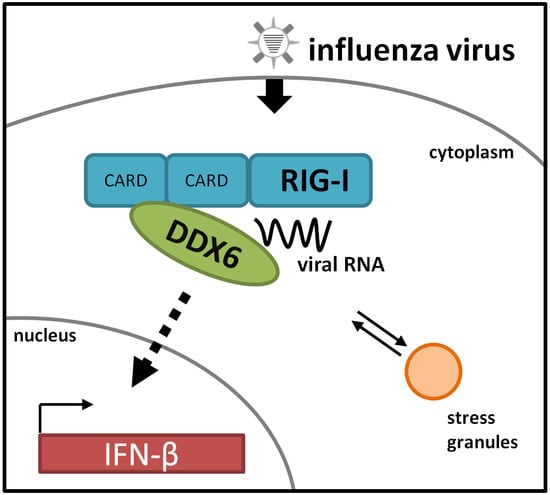
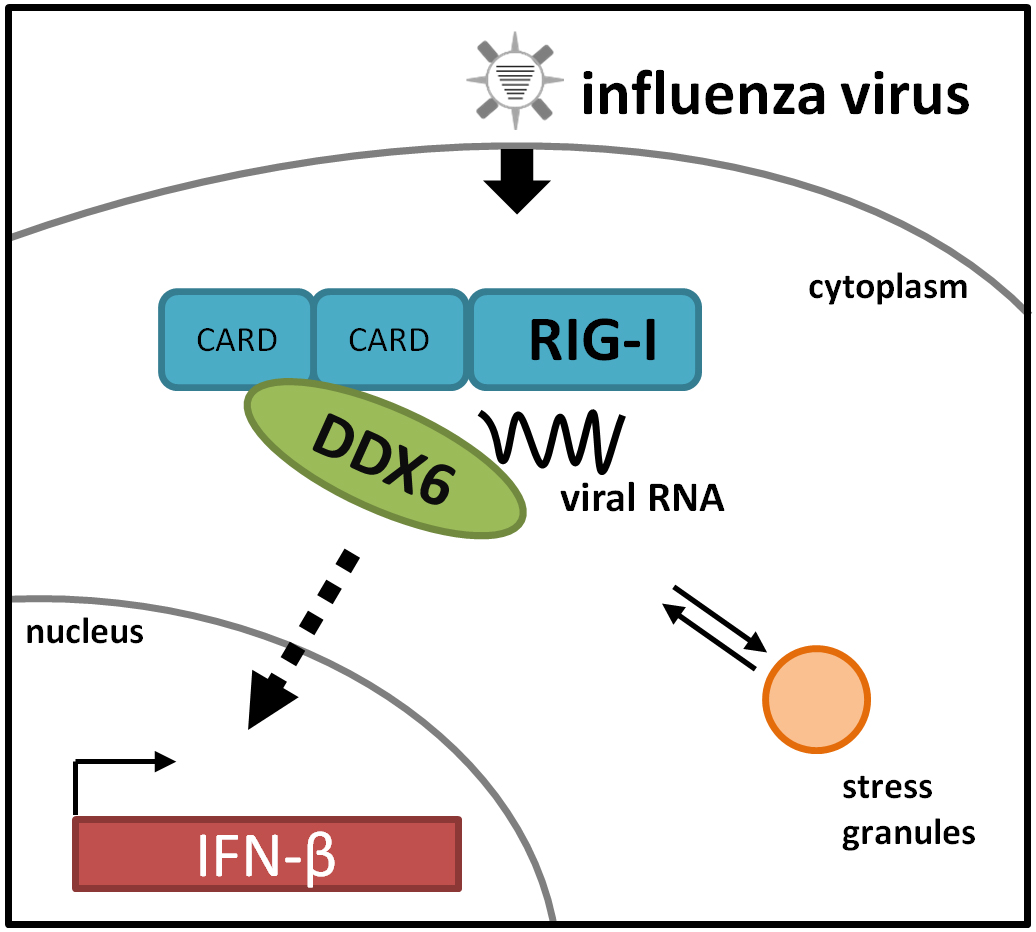
The RNA Helicase DDX6 Associates with RIG-I to Augment Induction of Antiviral Signaling

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
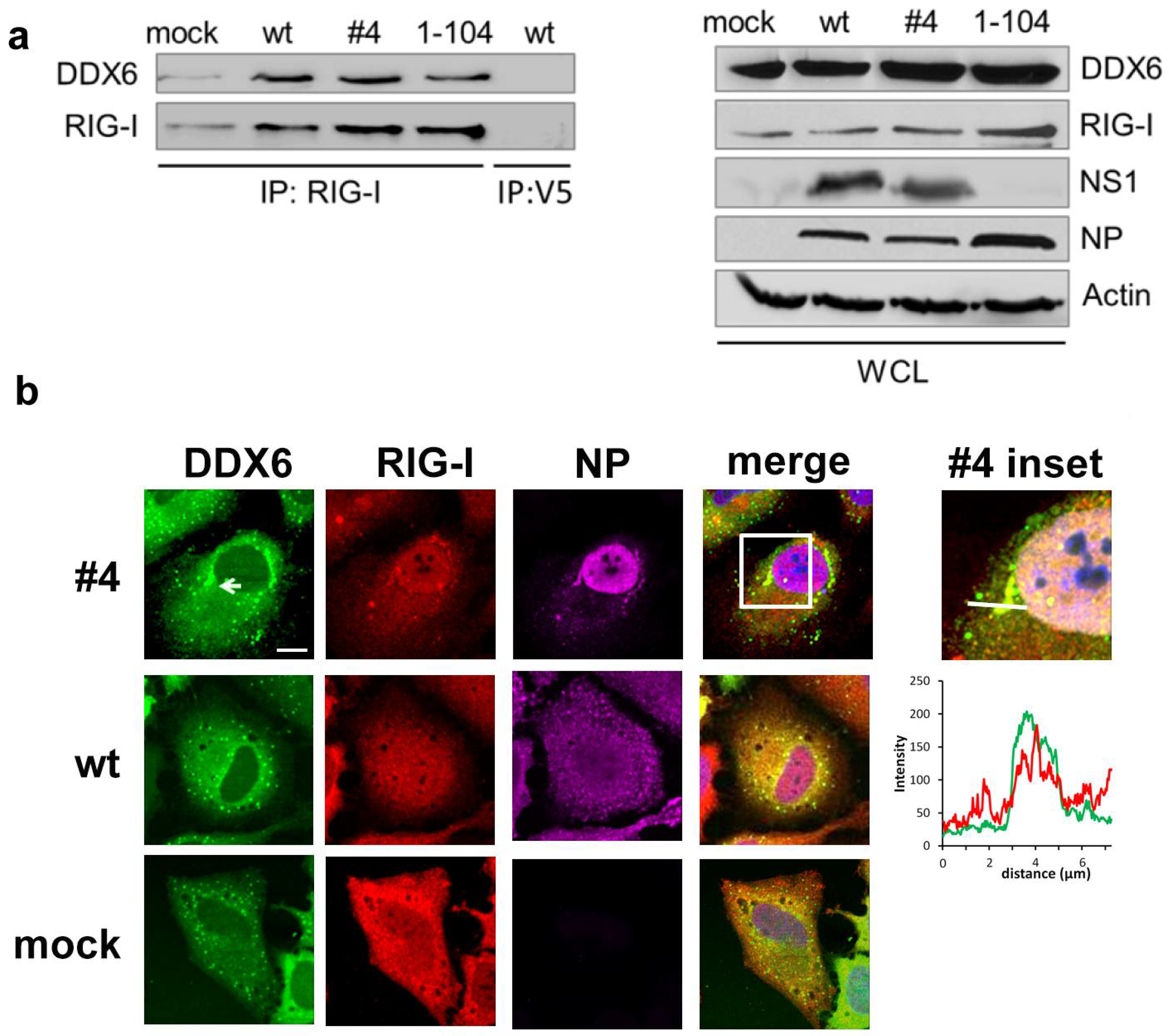
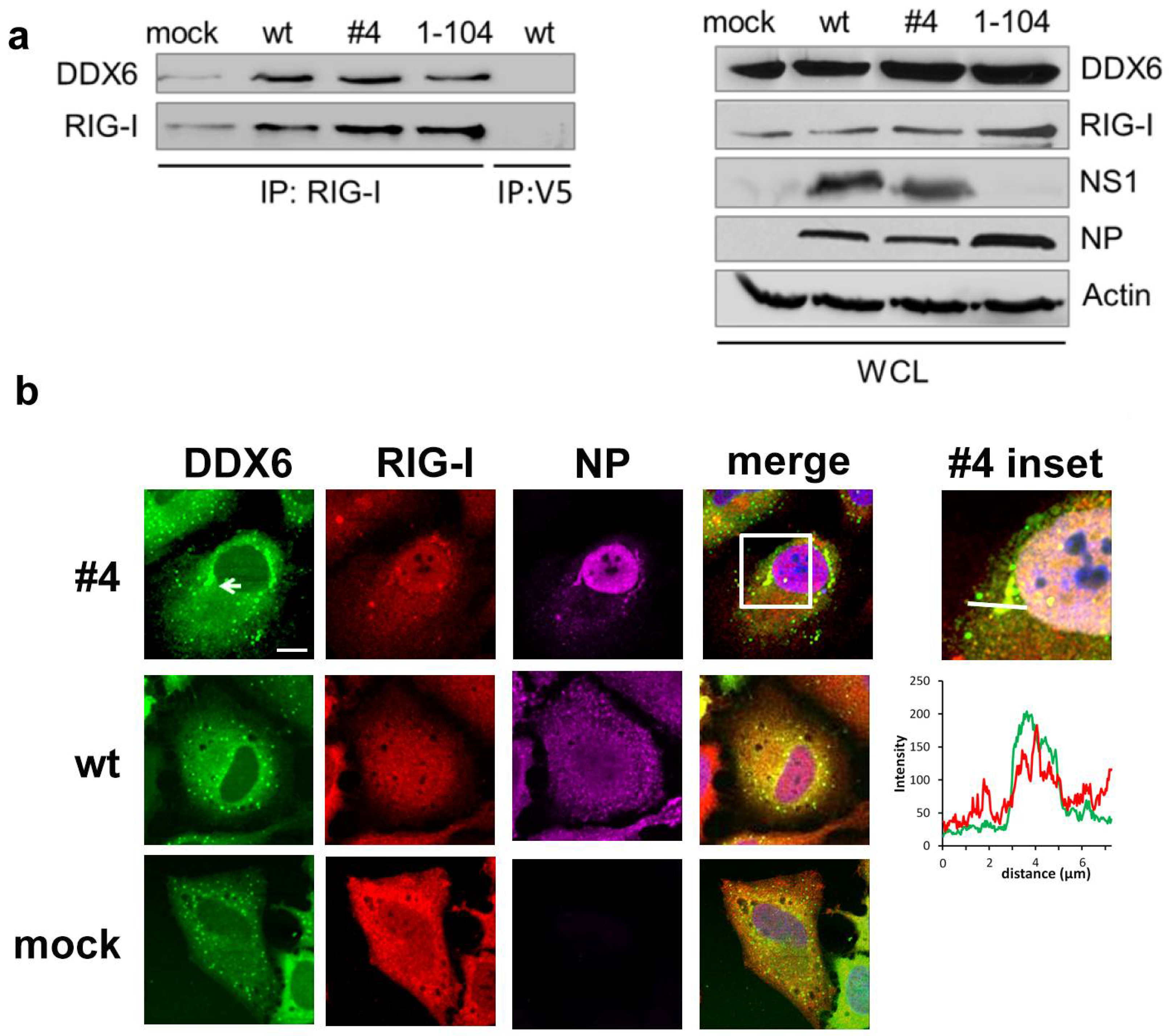
2.1. DDX6 Interacts with RIG-I
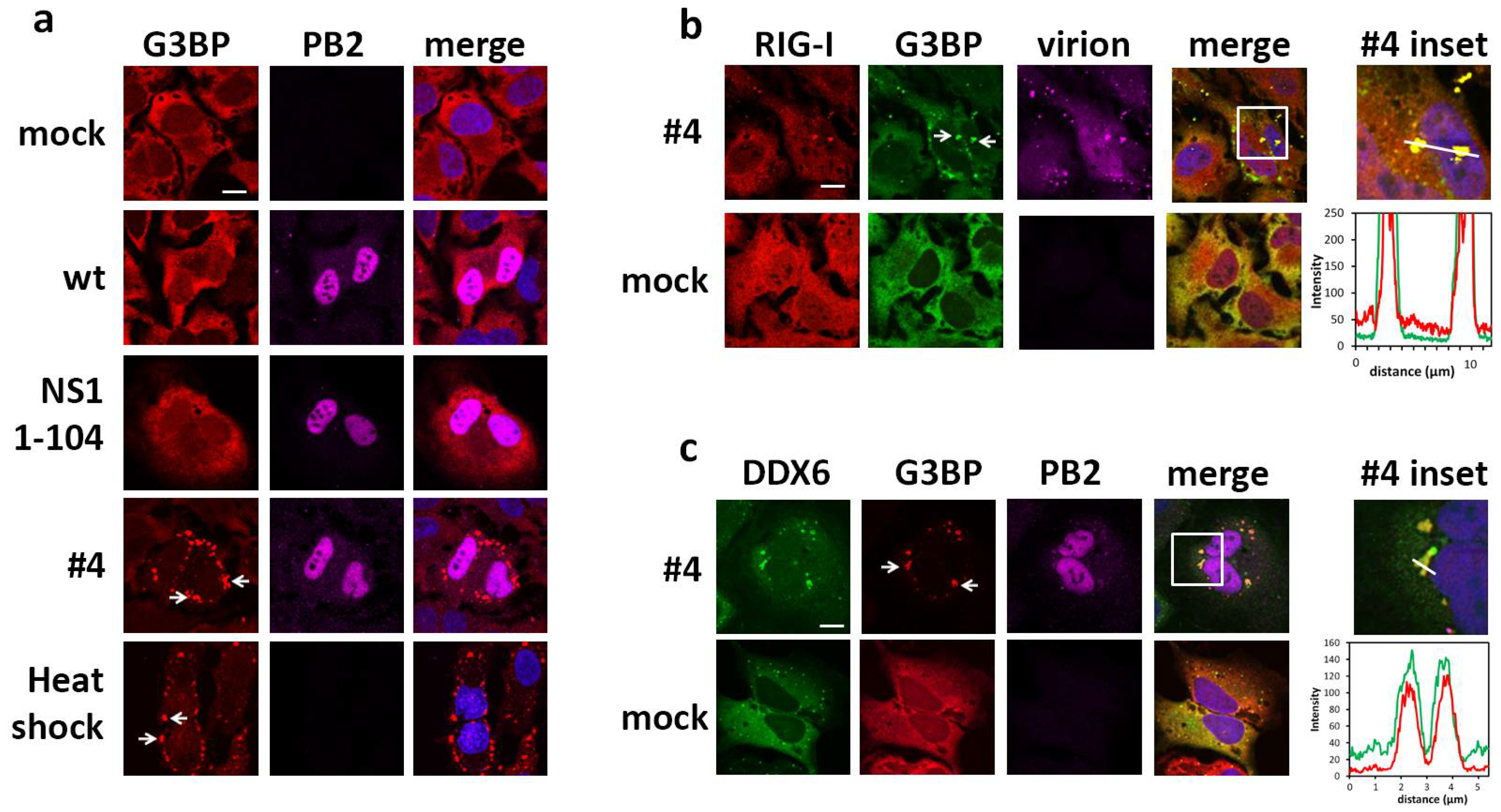
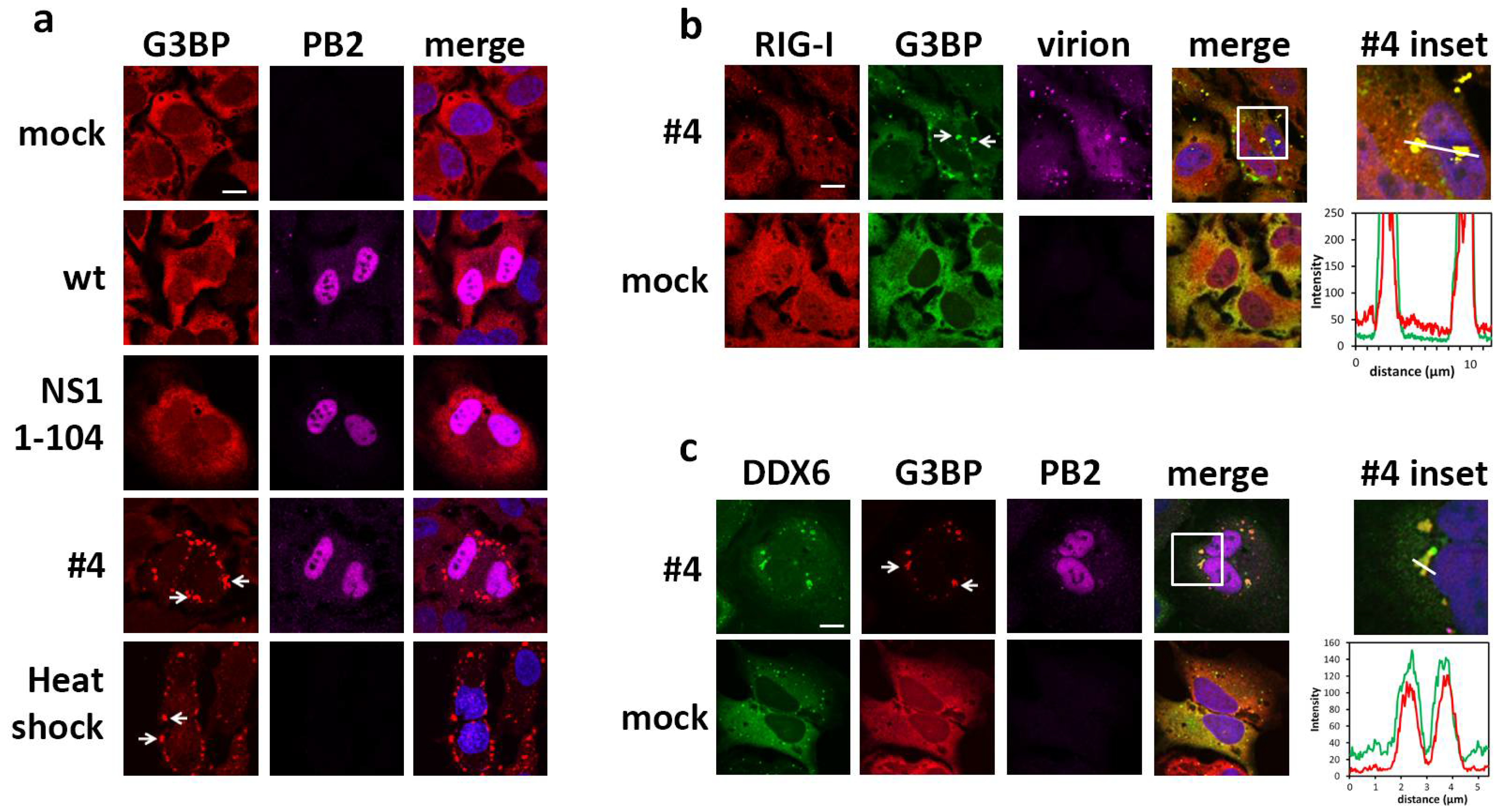
2.2. DDX6 Colocalizes with RIG-I in Stress Granules
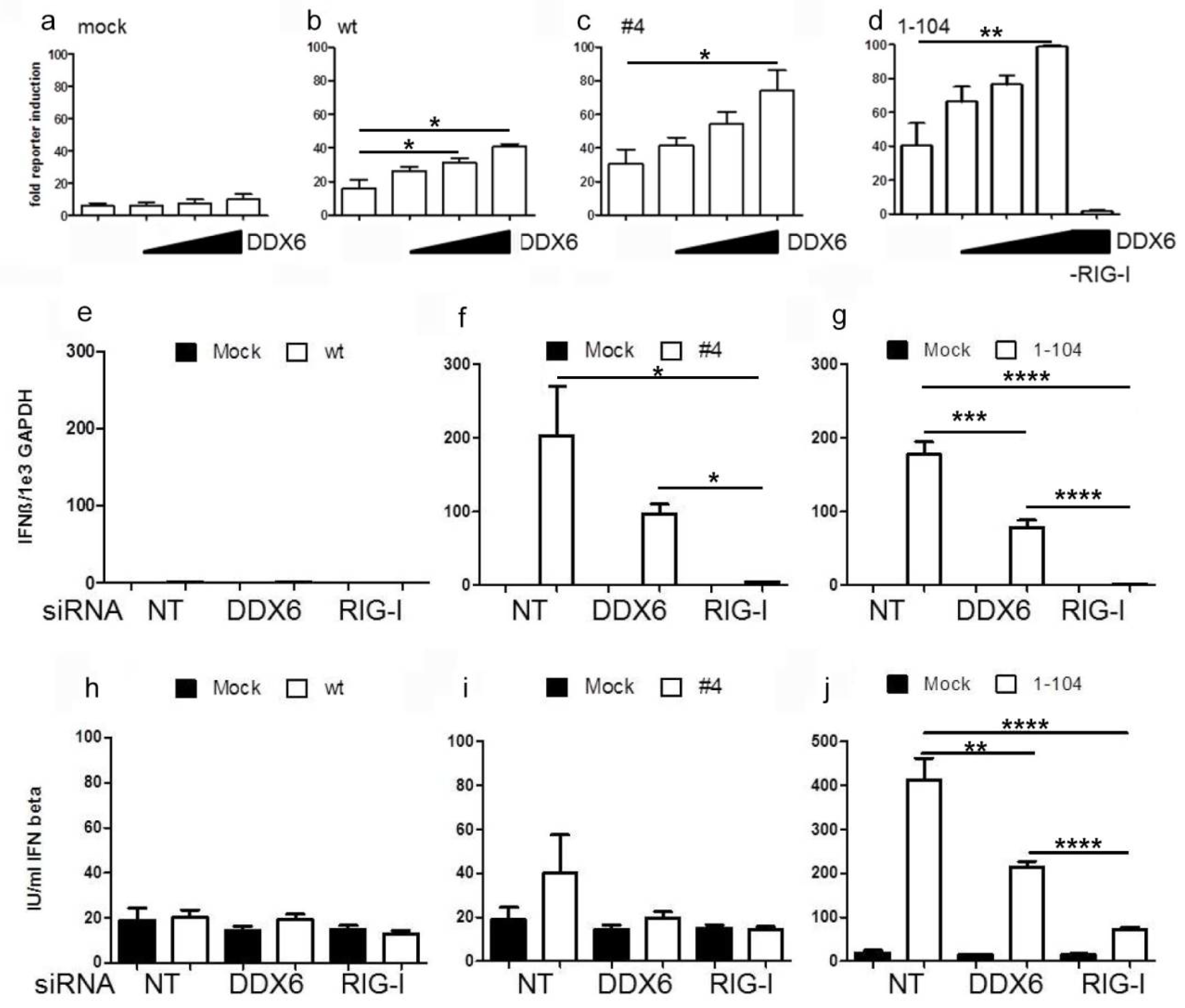
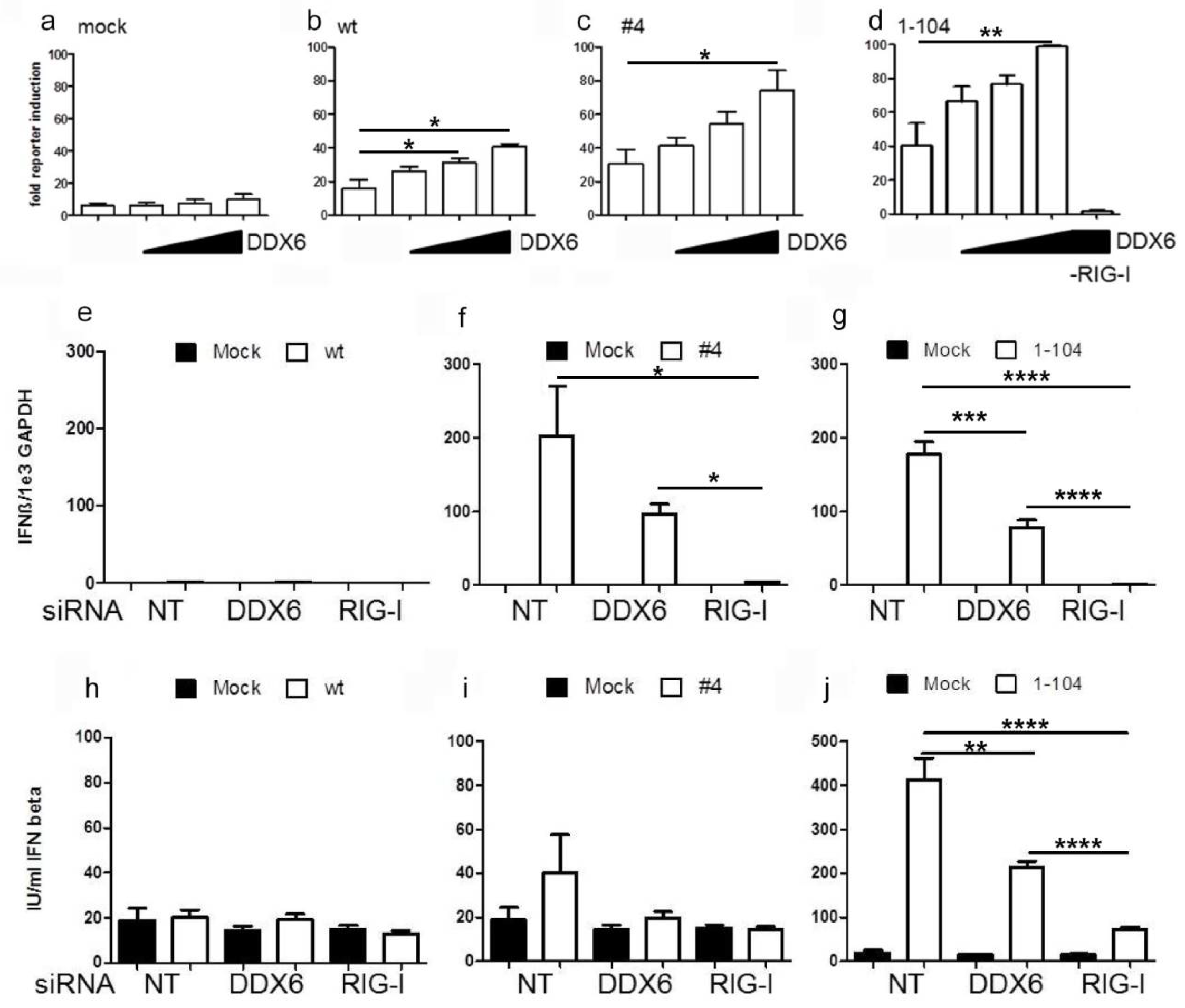
2.3. DDX6 Enhances RIG-I Mediated IFN-β Gene Expression
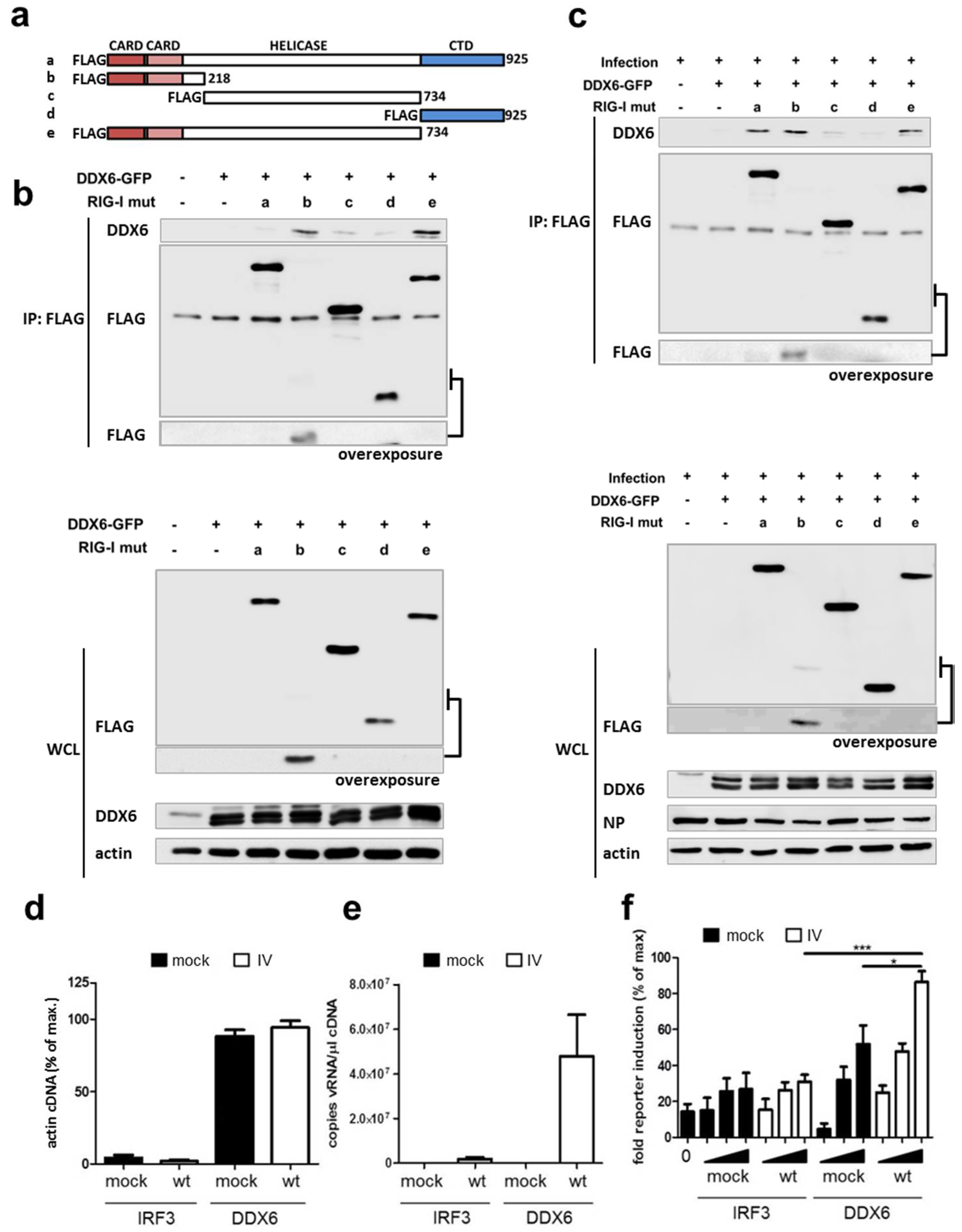
2.4. RIG-I Binds DDX6 via Its CARD Domains
2.5. DDX6 Binds Viral RNA That Activates RIG-I
3. Discussion
4. Materials and Methods
4.1. Cells and Viruses
4.2. Plasmids
4.3. RNA Interference
4.4. Immunoprecipitation and Antibodies
4.5. Confocal Microscopy
4.6. Quantitative RT-PCR
4.7. Statistical Analysis
4.8. IFN-β Luciferase Reporter Assay
4.9. Enyzme-Linked Immunosorbent Assay (ELISA)
4.10. Affinity Purification of RIG-I Complexes
4.11. Mass Spectrometric Analysis
4.12. Data Processing and Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guo, Z.; Chen, L.M.; Zeng, H.; Gomez, J.A.; Plowden, J.; Fujita, T.; Katz, J.M.; Donis, R.O.; Sambhara, S. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 2007, 36, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Mibayashi, M.; Martinez-Sobrido, L.; Loo, Y.M.; Cardenas, W.B.; Gale, M., Jr.; Garcia-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Opitz, B.; Rejaibi, A.; Dauber, B.; Eckhard, J.; Vinzing, M.; Schmeck, B.; Hippenstiel, S.; Suttorp, N.; Wolff, T. IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell. Microbiol. 