Arun Kumar, A.S.; Kumar, S.S.; Umamaheswaran, G.; Kesavan, R.; Balachandar, J.; Adithan, C. Association of CYP2C8, CYP2C9 and CYP2J2 gene polymorphisms with myocardial infarction in South Indian population. Pharmacol. Rep. 2015, 67, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhao, J.H.; Ma, P.J.; Su, L.L.; Tao, S.B.; Ji, S.B. Association of CYP2J2 gene polymorphisms with ischemic stroke. Int. J. Clin. Exp. Med. 2015, 8, 8163–8167. [Google Scholar] [PubMed]
- Wang, S.Y.; Xing, P.F.; Zhang, C.Y.; Deng, B.Q. Association of CYP2J2 gene polymorphisms with ischemic stroke and stroke subtypes in Chinese population. Medicine 2017, 96, e6266. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.R.; North, K.E.; Bray, M.S.; Couper, D.J.; Heiss, G.; Zeldin, D.C. CYP2J2 and CYP2C8 polymorphisms and coronary heart disease risk: The Atherosclerosis Risk in Communities (ARIC) study. Pharmacogenet. Genom. 2007, 17, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Spiecker, M.; Darius, H.; Hankeln, T.; Soufi, M.; Sattler, A.M.; Schaefer, J.R.; Node, K.; Borgel, J.; Mugge, A.; Lindpaintner, K.; et al. Risk of coronary artery disease associated with polymorphism of the cytochrome P450 epoxygenase CYP2J2. Circulation 2004, 110, 2132–2136. [Google Scholar] [CrossRef] [PubMed]
- Dreisbach, A.W.; Japa, S.; Sigel, A.; Parenti, M.B.; Hess, A.E.; Srinouanprachanh, S.L.; Rettie, A.E.; Kim, H.; Farin, F.M.; Hamm, L.L.; et al. The Prevalence of CYP2C8, 2C9, 2J2, and soluble epoxide hydrolase polymorphisms in African Americans with hypertension. Am. J. Hypertens. 2005, 18, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- King, L.M.; Gainer, J.V.; David, G.L.; Dai, D.; Goldstein, J.A.; Brown, N.J.; Zeldin, D.C. Single nucleotide polymorphisms in the CYP2J2 and CYP2C8 genes and the risk of hypertension. Pharmacogenet. Genom. 2005, 15, 7–13. [Google Scholar] [CrossRef]
- Wu, S.N.; Zhang, Y.; Gardner, C.O.; Chen, Q.; Li, Y.; Wang, G.L.; Gao, P.J.; Zhu, D.L. Evidence for association of polymorphisms in CYP2J2 and susceptibility to essential hypertension. Ann. Hum. Genet. 2007, 71, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Polonikov, A.V.; Ivanov, V.P.; Solodilova, M.A.; Khoroshaya, I.V.; Kozhuhov, M.A.; Ivakin, V.E.; Katargina, L.N.; Kolesnikova, O.E. A common polymorphism G-50T in cytochrome P450 2J2 gene is associated with increased risk of essential hypertension in a Russian population. Dis. Markers 2008, 24, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Alghasham, A.; Ali, A.; Ismail, H.; Dowaidar, M.; Settin, A.A. CYP2J2-50 G/T and ADRB2 G46A gene polymorphisms in Saudi subjects with hypertension. Genet. Test. Mol. Biomarkers 2012, 16, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Fava, C.; Montagnana, M.; Almgren, P.; Hedblad, B.; Engstrom, G.; Berglund, G.; Minuz, P.; Melander, O. The common functional polymorphism -50G>T of the CYP2J2 gene is not associated with ischemic coronary and cerebrovascular events in an urban-based sample of Swedes. J. Hypertens. 2010, 28, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.M.; Hullin, F.; Chap, H.; Douste-Blazy, L. Phosphatidylcholine is the major phospholipid providing arachidonic acid for prostacyclin synthesis in thrombin-stimulated human endothelial cells. Thromb. Res. 1984, 34, 117–123. [Google Scholar] [CrossRef]
- Nishikiori, M.; Iizuka, H.; Ichiba, H.; Sadamoto, K.; Fukushima, T. Determination of free fatty acids in human serum by HPLC with fluorescence detection. J. Chromatogr. Sci. 2015, 53, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Qin, S.; Li, L.; Chen, X.; Wang, Q.; Wei, J. An Optimized High Throughput Clean-Up Method Using Mixed-Mode SPE Plate for the Analysis of Free Arachidonic Acid in Plasma by LC-MS/MS. Int. J. Anal. Chem. 2015, 2015, 374819. [Google Scholar] [CrossRef] [PubMed]
- Purdon, A.D.; Rao, A.K. Interaction of albumin, arachidonic acid and prostanoids in platelets. Prostaglandins Leukot. Essent. Fatty Acids 1989, 35, 213–218. [Google Scholar] [CrossRef]
- Dobner, P.; Engelmann, B. Low-density lipoproteins supply phospholipid-bound arachidonic acid for platelet eicosanoid production. Am. J. Physiol. 1998, 275, E777–E784. [Google Scholar] [CrossRef] [PubMed]
