The Cyanobacterial Ribosomal-Associated Protein LrtA from Synechocystis sp. PCC 6803 Is an Oligomeric Protein in Solution with Chameleonic Sequence Properties


 ,
,  , , ,
, , , 
Abstract
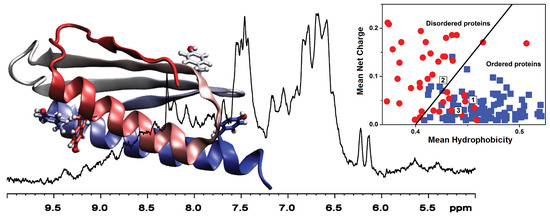
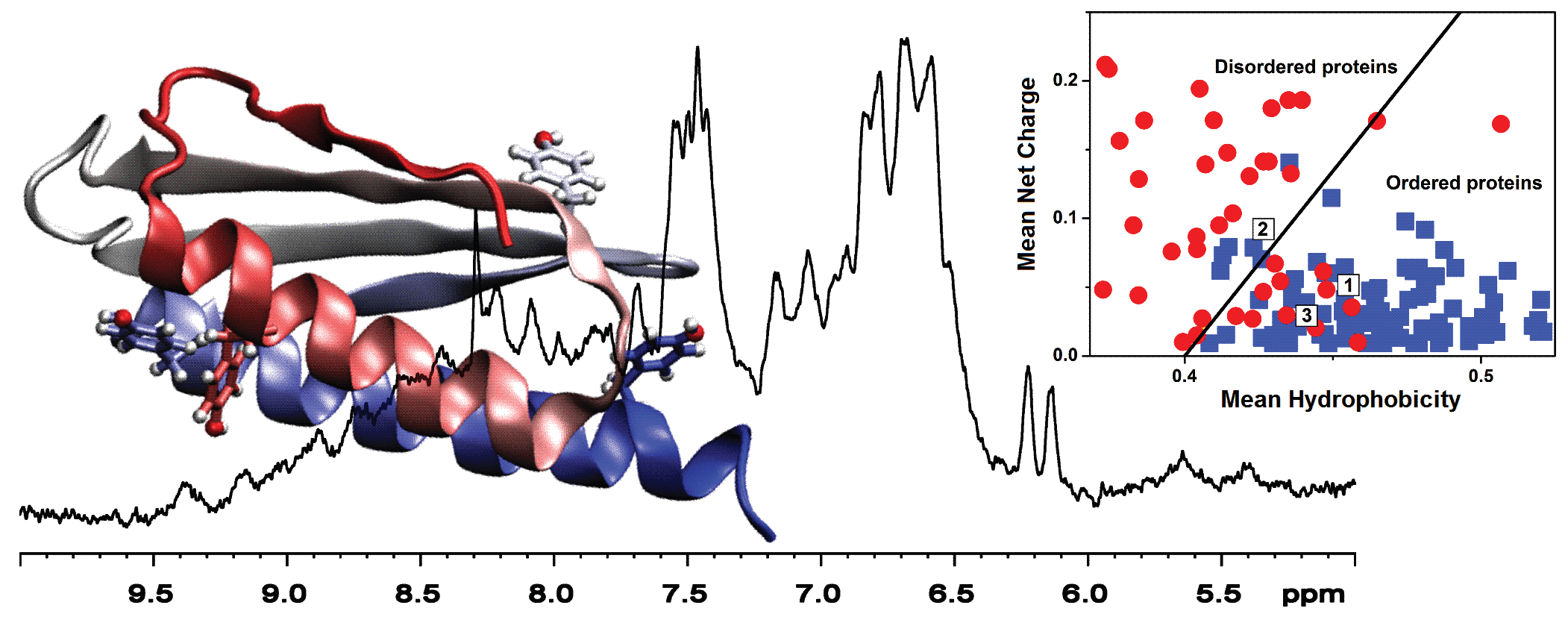
1. Introduction
2. Results
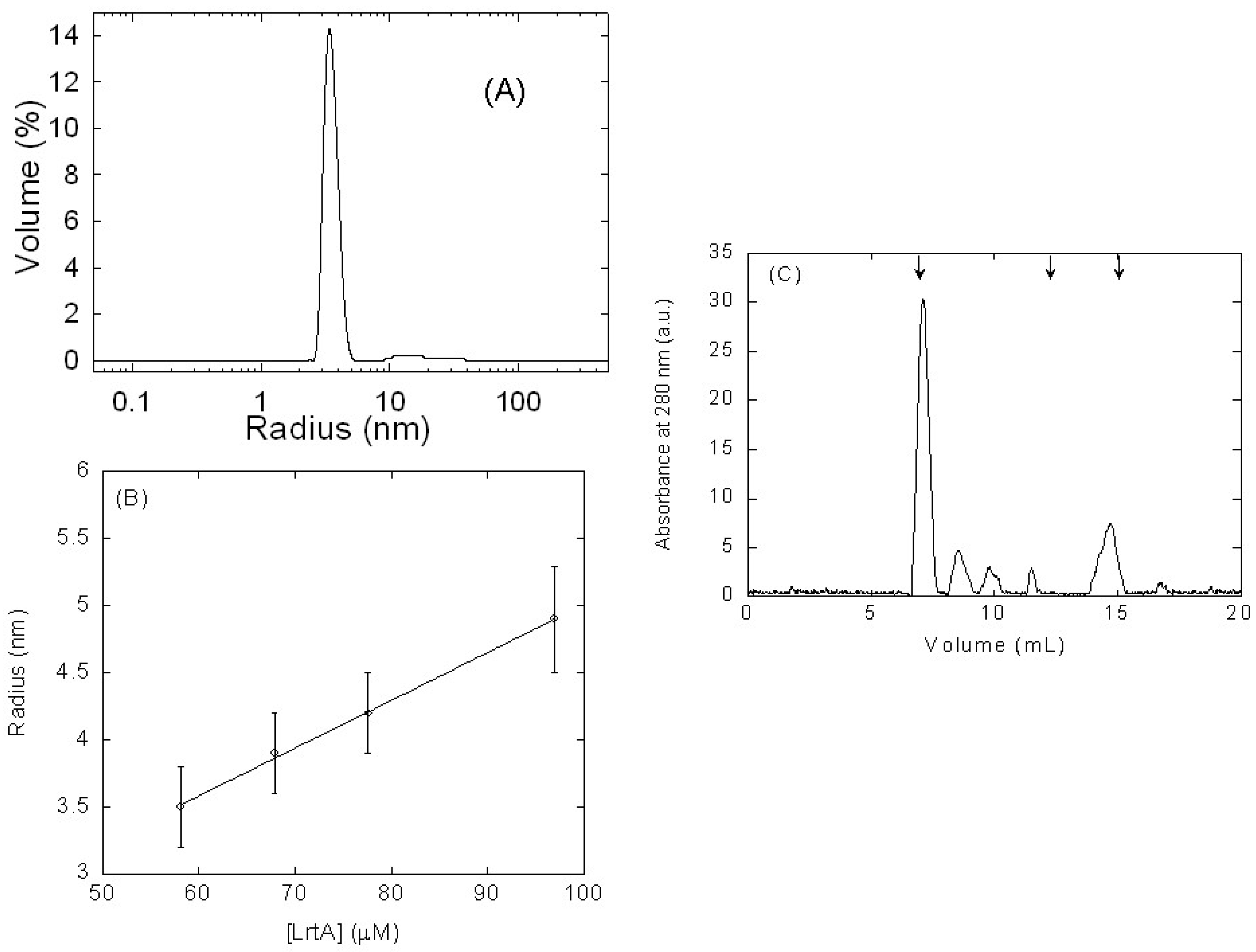
2.1. Isolated LrtA Was Involved in a Self-Association Equilibrium in Solution
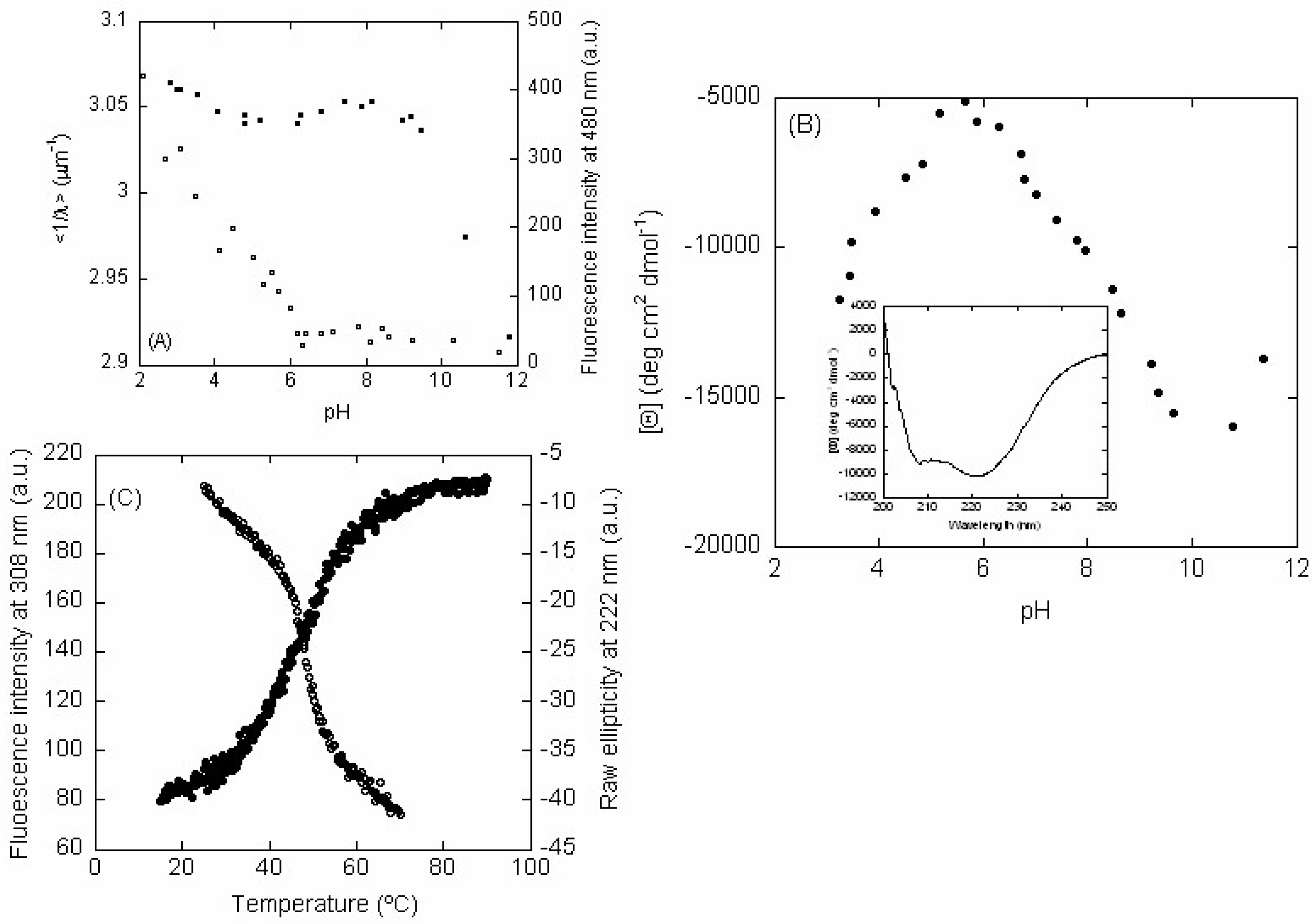
2.2. LrtA Acquired a Native-Like Conformation Between pH 6.0 and 9.0
2.2.1. Fluorescence
2.2.2. CD
2.3. LrtA Showed an Irreversible Complex Unfolding Equilibrium
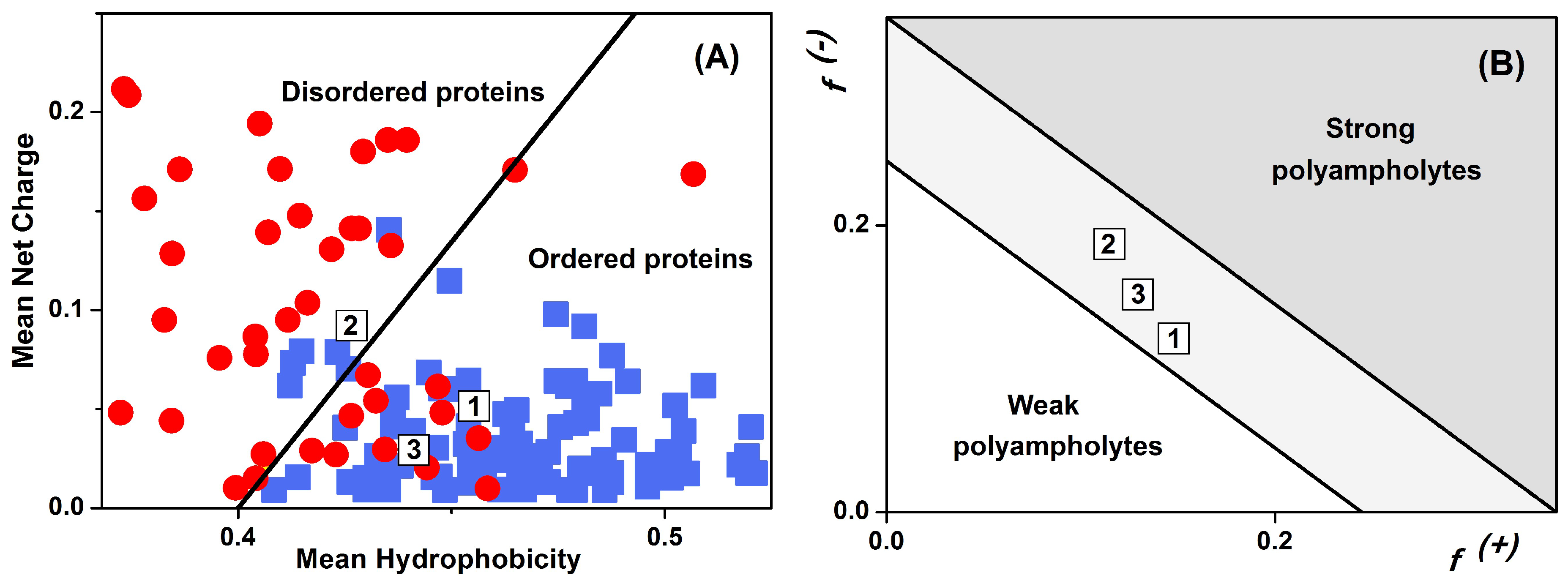
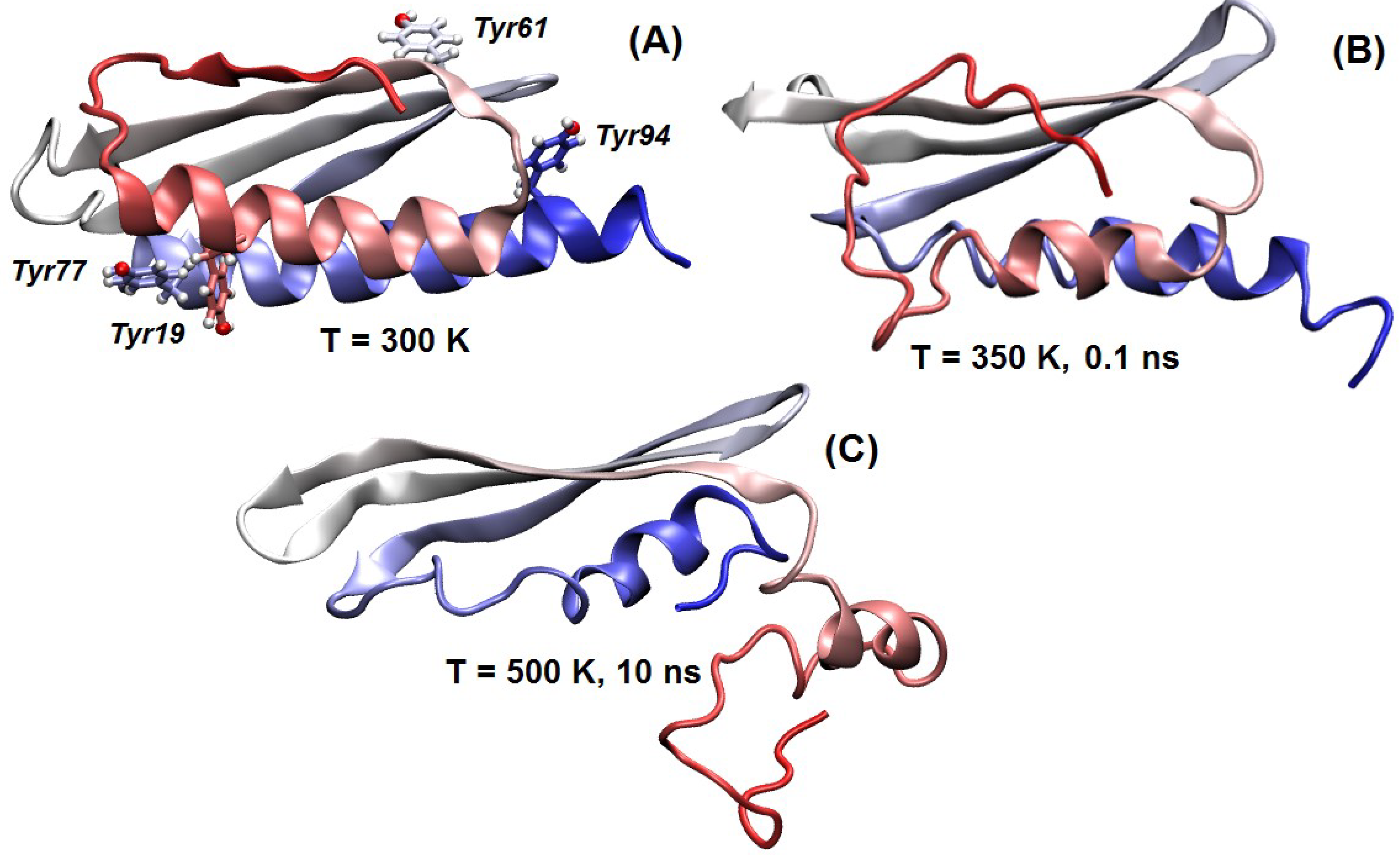
2.4. Sequence Properties and Molecular Modeling of LrtA
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Protein Expression and Purification
4.3. Fluorescence
4.4. Fluorescence Lifetimes
4.5. CD
4.6. NMR Spectroscopy
4.7. DLS
4.8. Molecular Modelling
4.9. SEC
4.10. Glutaraldehyde Cross-Linking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANS | 8-anilino-1-naphthalene sulfonic acid |
| β-ME | β-mercaptoethanol |
| CD | circular dichroism |
| DLS | dynamic light scattering |
| DOSY | diffusion ordered spectroscopy |
| EM | electron microscopy |
| GdmCl | guanidine hydrochloride |
| HPF | hibernating promoting factor |
| IDP | intrinsically disordered protein |
| IMAC | immobilized affinity chromatography |
| IRF | instrument response function |
| ITC | isothermal titration calorimetry |
| MD | molecular dynamics |
| NMR | nuclear magnetic resonance |
| PEI | polyethylenimine |
| RMF | ribosome modulation factor |
| RNase | ribonuclease |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SEC | size exclusion chromatography |