2007, 9, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Onomoto, K.; Jogi, M.; Akaboshi, T.; Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015, 32, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. Rig-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-triphosphate rna is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M. Master sensors of pathogenic RNA—RIG-I like receptors. Immunobiology 2013, 218, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Devarkar, S.C.; Wang, C.; Miller, M.T.; Ramanathan, A.; Jiang, F.; Khan, A.G.; Patel, S.S.; Marcotrigiano, J. Structural basis for m7G recognition and 2′-o-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. USA 2016, 113, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.W.; Lee, M.C.; Wang, J.; Chen, C.Y.; Cheng, Y.W.; Lee, H. DDX3 loss by p53 inactivation promotes tumor malignancy via the MDM2/Slug/E-cadherin pathway and poor patient outcome in non-small-cell lung cancer. Oncogene 2014, 33, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, J.; Wicht, O.; Wolanski, J.C.; Baur, N.; Bastian, S.; Haas, D.A.; Matula, P.; Knapp, B.; Meyniel-Schicklin, L.; Wang, C.; et al. Phosphorylation-dependent feedback inhibition of RIG-I by DAPK1 identified by kinome-wide siRNA screening. Mol. Cell 2017, 65, 403–415.e8. [Google Scholar] [CrossRef] [PubMed]
- Peisley, A.; Wu, B.; Xu, H.; Chen, Z.J.; Hur, S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 2014, 509, 110–114. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, N.P.; Wies, E.; Stoll, A.; Gack, M.U. Conventional protein kinase C-alpha (PKC-alpha) and PKC-beta negatively regulate RIG-I antiviral signal transduction. J. Virol. 2012, 86, 1358–1371. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yuan, B.; Zhu, W.; Zhang, R.; Li, L.; Hao, X.; Chen, S.; Hou, F. UBE2D3 and UBE2N are essential for RIG-I-mediated MAVS aggregation in antiviral innate immunity. Nat. Commun. 2017, 8, 15138. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Miyashita, M.; Matsumoto, M.; Seya, T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 2013, 9, e1003533. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Ren, H.; Liu, Y.; Teeling, J.L.; Gu, J. Phosphorylation of RIG-I by casein kinase II inhibits its antiviral response. J. Virol. 2011, 85, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Wies, E.; Wang, M.K.; Maharaj, N.P.; Chen, K.; Zhou, S.; Finberg, R.W.; Gack, M.U. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 2013, 38, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Kuniyoshi, K.; Takeuchi, O.; Pandey, S.; Satoh, T.; Iwasaki, H.; Akira, S.; Kawai, T. Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I-mediated antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2014, 111, 5646–5651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, S.; Shiratori, S.; Yamato, H.; Kameyama, T.; Kitatsuji, C.; Kashigi, F.; Goto, S.; Kameoka, S.; Fujikura, D.; Yamada, T.; et al. Zaps is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat. Immunol. 2011, 12, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Kok, K.H.; Lui, P.Y.; Ng, M.H.; Siu, K.L.; Au, S.W.; Jin, D.Y. The double-stranded RNA-binding protein pact functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 2011, 9, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, Y.; Ghosh, A.; Cuevas, R.A.; Forero, A.; Dhar, J.; Ibsen, M.S.; Schmid-Burgk, J.L.; Schmidt, T.; Ganapathiraju, M.K.; et al. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 2014, 40, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Sun, T.; Li, G.; Pan, W.; Wang, K.; Dai, J. DEAD-Box helicase DDX25 is a negative regulator of type I interferon pathway and facilitates RNA virus infection. Front. Cell. Infect. Microbiol. 2017, 7, 356. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Jia, M.; Song, H.; Yu, Z.; Wang, W.; Li, Q.; Zhang, L.; Zhao, W.; Cao, X. The E3 ubiquitin ligase TRIM40 attenuates antiviral immune responses by targeting MDA5 and RIG-I. Cell Rep. 2017, 21, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Pindel, A.; Sadler, A. The role of protein kinase R in the interferon response. J. Interferon Cytokine Res. 2011, 31, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Anderson, P. Regulation of translation by stress granules and processing bodies. Prog. Mol. Biol. Transl. Sci. 2009, 90, 155–185. [Google Scholar] [PubMed]
- Reineke, L.C.; Lloyd, R.E. Diversion of stress granules and p-bodies during viral infection. Virology 2013, 436, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Khaperskyy, D.A.; Hatchette, T.F.; McCormick, C. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.W.; Song, W.; Wang, P.; Tai, H.; Chen, Y.; Zheng, M.; Wen, X.; Lau, S.Y.; Wu, W.L.; Matsumoto, K.; et al. The NS1 protein of influenza A virus interacts with cellular processing bodies and stress granules through RNA-associated protein 55 (RAP55) during virus infection. J. Virol. 2012, 86, 12695–12707. [Google Scholar] [CrossRef] [PubMed]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS ONE 2012, 7, e43031. [Google Scholar] [CrossRef]
- Onomoto, K.; Yoneyama, M.; Fung, G.; Kato, H.; Fujita, T. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014, 35, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villan, E.; Garcia-Sastre, A.; Gack, M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012, 8, e1003059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauber, B.; Heins, G.; Wolff, T. The influenza B virus nonstructural NS1 protein is essential for efficient viral growth and antagonizes beta interferon induction. J. Virol. 2004, 78, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Donelan, N.R.; Dauber, B.; Wang, X.; Basler, C.F.; Wolff, T.; Garcia-Sastre, A. The N- and C-terminal domains of the NS1 protein of influenza B virus can independently inhibit IRF-3 and beta interferon promoter activation. J. Virol. 2004, 78, 11574–11582. [Google Scholar] [CrossRef] [PubMed]