- Veerkamp, J.H.; van Moerkerk, H.T.; Prinsen, C.F.; van Kuppevelt, T.H. Structural and functional studies on different human FABP types. Mol. Cell. Biochem. 1999, 192, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Gross, R.; Turk, J. Arachidonic acid induces an increase in the cytosolic calcium concentration in single pancreatic islet beta cells. Biochem. Biophys. Res. Commun. 1992, 184, 647–653. [Google Scholar] [CrossRef]
- Chilton, F.H.; Fonteh, A.N.; Surette, M.E.; Triggiani, M.; Winkler, J.D. Control of arachidonate levels within inflammatory cells. Biochim. Biophys. Acta 1996, 1299, 1–15. [Google Scholar] [CrossRef]
- Brash, A.R. Arachidonic acid as a bioactive molecule. J. Clin. Investig. 2001, 107, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.; Bertolino, S.; Abbot, S.E.; Aaronson, P.I.; Smirnov, S.V. Modulation of arachidonic acid release and membrane fluidity by albumin in vascular smooth muscle and endothelial cells. Circ. Res. 1998, 83, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, M.; Zaccagnino, P.; Oliveros-Celis, C.; Lorusso, M. Arachidonic acid induces specific membrane permeability increase in heart mitochondria. FEBS Lett. 2006, 580, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, H.; Kontani, M.; Kawashima, H.; Akimoto, K.; Kusumoto, A.; Kiso, Y.; Koga, Y.; Shibata, H. Arachidonic acid-enriched triacylglycerol improves cognitive function in elderly with low serum levels of arachidonic acid. J. Oleo Sci. 2014, 63, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Kotani, S.; Nakazawa, H.; Tokimasa, T.; Akimoto, K.; Kawashima, H.; Toyoda-Ono, Y.; Kiso, Y.; Okaichi, H.; Sakakibara, M. Synaptic plasticity preserved with arachidonic acid diet in aged rats. Neurosci. Res. 2003, 46, 453–461. [Google Scholar] [CrossRef]
- Meves, H. Modulation of ion channels by arachidonic acid. Prog. Neurobiol. 1994, 43, 175–186. [Google Scholar] [CrossRef]
- Cao, Y.; Pearman, A.T.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Intracellular unesterified arachidonic acid signals apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 11280–11285. [Google Scholar] [CrossRef] [PubMed]
- Kaspera, R.; Totah, R.A. Epoxyeicosatrienoic acids: Formation, metabolism and potential role in tissue physiology and pathophysiology. Expert Opin. Drug Metab. Toxicol. 2009, 5, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Zeldin, D.C.; Wei, S.; Falck, J.R.; Hammock, B.D.; Snapper, J.R.; Capdevila, J.H. Metabolism of epoxyeicosatrienoic acids by cytosolic epoxide hydrolase: Substrate structural determinants of asymmetric catalysis. Arch. Biochem. Biophys. 1995, 316, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, N.L.; Fang, X.; Kaduce, T.L.; VanRollins, M.; Chatterjee, P.; Spector, A.A. Potentiation of endothelium-dependent relaxation by epoxyeicosatrienoic acids. Circ. Res. 1997, 81, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Widstrom, R.L.; Norris, A.W.; Spector, A.A. Binding of cytochrome P450 monooxygenase and lipoxygenase pathway products by heart fatty acid-binding protein. Biochemistry 2001, 40, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Macias-Perez, I.; Abair, T.; Wei, S.; Su, Y.; Zent, R.; Falck, J.R.; Capdevila, J.H. Characterization of 5,6- and 8,9-epoxyeicosatrienoic acids (5,6- and 8,9-EET) as potent in vivo angiogenic lipids. J. Biol. Chem. 2005, 280, 27138–27146. [Google Scholar] [CrossRef] [PubMed]
- Campbell, W.B.; Fleming, I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch. Eur. J. Physiol. 2010, 459, 881–895. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Maki-Petaja, K.; Cheriyan, J.; McEniery, C.; Wilkinson, I.B. The role of epoxyeicosatrienoic acids in the cardiovascular system. Br. J. Clin. Pharmacol. 2015, 80, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, L.; Han, W.; Shen, T.; Ma, C.; Liu, Y.; Nie, X.; Liu, M.; Ran, Y.; Zhu, D. Activation of JNK/c-Jun is required for the proliferation, survival, and angiogenesis induced by EET in pulmonary artery endothelial cells. J. Lipid Res. 2012, 53, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Lin, L.; Chen, J.X.; Lee, C.R.; Seubert, J.M.; Wang, Y.; Wang, H.; Chao, Z.R.; Tao, D.D.; Gong, J.P.; et al. Cytochrome P-450 epoxygenases protect endothelial cells from apoptosis induced by tumor necrosis factor-alpha via MAPK and PI3K/Akt signaling pathways. Am. J. Phys. Heart Circ. Physiol. 2007, 293, h142–h151. [Google Scholar] [CrossRef] [PubMed]
