Gene Profiling in Late Blight Resistance in Potato Genotype SD20



Abstract
1. Introduction
2. Results
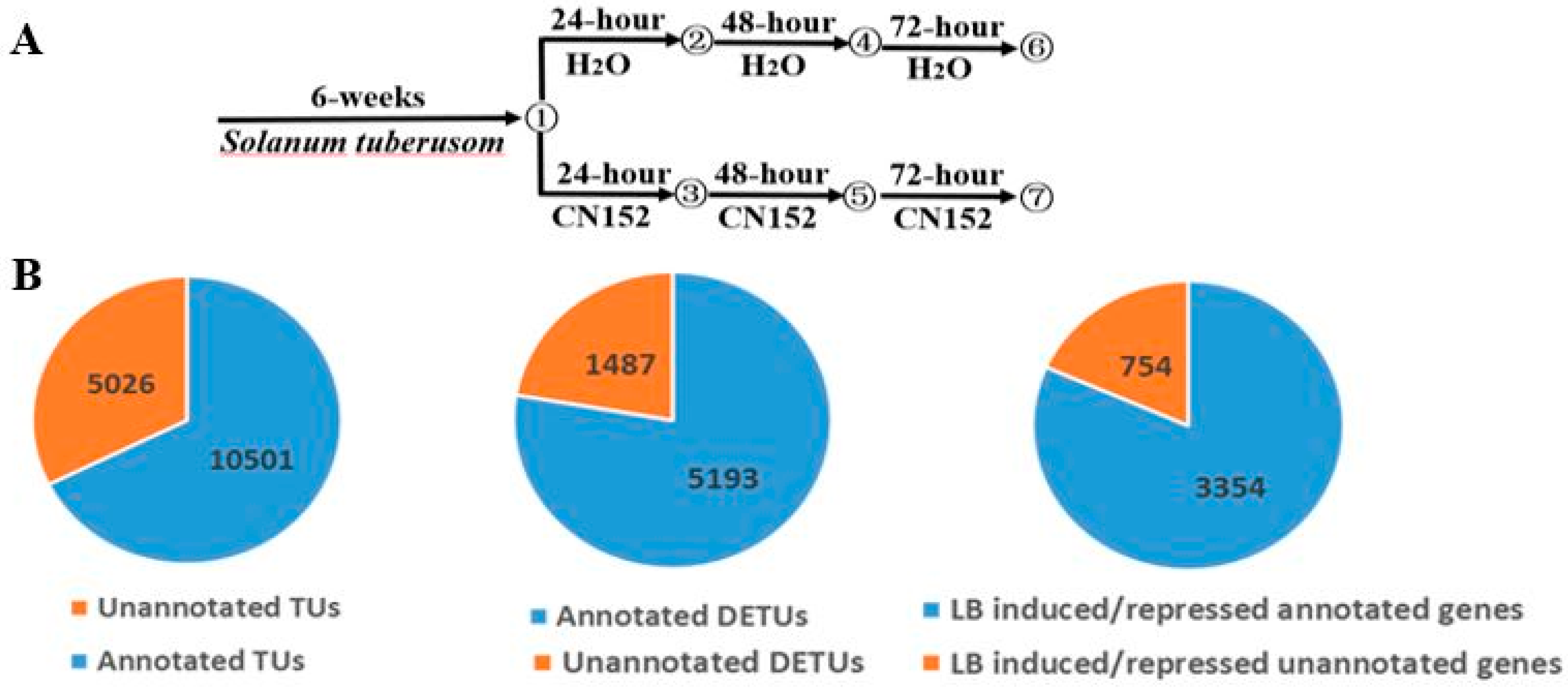