- Dauber, B.; Schneider, J.; Wolff, T. Double-stranded RNA binding of influenza B virus nonstructural NS1 protein inhibits protein kinase R but is not essential to antagonize production of alpha/beta interferon. J. Virol. 2006, 80, 11667–11677. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Loo, Y.M.; Horner, S.M.; Zornetzer, G.A.; Katze, M.G.; Gale, M., Jr. The mitochondrial targeting chaperone 14-3-3ε regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe 2012, 11, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010, 141, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Panas, M.D.; Kedersha, N.; McInerney, G.M. Methods for the characterization of stress granules in virus infected cells. Methods 2015, 90, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Khaperskyy, D.A.; Emara, M.M.; Johnston, B.P.; Anderson, P.; Hatchette, T.F.; McCormick, C. Influenza A virus host shutoff disables antiviral stress-induced translation arrest. PLoS Pathog. 2014, 10, e1004217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostareck, D.H.; Naarmann-de Vries, I.S.; Ostareck-Lederer, A. DDX6 and its orthologs as modulators of cellular and viral RNA expression. Wiley Interdiscip. Rev. RNA 2014, 5, 659–678. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.M.; Bidet, K.; Yinglin, A.; Ler, S.G.; Hogue, K.; Blackstock, W.; Gunaratne, J.; Garcia-Blanco, M.A. Quantitative mass spectrometry of denv-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biol. 2011, 8, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Boland, A.; Kuzuoglu-Ozturk, D.; Bawankar, P.; Loh, B.; Chang, C.T.; Weichenrieder, O.; Izaurralde, E. A