- Ng, V.Y.; Huang, Y.; Reddy, L.M.; Falck, J.R.; Lin, E.T.; Kroetz, D.L. Cytochrome P450 eicosanoids are activators of peroxisome proliferator-activated receptor alpha. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Wray, J.A.; Sugden, M.C.; Zeldin, D.C.; Greenwood, G.K.; Samsuddin, S.; Miller-Degraff, L.; Bradbury, J.A.; Holness, M.J.; Warner, T.D.; Bishop-Bailey, D. The epoxygenases CYP2J2 activates the nuclear receptor PPARalpha in vitro and in vivo. PLoS ONE 2009, 4, e7421. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Herrnreiter, A.; Pfister, S.L.; Gauthier, K.M.; Falck, B.A.; Falck, J.R.; Campbell, W.B. GPR40 is a Low Affinity Epoxyeicosatrienoic Acid Receptor in Vascular Cells. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, L.A.; Smith, N.J.; Milligan, G. International Union of Pharmacology. LXXI. Free fatty acid receptors FFA1, -2, and -3: Pharmacology and pathophysiological functions. Pharmacol. Rev. 2008, 60, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.D.; Poitout, V. The fatty acid receptor FFA1/GPR40 a decade later: How much do we know? Trends Endocrinol. Metab. 2013, 24, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Kotarsky, K.; Nilsson, N.E.; Flodgren, E.; Owman, C.; Olde, B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem. Biophys. Res. Commun. 2003, 301, 406–410. [Google Scholar] [CrossRef]
- Delozier, T.C.; Kissling, G.E.; Coulter, S.J.; Dai, D.; Foley, J.F.; Bradbury, J.A.; Murphy, E.; Steenbergen, C.; Zeldin, D.C.; Goldstein, J.A. Detection of human CYP2C8, CYP2C9, and CYP2J2 in cardiovascular tissues. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Michaud, V.; Frappier, M.; Dumas, M.C.; Turgeon, J. Metabolic activity and mRNA levels of human cardiac CYP450s involved in drug metabolism. PLoS ONE 2010, 5, e15666. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, E.A.; Kaspera, R.; Mokadam, N.A.; Jones, J.P., 3rd; Totah, R.A. Activity, inhibition, and induction of cytochrome P450 2J2 in adult human primary cardiomyocytes. Drug Metab. Dispos. Biol. Fate Chem. 2013, 41, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Askari, A.; Thomson, S.J.; Edin, M.L.; Zeldin, D.C.; Bishop-Bailey, D. Roles of the epoxygenase CYP2J2 in the endothelium. Prostaglandins Other Lipid Mediat. 2013, 107, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Marden, N.Y.; Murray, M. Characterization of a c-Jun-responsive module in the 5′-flank of the human CYP2J2 gene that regulates transactivation. Biochem. J. 2005, 391, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.; Murray, M. Up-regulation of human CYP2J2 in HepG2 cells by butylated hydroxyanisole is mediated by c-Jun and Nrf2. Mol. Pharmacol. 2010, 77, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Bystrom, J.; Thomson, S.J.; Johansson, J.; Edin, M.L.; Zeldin, D.C.; Gilroy, D.W.; Smith, A.M.; Bishop-Bailey, D. Inducible CYP2J2 and its product 11,12-EET promotes bacterial phagocytosis: A role for CYP2J2 deficiency in the pathogenesis of Crohn’s disease? PLoS ONE 2013, 8, e75107. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, E.A.; Lemaitre, R.N.; Sotoodehnia, N.; Gharib, S.A.; Totah, R.A. CYP2J2 Expression in Adult Ventricular Myocytes Protects Against Reactive Oxygen Species Toxicity. Drug Metab. Dispos. Biol. Fate Chem. 2018, 46, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Herse, F.; Lamarca, B.; Hubel, C.A.; Kaartokallio, T.; Lokki, A.I.; Ekholm, E.; Laivuori, H.; Gauster, M.; Huppertz, B.; Sugulle, M.; et al. Cytochrome P450 subfamily 2J polypeptide 2 expression and circulating epoxyeicosatrienoic metabolites in preeclampsia. Circulation 2012, 126, 2990–2999. [Google Scholar] [CrossRef] [PubMed]