2.1. RNA-Sequencing and Transcriptome Assembly
2.2. Differential Expression Analysis
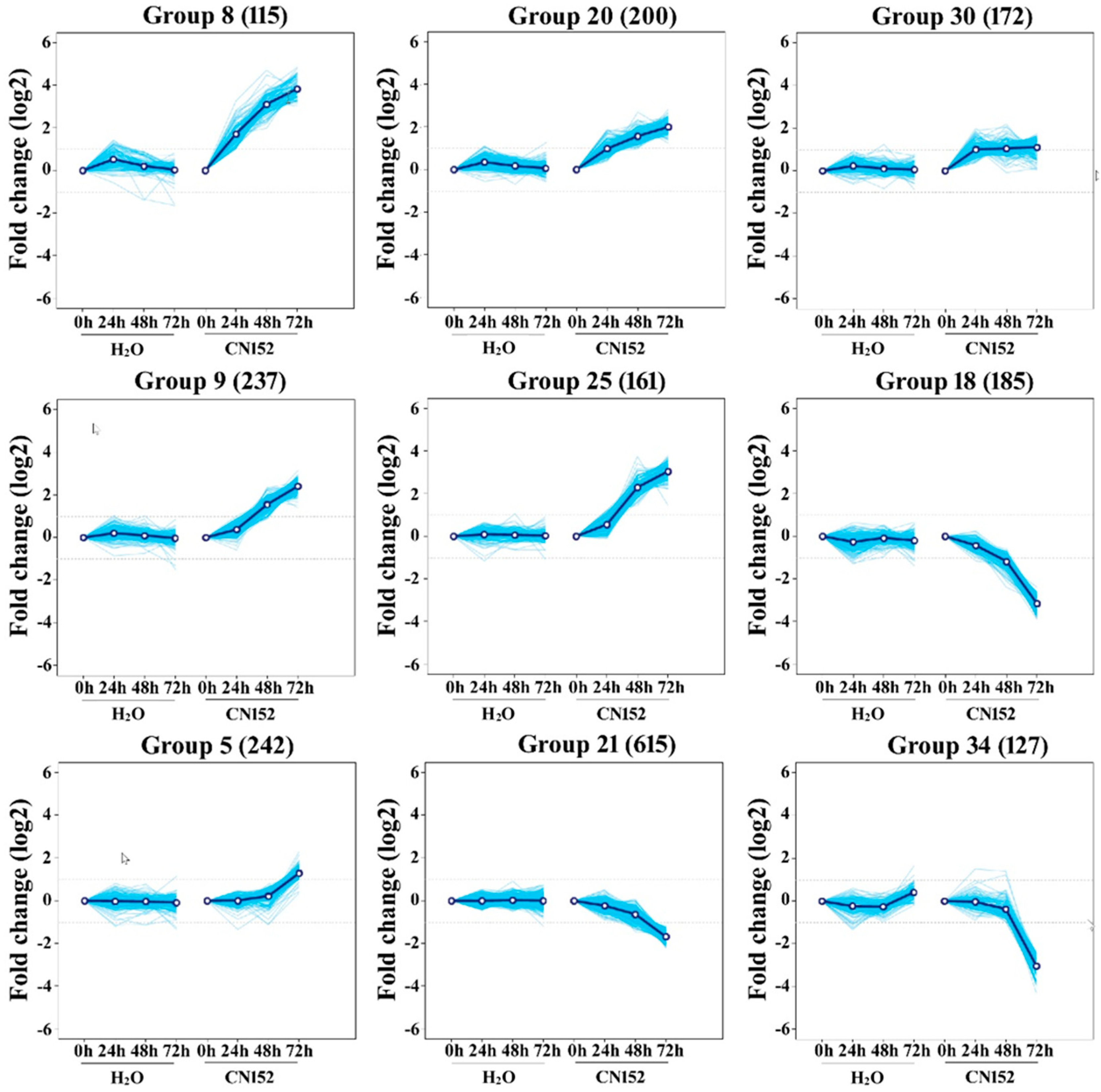