| UV | ultraviolet |
References
- Tan, X.; Varughese, M.; Widger, W.R. A light-repressed transcript found in Synechococcus sp. PCC 7002 is similar to a chloroplast-specific small subunit ribosomal protein and to a transcription modulator protein associated with sigma 54. J. Biol. Chem. 1994, 269, 20905–20912. [Google Scholar] [PubMed]
- Samartzidou, H.; Widger, W.R. Transcriptional and post-transcriptional control of mRNA from lrtA, a light-repressed transcript in Synechococcus sp. PC 7002. Plant Physiol. 1998, 117, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Galmozzi, C.V.; Florencio, F.J.; Muro-Pastor, M.I. The cyanobacterial ribosomal-associated protein LrtA is involved in post-stress survival in Synechocystis sp. PCC 6803. PLoS ONE 2016. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Wada, A. The 100S ribosome: Ribosomal hibernation induced by stress. Wiley Interdiscip. Rev. RNA 2014, 5, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Khusainov, I.; Vicens, Q.; Ayupov, R.; Usachev, K.; Myasnikov, A.; Simonetti, A.; Validov, S.; Kieffer, B.; Yuspova, G.; Yusupov, M.; et al. Structures and dynamics of hibernating ribosomes from Staphylococcus aureus mediated by intermolecular interactions of HPF. EMBO J. 2017, 36, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Starosta, A.L.; Lasak, J.; Jung, K.; Wilson, D.N. The bacterial translation stress response. FEMS Microbiol. Rev. 2014, 38, 1172–1201. [Google Scholar] [CrossRef] [PubMed]