- Seubert, J.; Yang, B.; Bradbury, J.A.; Graves, J.; Degraff, L.M.; Gabel, S.; Gooch, R.; Foley, J.; Newman, J.; Mao, L.; et al. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ. Res. 2004, 95, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Edin, M.L.; Wang, Z.; Bradbury, J.A.; Graves, J.P.; Lih, F.B.; DeGraff, L.M.; Foley, J.F.; Torphy, R.; Ronnekleiv, O.K.; Tomer, K.B.; et al. Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. FASEB J. 2011, 25, 3436–3447. [Google Scholar] [CrossRef] [PubMed]
- Islam, O.; Patil, P.; Goswami, S.K.; Razdan, R.; Inamdar, M.N.; Rizwan, M.; Mathew, J.; Inceoglu, B.; Stephen Lee, K.S.; Hwang, S.H.; et al. Inhibitors of soluble epoxide hydrolase minimize ischemia-reperfusion-induced cardiac damage in normal, hypertensive, and diabetic rats. Cardiovasc. Ther. 2017, 35. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, T.; He, X.; Liu, X.; Wang, B.; Liu, Y.; Li, Z.; Tan, R.; Ding, C.; Wang, H.; et al. CYP2J2 Overexpression Increases EETs and Protects against HFD-Induced Atherosclerosis in ApoE−/− Mice. J. Cardiovasc. Pharmacol. 2016, 67, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; El-Sikhry, H.; Chaudhary, K.R.; Batchu, S.N.; Shayeganpour, A.; Jukar, T.O.; Bradbury, J.A.; Graves, J.P.; DeGraff, L.M.; Myers, P.; et al. Overexpression of CYP2J2 provides protection against doxorubicin-induced cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H37–H46. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zeng, H.; Wen, Z.; Chen, C.; Wang, D.W. CYP2J2 and its metabolites (epoxyeicosatrienoic acids) attenuate cardiac hypertrophy by activating AMPKalpha2 and enhancing nuclear translocation of Akt1. Aging Cell 2016, 15, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Westphal, C.; Spallek, B.; Konkel, A.; Marko, L.; Qadri, F.; DeGraff, L.M.; Schubert, C.; Bradbury, J.A.; Regitz-Zagrosek, V.; Falck, J.R.; et al. CYP2J2 overexpression protects against arrhythmia susceptibility in cardiac hypertrophy. PLoS ONE 2013, 8, e73490. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ni, L.; Yang, L.; Duan, Q.; Chen, C.; Edin, M.L.; Zeldin, D.C.; Wang, D.W. CYP2J2-derived epoxyeicosatrienoic acids suppress endoplasmic reticulum stress in heart failure. Mol. Pharmacol. 2014, 85, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Li, X.; Yan, J.; Yu, X.; Yang, G.; Xiao, X.; Voltz, J.W.; Zeldin, D.C.; Wang, D.W. Overexpression of cytochrome P450 epoxygenases prevents development of hypertension in spontaneously hypertensive rats by enhancing atrial natriuretic peptide. J. Pharmacol. Exp. Ther. 2010, 334, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Wang, L.; Li, R.; Ma, B.; Tu, L.; Xu, X.; Dackor, R.T.; Zeldin, D.C.; Wang, D.W. Gene delivery of cytochrome p450 epoxygenase ameliorates monocrotaline-induced pulmonary artery hypertension in rats. Am. J. Respir. Cell Mol. Biol. 2010, 43, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Tu, L.; Li, X.; Yang, S.; Chen, C.; Xu, X.; Wang, P.; Wang, D.W. Delivery of AAV2-CYP2J2 protects remnant kidney in the 5/6-nephrectomized rat via inhibition of apoptosis and fibrosis. Hum. Gene Ther. 2012, 23, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhao, G.; Yan, J.; Liu, W.; Feng, W.; Ma, B.; Yang, L.; Wang, J.A.; Tu, L.; Wang, D.W. CYP2J2 overexpression increases EETs and protects against angiotensin II-induced abdominal aortic aneurysm in mice. J. Lipid Res. 2013, 54, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Xiong, X.; Chen, C.; Li, H.; Xu, X.; Li, X.; Li, R.; Chen, G.; Dackor, R.T.; Zeldin, D.C.; et al. Cardiac-specific overexpression of CYP2J2 attenuates diabetic cardiomyopathy in male streptozotocin-induced diabetic mice. Endocrinology 2013, 154, 2843–2856. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xu, X.; Chen, C.; Wang, Y.; Gruzdev, A.; Zeldin, D.C.; Wang, D.W. CYP2J2 attenuates metabolic dysfunction in diabetic mice by reducing hepatic inflammation via the PPARgamma. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E270–E282. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, P.; Zhao, G.; Xu, G.; Gruzdev, A.; Zeldin, D.C.; Wang, D.W. Cytochrome P450 epoxygenase CYP2J2 attenuates nephropathy in streptozotocin-induced diabetic mice. Prostaglandins Other Lipid Mediat. 