DDX6-CNOT1 complex and W-binding pockets in CNOT9 reveal direct links between miRNA target recognition and silencing. Mol. Cell 2014, 54, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Aparicio, M.T.; Ayllon, J.; Leo-Macias, A.; Wolff, T.; Garcia-Sastre, A. Subcellular localizations of RIG-I, TRIM25, and MAVS complexes. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Arribas-Layton, M.; Chen, Y.; Lykke-Andersen, J.; Sen, G.L. Ddx6 orchestrates mammalian progenitor function through the mRNA degradation and translation pathways. Mol. Cell 2015, 60, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Ramanathan, A.; Miller, M.T.; Tang, G.Q.; Gale, M., Jr.; Patel, S.S.; Marcotrigiano, J. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 2011, 479, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Ernoult-Lange, M.; Baconnais, S.; Harper, M.; Minshall, N.; Souquere, S.; Boudier, T.; Benard, M.; Andrey, P.; Pierron, G.; Kress, M.; et al. Multiple binding of repressed mRNAs by the P-body protein Rck/p54. RNA 2012, 18, 1702–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Hogetsu, K.; Usukura, J.; Sato, T.; Kumasaka, T.; Akao, Y.; Tanaka, N. Structural insight of human DEAD-box protein Rck/p54 into its substrate recognition with conformational changes. Genes Cells 2006, 11, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Sediri, H.; Felgenhauer, U.; Binzen, I.; Banfer, S.; Jacob, R.; Brunotte, L.; Garcia-Sastre, A.; Schmid-Burgk, J.L.; Schmidt, T.; et al. Influenza virus adaptation PB2-627K modulates nucleocapsid inhibition by the pathogen sensor RIG-I. Cell Host Microbe 2015, 17, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Makela, S.M.; Osterlund, P.; Westenius, V.; Latvala, S.; Diamond, M.S.; Gale, M., Jr.; Julkunen, I. RIG-I signaling is essential for influenza B virus-induced rapid interferon gene expression. J. Virol. 