- Agafonov, D.E.; Spirin, A.S. The ribosome-associated inhibitor A reduces translation errors. Biochem. Biophys. Res. Commun. 2004, 320, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Polikanov, Y.S.; Blaha, G.M.; Steitz, T.A. How hibernation factors RMF, HPF and YfiA turn off protein synthesis. Science 2012, 336, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Ueta, M.; Yoshida, H.; Wada, C.; Baba, T.; Mori, H.; Wada, A. Ribosome binding proteins YHbH and YfiA have opposite functions during 100S formation in the stationary phase of Escherichia coli. Genes Cells 2005, 10, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Ueta, M.; Ohniwa, R.L.; Yoshida, H.; Maki, Y.; Wada, C.; Wada, A. Role of HPF (hibernation promoting factor) in translational activity in Escherichia coli. J. Biochem. 2008, 143, 425–433. [Google Scholar] [CrossRef] [PubMed]
- De Bari, H.; Berry, E.A. Structure of Vibrio cholerae ribosome hibernation factor. Acta Crystallogr. Sect. F 2013, 69, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 2nd ed.; Plenum Press: New York, NY, USA, 1999. [Google Scholar]
- Albani, J.R. Principle and Applications of Fluorescence Spectroscopy; Blackwell Publishing: Oxford, UK, 2007. [Google Scholar]
- Cantor, C.R.; Schimmel, P.R. Biophysical Chemistry; W. H. Freeman: New York, NY, USA, 1980. [Google Scholar]
- Cavanagh, J.F.; Wayne, J.; Palmer, A.G., III; Skelton, N.J. Protein NMR Spectroscopy: Principles and Practice, 1st ed.; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef] [PubMed]
- Nobbmann, U.; Connah, M.; Fish, B.; Varley, P.; Gee, C.; Mulot, S.; Chen, J.; Zhou, L.; Lu, Y.; Sheng, F.; et al. Dynamic light scattering as a relative tool for assessing the molecular integrity and stability of monoclonal antibodies. Biotechnol. Genet. Eng. Rev. 2007, 24, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Grimsley, G.R.; Scholtz, J.M. Protein ionizable groups: pK values and their contribution to protein stability and solubility. J. Biol. Chem. 2009, 284, 13285–13289. [Google Scholar] [CrossRef] [PubMed]