2011, 96, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.G.; Sodhi, K.; Silvis, A.M.; Vanella, L.; Favero, G.; Rezzani, R.; Lee, C.; Zeldin, D.C.; Schwartzman, M.L. CYP2J2 targeting to endothelial cells attenuates adiposity and vascular dysfunction in mice fed a high-fat diet by reprogramming adipocyte phenotype. Hypertension 2014, 64, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.R.; Zordoky, B.N.; Edin, M.L.; Alsaleh, N.; El-Kadi, A.O.; Zeldin, D.C.; Seubert, J.M. Differential effects of soluble epoxide hydrolase inhibition and CYP2J2 overexpression on postischemic cardiac function in aged mice. Prostaglandins Other Lipid Mediat. 2013, 104–105, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Theken, K.N.; Schuck, R.N.; Edin, M.L.; Tran, B.; Ellis, K.; Bass, A.; Lih, F.B.; Tomer, K.B.; Poloyac, S.M.; Wu, M.C.; et al. Evaluation of cytochrome P450-derived eicosanoids in humans with stable atherosclerotic cardiovascular disease. Atherosclerosis 2012, 222, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Schuck, R.N.; Theken, K.N.; Edin, M.L.; Caughey, M.; Bass, A.; Ellis, K.; Tran, B.; Steele, S.; Simmons, B.P.; Lih, F.B.; et al. Cytochrome P450-derived eicosanoids and vascular dysfunction in coronary artery disease patients. Atherosclerosis 2013, 227, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Oni-Orisan, A.; Edin, M.L.; Lee, J.A.; Wells, M.A.; Christensen, E.S.; Vendrov, K.C.; Lih, F.B.; Tomer, K.B.; Bai, X.; Taylor, J.M.; et al. Cytochrome P450-derived epoxyeicosatrienoic acids and coronary artery disease in humans: A targeted metabolomics study. J. Lipid Res. 2016, 57, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Spiecker, M.; Liao, J. Cytochrome P450 epoxygenase CYP2J2 and the risk of coronary artery disease. Trends Cardiovasc. Med. 2006, 16, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; American Heart Association; et al. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [PubMed]
- Arnold, W.R.; Baylon, J.L.; Tajkhorshid, E.; Das, A. Arachidonic Acid Metabolism by Human Cardiovascular CYP2J2 Is Modulated by Doxorubicin. Biochemistry 2017, 56, 6700–6712. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.R.; Das, A. An Emerging Pathway of Doxorubicin Cardiotoxicity Mediated through CYP2J2. Biochemistry 2018, 57, 2294–2296. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhang, X.; Chen, C.; Wen, Z.; Hoopes, S.L.; Zeldin, D.C.; Wang, D.W. Cardiomyocyte-specific expression of CYP2J2 prevents development of cardiac remodelling induced by angiotensin II. Cardiovasc. Res. 2015, 105, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Olson, E.N. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hill, J.A. Electrophysiological remodeling in heart failure. J. Mol. Cell. Cardiol. 2010, 48, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Capdevila, J.H.; Zeldin, D.C.; Rosenberg, R.L. Inhibition of cardiac L-type calcium channels by epoxyeicosatrienoic acids. Mol. Pharmacol. 1999, 55, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Lu, T.; Weintraub, N.L.; VanRollins, M.; Spector, A.A.; Shibata, E.F. Effects of epoxyeicosatrienoic acids on the cardiac sodium channels in isolated rat ventricular myocytes. J. Physiol. 1999, 519, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Xiao, Y.F.; Bradbury, J.A.; Graves, J.P.; Degraff, L.M.; Seubert, J.M.; Zeldin, D.C. Electrophysiological properties of cardiomyocytes isolated from CYP2J2 transgenic mice. Mol. Pharmacol. 2007, 72, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Bodiga, S.; Zhang, R.; Jacobs, D.E.; Larsen, B.T.; Tampo, A.; Manthati, V.L.; Kwok, W.M.; Zeldin, D.C.; Falck, J.R.; Gutterman, D.D.; et al. Protective actions of epoxyeicosatrienoic acid: Dual targeting of cardiovascular PI3K and KATP channels. J. Mol. Cell. Cardiol. 2009, 46, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Batchu, S.N.; Chaudhary, K.R.; El-Sikhry, H.; Yang, W.; Light, P.E.; Oudit, G.Y.; Seubert, J.M. Role of PI3Kalpha and sarcolemmal ATP-sensitive potassium channels in epoxyeicosatrienoic acid mediated cardioprotection. J. Mol. Cell. Cardiol. 