2015, 89, 12014–12025. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, H.; Sutton, T.; Obadan, A.; Perez, D.R. Interactions between the influenza A virus RNA polymerase components and retinoic acid-inducible gene I. J. Virol. 2014, 88, 10432–10447. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, R.; Zhou, Q.; Xu, Z.; Li, C.; Wang, S.; Mao, A.; Zhang, X.; He, W.; Shu, H.B. LSm14A is a processing body-associated sensor of viral nucleic acids that initiates cellular antiviral response in the early phase of viral infection. Proc. Natl. Acad. Sci. USA 2012, 109, 11770–11775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumb, J.H.; Li, Q.; Popov, L.M.; Ding, S.; Keith, M.T.; Merrill, B.D.; Greenberg, H.B.; Li, J.B.; Carette, J.E. DDX6 represses aberrant activation of interferon-stimulated genes. Cell Rep. 2017, 20, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.S.; Takahasi, K.; Ng, C.S.; Ouda, R.; Onomoto, K.; Yoneyama, M.; Lai, J.C.; Lattmann, S.; Nagamine, Y.; Matsui, T.; et al. DHX36 enhances RIG-I signaling by facilitating PKR-mediated antiviral stress granule formation. PLoS Pathog. 2014, 10, e1004012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, M.; Oshiumi, H.; Matsumoto, M.; Seya, T. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell. Biol. 2011, 31, 3802–3819. [Google Scholar] [CrossRef] [PubMed]
- Thulasi Raman, S.N.; Liu, G.; Pyo, H.M.; Cui, Y.C.; Xu, F.; Ayalew, L.E.; Tikoo, S.K.; Zhou, Y. DDX3 interacts with influenza A virus NS1 and NP proteins and exerts antiviral function through regulation of stress granule formation. J. Virol. 2016, 90, 3661–3675. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liu, C.H.; Zhou, L.; Krug, R.M. Cellular DDX21 RNA helicase inhibits influenza A virus replication but is counteracted by the viral NS1 protein. Cell Host Microbe 2014, 15, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Y.; Segovia, J.A.; Chang, T.H.; Morris, I.R.; Berton, M.T.; Tessier, P.A.; Tardif, M.R.; Cesaro, A.; Bose, S. Damp molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: Role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog. 2014, 10, e1003848. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.Y.; Rana, T.M. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol. 2006, 4, e210. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, M.; Budt, M.; Wolff, T. Highly pathogenic H5N1 influenza A virus strains provoke heterogeneous IFN-α/β responses that distinctively affect viral propagation in human cells. PLoS ONE 2013, 8, e56659. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Núñez, R.D.; Budt, M.; Saenger, S.; Paki, K.; Arnold, U.; Sadewasser, A.; Wolff, T. The RNA Helicase DDX6 Associates with RIG-I to Augment Induction of Antiviral Signaling. Int. J. Mol. Sci. 2018, 19, 1877. https://doi.org/10.3390/ijms19071877
Núñez RD, Budt M, Saenger S, Paki K, Arnold U, Sadewasser A, Wolff T. The RNA Helicase DDX6 Associates with RIG-I to Augment Induction of Antiviral Signaling. International Journal of Molecular Sciences. 2018; 19(7):1877. https://doi.org/10.3390/ijms19071877
Chicago/Turabian StyleNúñez, Rocío Daviña, Matthias Budt, Sandra Saenger, Katharina Paki, Ulrike Arnold, Anne Sadewasser, and Thorsten Wolff. 2018. "The RNA Helicase DDX6 Associates with RIG-I to Augment Induction of Antiviral Signaling" International Journal of Molecular Sciences 19, no. 7: 1877. https://doi.org/10.3390/ijms19071877
APA StyleNúñez, R. D., Budt, M., Saenger, S., Paki, K., Arnold, U., Sadewasser, A., & Wolff, T. (2018). The RNA Helicase DDX6 Associates with RIG-I to Augment Induction of Antiviral Signaling. International Journal of Molecular Sciences, 19(7), 1877. https://doi.org/10.3390/ijms19071877