- Grimsley, G.R.; Scholtz, J.M.; Pace, C.N. A summary of the measured pK values of the ionizable groups in folded proteins. Protein Sci. 2009, 18, 247–251. [Google Scholar] [PubMed]
- Lux, B.; Gerard, D.; Laustriat, G. Tyrosine fluorescence of S8 and S15 Escherichia coli ribosomal proteins. FEBS Lett. 1977, 80, 66–70. [Google Scholar] [CrossRef]
- Soengas, M.S.; Mateo, C.R.; Salas, M.; Acuña, A.U.; Gutiérrez, C. Structural features of φ29 single-stranded DNA-binding protein. Environment of tyrosines in terms of complex formation with DNA. J. Biol. Chem. 1997, 272, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analysis from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef] [PubMed]
- Woody, R.W. Circular dichroism. Methods Enzymol. 1995, 246, 34–71. [Google Scholar] [PubMed]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta 2005, 1751, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmók, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.R.; Thomson, R.; McNeil, P.; Esnouf, R.M. RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, M.Y.; Sokolovskiy, I.V.; Galzitskaya, O.V. IsUnstruct: Prediction of the residue status to be ordered or disordered in the protein chain by a method based on the Ising model. J. Biomol. Struct. Dyn. 2013, 31, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.; Gillespie, J.; Fink, A. Why are “natively unfolded” proteins unstructured under physiological conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered proteins. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, H.; Zheng, W.M.; Xu, D.; Zhu, J.; Wang, B.; Ning, K.; Sun, S.; Li, S.C.; Bu, D. FALCON@home: A high-throughput protein structure prediction server based on remote homologue recognition. Bioinformatics 2016, 32, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; Wiley and Sons: New York, NY, USA, 1986. [Google Scholar]
- Pantoja-Uceda, D.; Neira, J.L.; Saelices, L.; Robles-Rengel, L.; Florencio, F.J.; Muro-Pastor, M.I.; Santoro, J. Dissecting the binding between glutamine synthetase and its tow natively unfolded protein inhibitors. Biochemistry 2016, 55, 3370–3382. [Google Scholar] [CrossRef] [PubMed]
- Rizzuti, B.; Daggett, V. Using simulations to provide the framework for experimental protein folding studies. Arch. Biochem. Biophys. 2013, 531, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Miroux, B.; Walker, J.E. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996, 260, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.R. Protein precipitation techniques. Methods Enzymol. 2009, 463, 331–342. [Google Scholar] [PubMed]
- Dunn, M.J. Initial planning: Determination of total protein concentration. In Protein Purification Methods; Harris, E.L.V., Angal, S., Eds.; Oxford University Press: Oxford, UK, 1995; pp. 10–20. [Google Scholar]
- Neira, J.L. Fluorescence, circular dichroism and mass spectrometry as tools to study virus structure. Subcell. Biochem. 2013, 68, 177–202. [Google Scholar] [PubMed]
- Tinoco, I., Jr.; Sauer, K.; Wang, J.C. Physical Chemistry: Principles and Applications in Biological Sciences, 3rd ed.; Prentice-Hall: New York, NY, USA, 1995; pp. 585–588. [Google Scholar]