2012, 53, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Cazade, M.; Bidaud, I.; Hansen, P.B.; Lory, P.; Chemin, J. 5,6-EET potently inhibits T-type calcium channels: Implication in the regulation of the vascular tone. Pflugers Arch. Eur. J. Physiol. 2014, 466, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Ye, D.; Wang, X.; Seubert, J.M.; Graves, J.P.; Bradbury, J.A.; Zeldin, D.C.; Lee, H.C. Cardiac and vascular KATP channels in rats are activated by endogenous epoxyeicosatrienoic acids through different mechanisms. J. Physiol. 2006, 575, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yoon, S.P.; Toews, M.L.; Imig, J.D.; Hwang, S.H.; Hammock, B.D.; Padanilam, B.J. Pharmacological inhibition of soluble epoxide hydrolase prevents renal interstitial fibrogenesis in obstructive nephropathy. Am. J. Physiol. Ren. Physiol. 2015, 308, F131–F139. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Masaki, T.; Doi, S.; Arakawa, T.; Yokoyama, Y.; Doi, T.; Kohno, N.; Yorioka, N. PPAR-gamma agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF-beta. Lab. Investig. J. Tech. Methods Pathol. 2009, 89, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Huang, J.; Chen, J.; Lai, J.; Zhu, F.; Xu, X.; Wang, D.W. CYP2J2-Derived EETs Attenuated Angiotensin II-Induced Adventitial Remodeling via Reduced Inflammatory Response. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 39, 721–739. [Google Scholar] [CrossRef] [PubMed]
- Minuz, P.; Jiang, H.; Fava, C.; Turolo, L.; Tacconelli, S.; Ricci, M.; Patrignani, P.; Morganti, A.; Lechi, A.; McGiff, J.C. Altered release of cytochrome p450 metabolites of arachidonic acid in renovascular disease. Hypertension 2008, 51, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Liu, J.; Kumar, G.; Skapek, S.X.; Falck, J.R.; Imig, J.D. Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J. 2013, 27, 2946–2956. [Google Scholar] [CrossRef] [PubMed]
- Wahl, G.; Khan, A.; Fish, B.; Cohen, E.; Falck, J.; Imig, J. Novel epoxyeicosatrienoic acid analog ameliorates renal injury in a rat model of radiation nephropathy (690.7). FASEB J. 2014, 28 (Suppl. 1), 690–697. [Google Scholar]
- Skibba, M.; Hye Khan, M.A.; Kolb, L.L.; Yeboah, M.M.; Falck, J.R.; Amaradhi, R.; Imig, J.D. Epoxyeicosatrienoic Acid Analog Decreases Renal Fibrosis by Reducing Epithelial-to-Mesenchymal Transition. Front. Pharmacol. 2017, 8, 406. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yamamoto, T.; Newman, J.W.; Kim, I.H.; Watanabe, T.; Hammock, B.D.; Stewart, J.; Pollock, J.S.; Pollock, D.M.; Imig, J.D. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J. Am. Soc. Nephrol. 2004, 15, 1244–1253. [Google Scholar] [PubMed]
- Imig, J.D.; Carpenter, M.A.; Shaw, S. The Soluble Epoxide Hydrolase Inhibitor AR9281 Decreases Blood Pressure, Ameliorates Renal Injury and Improves Vascular Function in Hypertension. Pharmaceuticals 2009, 2, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Jung, O.; Jansen, F.; Mieth, A.; Barbosa-Sicard, E.; Pliquett, R.U.; Babelova, A.; Morisseau, C.; Hwang, S.H.; Tsai, C.; Hammock, B.D.; et al. Inhibition of the soluble epoxide hydrolase promotes albuminuria in mice with progressive renal disease. PLoS ONE 2010, 5, e11979. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Amjad, A.; Fu, Z.; Ma, Y.; Huang, D.; Xie, X.; Liu, F. Single Nucleotide Polymorphism of the CYP2J2 Gene is Associated with Essential Hypertension in Uygur Population in China. Biochem. Anal. Biochem. 2015, 4, 159. [Google Scholar]
- Genvigir, F.D.V.; Nishikawa, A.M.; Felipe, C.R.; Tedesco-Silva, H., Jr.; Oliveira, N.; Salazar, A.B.C.; Medina-Pestana, J.O.; Doi, S.Q.; Hirata, M.H.; Hirata, R.D.C. Influence of ABCC2, CYP2C8, and CYP2J2 Polymorphisms on Tacrolimus and Mycophenolate Sodium-Based Treatment in Brazilian Kidney Transplant Recipients. Pharmacotherapy 2017, 37, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Wang, M.H. Eicosanoids, beta-cell function, and diabetes. Prostaglandins Other Lipid Mediat. 2011, 95, 1–10. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Statistics about Diabetes. Available online: http://www.diabetes.org/diabetes-basics/statistics/ (accessed on 16 April 2018).
- World Health Organization Diabetes Fact Sheet. Available online: http://www.who.int/mediacentre/factsheets/fs312/en/ (accessed on 16 April 2018).
- Dai, M.; Wu, L.; Wang, P.; Wen, Z.; Xu, X.; Wang, D.W. CYP2J2 and Its Metabolites EETs Attenuate Insulin Resistance via Regulating Macrophage Polarization in Adipose Tissue. Sci. Rep. 2017, 7, 46743. [Google Scholar] [CrossRef] [PubMed]
- Spranger, J.; Kroke, A.; Mohlig, M.; Hoffmann, K.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Inflammatory cytokines and the risk to develop type 2 diabetes: Results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes 2003, 52, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Henquin, J.C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 2000, 49, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jorns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54 (Suppl. 2), S97–S107. [Google Scholar] [CrossRef] [PubMed]
- Zeldin, D.C.; Foley, J.; Boyle, J.E.; Moomaw, C.R.; Tomer, K.B.; Parker, C.; Steenbergen, C.; Wu, S. Predominant expression of an arachidonate epoxygenase in islets of Langerhans cells in human and rat pancreas. Endocrinology 1997, 138, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Chang, H.H.; Zhou, Y.; Zhang, S.; Hwang, S.H.; Morisseau, C.; Wang, C.Y.; Inscho, E.W.; Hammock, B.D.; Wang, M.H. Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J. Pharmacol. Exp. Ther. 2010, 334, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Dziumbla, S.; Lin, J.; Bibli, S.I.; Zukunft, S.; de Mos, J.; Awwad, K.; Fromel, T.; Jungmann, A.; Devraj, K.; et al. Inhibition of soluble epoxide hydrolase prevents diabetic retinopathy. Nature 2017, 552, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Birschbach, J.; Khan, M.A.H.; Sharma, A.; Hartmann, M.; Blöcher, R.; Proschak, E.; Imig, J.D. Dual Soluble Epoxide Hydrolase Inhibitor/PPAR-γ Agonist Reduces Kidney Injury in Metabolic Syndrome Rat. FASEB J. 2016, 30 (Suppl. 1), 740–745. [Google Scholar]


{kind=link}
| Disease State | Population | Risk | Significant Association | References |
|---|---|---|---|---|
| Premature myocardial infarction | Taiwanese | Increased | Yes | [14] |
| Myocardial infarction (MI) | Germanic | None | / | [15] |
| Caucasian in western Washington state | Increased | Yes | [16] | |
| South Indian | Increased | Yes | [17] | |
| Ischemic stroke | Chinese Han | Increased | Yes | [18,19] |
| Atherosclerosis | African-American | Decreased | Yes | [20] |
| Caucasian from central Germany | Increased | Yes | [21] | |
| Hypertension | African-American | None | / | [22,23] |
| Caucasian in Tennessee | Increased | Yes | [23] | |
| Chinese Han | Increased | Yes | [24] | |
| Russian | Increased | Yes | [25] | |
| Saudi Arabian | Increased | Yes | [26] | |
| Middle-aged Swedes | None | / | [27] | |
| South Indian | None | / | [17] |
| Source | Type of Effectors | Effect on CYP2J2 Expression | References |
|---|---|---|---|
| Primary human ventricular myocytes | Chemical | Little * | [61] |
| Reactive oxygen species | Increased * | [66] | |
| HepG2 cells | Chemical | Increased | [63,64] |
| Peripheral human mononuclear cells | Bacterial lipopolysaccharides | Increased | [65] |
| Human, first-trimester trophoblast-derived cells | Angiotensin-II | None | [67] |
| Hypoxia | None | [67] | |
| TNF-α | Increased | [67] |
| Disease State | Model | Condition or Treatment | Effects | References |
|---|---|---|---|---|
| Ischemic-reperfusion injury | Mouse overexpressing cardiac-specific CYP2J2 | Isolated perfused heart | Improved left ventricular recovery | [68] |
| Mouse overexpressing endothelial-specific CYP2J2 | Isolated perfused heart | No improvement on left ventricular function | [69] | |
| Ischemic-reperfusion injury and hypertension | Wistar rat | sEH inhibitor | Minimized cardiac damage | [70] |