- Bokma, J.T.; Johnson, W.C., Jr.; Blok, J. CD of the Li-salt of DNA in ethanol/water mixtures: Evidence for the B- to C-form transition in solution. Biopolymers 1987, 26, 893–909. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Johnson, W.C., Jr. Comparison of base inclination in Ribo-AU and Deoxyribo-AT polymers. Biopolymers 1995, 36, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Charnavets, T.; Nunvar, J.; Necasova, I.; Völker, J.; Breslauer, K.J.; Schneider, B. Conformational diversity of the single-stranded DNA from bacterial repetitive extragenic palindromes: Implications for the DNA recognition elements of transposases. Biopolymers 2015, 103, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Atkins, P.; De Paula, J. Physical Chemistry for the Life Sciences, 1st ed.; W. H. Freeman: Oxford, UK, 2006; pp. 565–566. [Google Scholar]
- Neira, J.L.; Hornos, F.; Bacarizo, J.; Cámara-Artigas, A.; Gómez, J. The monomeric species of the regulatory domain of Tyrosine Hydroxylase has a low conformational stability. Biochemistry 2016, 55, 6209–6220. [Google Scholar] [CrossRef] [PubMed]
- Bevington, P.R.; Robinson, K.D. Data Reduction and Error Analysis for the Physical Sciences, 3rd ed.; McGraw-Hill: New York, NY, USA, 2003. [Google Scholar]
- James, D.R.; Siemiarczuk, A.; Ware, W.R. Stroboscopic optical boxcar technique for the determination of fluorescence lifetimes. Rev. Sci. Instrum. 1992, 63, 1710–1716. [Google Scholar] [CrossRef]
- Piotto, M.; Saudek, V.; Sklenar, V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 1993, 2, 661–665. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, J. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Evoli, S.; Guzzi, R.; Rizzuti, B. Molecular simulations of β-lactoglobulin complexed with fatty acids reveal the structural basis of ligand affinity to internal and possible external binding sites. Proteins 2014, 82, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Rizzuti, B.; Bartucci, R.; Sportelli, L.; Guzzi, R. Fatty acid binding into the highest affinity site of human serum albumin observed in molecular dynamics simulation. Arch. Biochem. Biophys. 2015, 579, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, A.; Rizzuti, B.; Guzzi, R. Stereoselective and domain-specific effects of ibuprofen on the thermal stability of human serum albumin. Eur. J. Pharm. Sci. 2018, 112, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, R.; Böhm, G.; Lilie, H.; Jaenicke, R. Folding proteins. In Protein Function: A Practical Approach, 2nd ed.; Creighton, T.E., Ed.; Oxford University Press: Oxford, UK, 1997; pp. 57–99. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (μM) | τ1 (ns) | a1 | τ2 (ns) | a2 | <τ> (ns) | χ2 |
|---|---|---|---|---|---|---|
| 98 | 0.49 ± 0.08 | 1.9 ± 0.2 | 3.4 ± 0.1 | 0.072 ± 0.005 | 1.101 | 1.384 |
| 9.8 | 0.46 ± 0.07 | 2.0 ± 0.2 | 3.6 ± 0.2 | 0.048 ± 0.006 | 0.9622 | 1.528 |
| 7.8 | 0.54 ± 0.08 | 1.8 ± 0.2 | 2.7 ± 0. 2 | 0.07 ± 0.01 | 0.9037 | 1.35 |
| 1.9 | 0.69 ± 0.09 | 0.71 ± 0.09 | 14.9 ± 0. 8 | 0.0156 ± 0.0008 | 5.233 | 1.22 |
| 0.98 | 0.48 ± 0.07 | 1.0 ± 0.1 | 19.5 ± 0.4 | 0.0104 ± 0.0005 | 5.996 | 1.238 |
| Solution Conditions | KI | Acrylamide | |
|---|---|---|---|
| Ksv (M−1) | Ksv (M−1) | υ (M−1) | |
| pH 3.0 | - a | 7.7 ± 0.5 | 2.1 ± 0.1 |
| pH 7.0 | 1.63 ± 0.03 | 11 ± 1 | 2.2 ± 0.3 |
| pH 11 | 0.78 ± 0.02 | 9.7 ± 0.5 | 0.5 ± 0.1 |