| Ischemic-reperfusion injury and diabetes | Wistar rat | sEH inhibitor | Minimized cardiac damage | [70] |
| Atherosclerosis | Apolipoprotein-E deficient mouse | High-fat diet and recombinant adeno-associated virus mediated CYP2J2 expression | Reduced vascular apoptosis | [71] |
| Doxorubicin-induced cardiotoxicity | Mouse overexpressing cardiac-specific CYP2J2 | Acute and chronic doxorubin | Lower cardiomyocyte apoptosis and less damage to left ventricular function | [72] |
| Hypertrophy | AMPKα2 knockout mouse | Angiotensin-II to induce hypertension, and recombinant adeno-associated virus mediated CYP2J2 expression | Mitigated cardiac hypertrophic effect of hypertension | [73] |
| Mouse overexpressing cardiac-specific CYP2J2 | Chronic pressure induced hypertrophy via transverse aortic constriction surgery | Reduced ventricular arrhythmia | [74] | |
| Mouse overexpressing cardiac-specific CYP2J2 | Chronic β-adrenergic stimulation by infusion of isoproterenol | Reduced atrial arrhythmia | [74] | |
| Hypertrophy and heart failure | Mouse overexpressing cardiac-specific CYP2J2 | Infusion of isoproterenol or angiotension-II | Reduced damage associated with hypertrophy and heart failure | [75] |
| Hypertension | Spontaneously hypertensive mouse | Recombinant adeno-associated virus mediated CYP2J2 expression | Improved systolic blood pressure | [76] |
| Pulmonary arterial hypertension | Sprague-Dawley rat | Monocrotaline to induce pulmonary arterial hypertension and CYP2J2 gene delivery | Attenuated development and vascular remodeling | [77] |
| Chronic kidney failure | 5/6 nephrectomized rat | Recombinant adeno-associated virus mediated CYP2J2 expression | Protected remaining renal function | [78] |
| Abdominal aortic aneurysm | Apolipoprotein-E deficient mouse | Angiotensin-II to induce abdominal aortic aneurysm and recombinant adeno-associated virus mediated aortic-specific CYP2J2 expression | Activated PPAR and inhibited inflammatory responses | [79] |
| Diabetes | Mouse overexpressing cardiac-specific CYP2J2 | Streptozotocin to induce diabetes and high-fat diet | Improved blood glucose and insulin levels, glucose tolerance and uptake, and protected against myocardial hypertrophy | [80] |
| Diabetic mouse | Recombinant adeno-associated virus mediated CYP2J2 expression | Improved metabolic function and attenuated inflammatory responses | [81] | |
| Diabetic nephropathy | Mouse overexpressing endothelial-specific CYP2J2 | Streptozotocin to induce diabetes | Attenuated renal damage | [82] |
| Obesity | Mouse overexpressing endothelial-specific CYP2J2 | High-fat diet to induce obesity | Improved blood glucose and insulin levels and inflammation markers | [83] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aliwarga, T.; Evangelista, E.A.; Sotoodehnia, N.; Lemaitre, R.N.; Totah, R.A. Regulation of CYP2J2 and EET Levels in Cardiac Disease and Diabetes. Int. J. Mol. Sci. 2018, 19, 1916. https://doi.org/10.3390/ijms19071916
Aliwarga T, Evangelista EA, Sotoodehnia N, Lemaitre RN, Totah RA. Regulation of CYP2J2 and EET Levels in Cardiac Disease and Diabetes. International Journal of Molecular Sciences. 2018; 19(7):1916. https://doi.org/10.3390/ijms19071916
Chicago/Turabian StyleAliwarga, Theresa, Eric A. Evangelista, Nona Sotoodehnia, Rozenn N. Lemaitre, and Rheem A. Totah. 2018. "Regulation of CYP2J2 and EET Levels in Cardiac Disease and Diabetes" International Journal of Molecular Sciences 19, no. 7: 1916. https://doi.org/10.3390/ijms19071916
APA StyleAliwarga, T., Evangelista, E. A., Sotoodehnia, N., Lemaitre, R. N., & Totah, R. A. (2018). Regulation of CYP2J2 and EET Levels in Cardiac Disease and Diabetes. International Journal of Molecular Sciences, 19(7), 1916. https://doi.org/10.3390/ijms19071916