| GdmCl (pH 7.0) | 3.1 ± 0.1 | 22 ± 4 | 2.0 ± 0.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contreras, L.M.; Sevilla, P.; Cámara-Artigas, A.; Hernández-Cifre, J.G.; Rizzuti, B.; Florencio, F.J.; Muro-Pastor, M.I.; García de la Torre, J.; Neira, J.L. The Cyanobacterial Ribosomal-Associated Protein LrtA from Synechocystis sp. PCC 6803 Is an Oligomeric Protein in Solution with Chameleonic Sequence Properties. Int. J. Mol. Sci. 2018, 19, 1857. https://doi.org/10.3390/ijms19071857
Contreras LM, Sevilla P, Cámara-Artigas A, Hernández-Cifre JG, Rizzuti B, Florencio FJ, Muro-Pastor MI, García de la Torre J, Neira JL. The Cyanobacterial Ribosomal-Associated Protein LrtA from Synechocystis sp. PCC 6803 Is an Oligomeric Protein in Solution with Chameleonic Sequence Properties. International Journal of Molecular Sciences. 2018; 19(7):1857. https://doi.org/10.3390/ijms19071857
Chicago/Turabian StyleContreras, Lellys M., Paz Sevilla, Ana Cámara-Artigas, José G. Hernández-Cifre, Bruno Rizzuti, Francisco J. Florencio, María Isabel Muro-Pastor, José García de la Torre, and José L. Neira. 2018. "The Cyanobacterial Ribosomal-Associated Protein LrtA from Synechocystis sp. PCC 6803 Is an Oligomeric Protein in Solution with Chameleonic Sequence Properties" International Journal of Molecular Sciences 19, no. 7: 1857. https://doi.org/10.3390/ijms19071857
APA StyleContreras, L. M., Sevilla, P., Cámara-Artigas, A., Hernández-Cifre, J. G., Rizzuti, B., Florencio, F. J., Muro-Pastor, M. I., García de la Torre, J., & Neira, J. L. (2018). The Cyanobacterial Ribosomal-Associated Protein LrtA from Synechocystis sp. PCC 6803 Is an Oligomeric Protein in Solution with Chameleonic Sequence Properties. International Journal of Molecular Sciences, 19(7), 1857. https://doi.org/10.3390/ijms19071857