2.3. Clustering Analysis of Differential Expression TUs
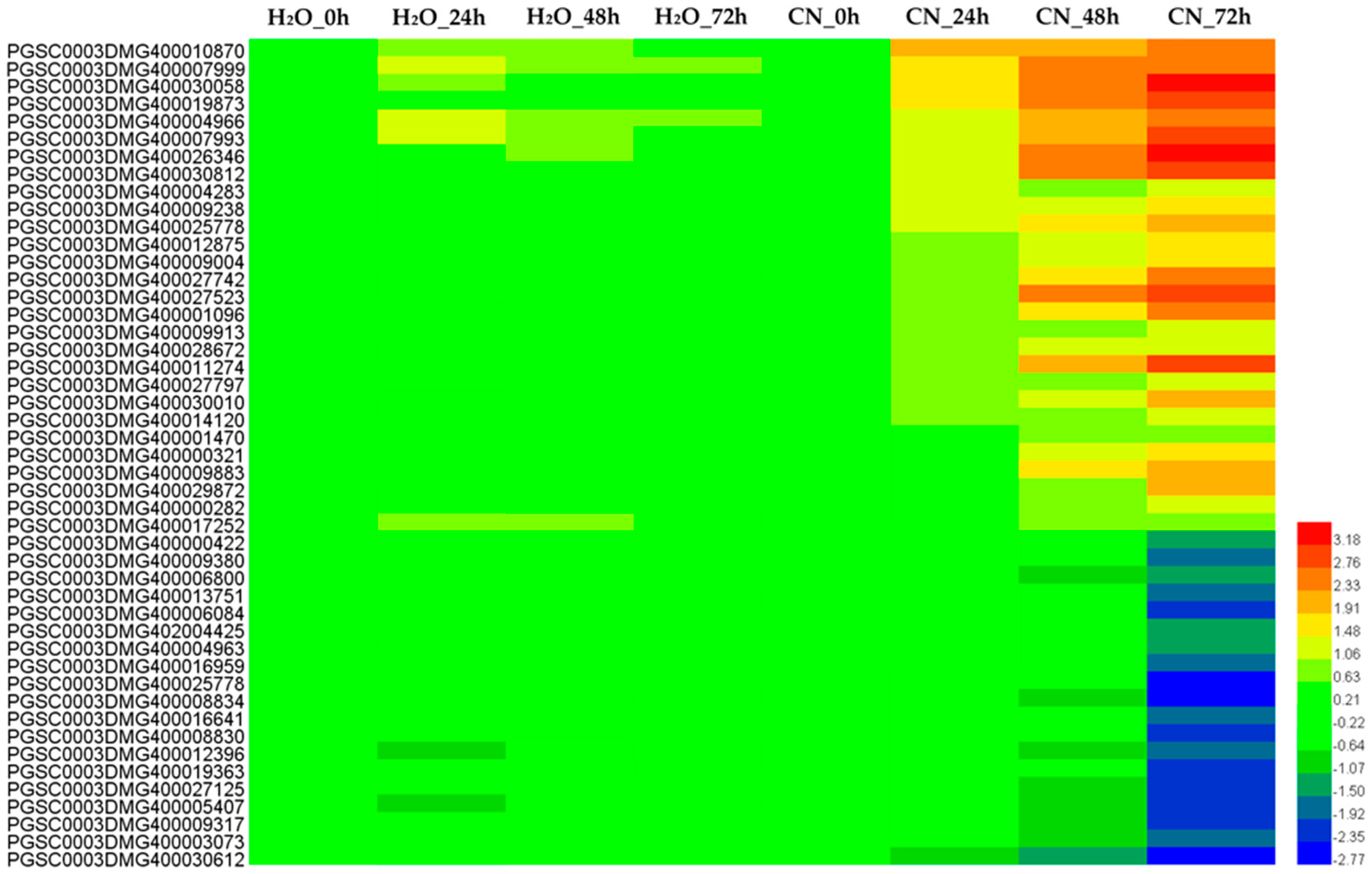
2.4. Analysis of Annotated DEGs
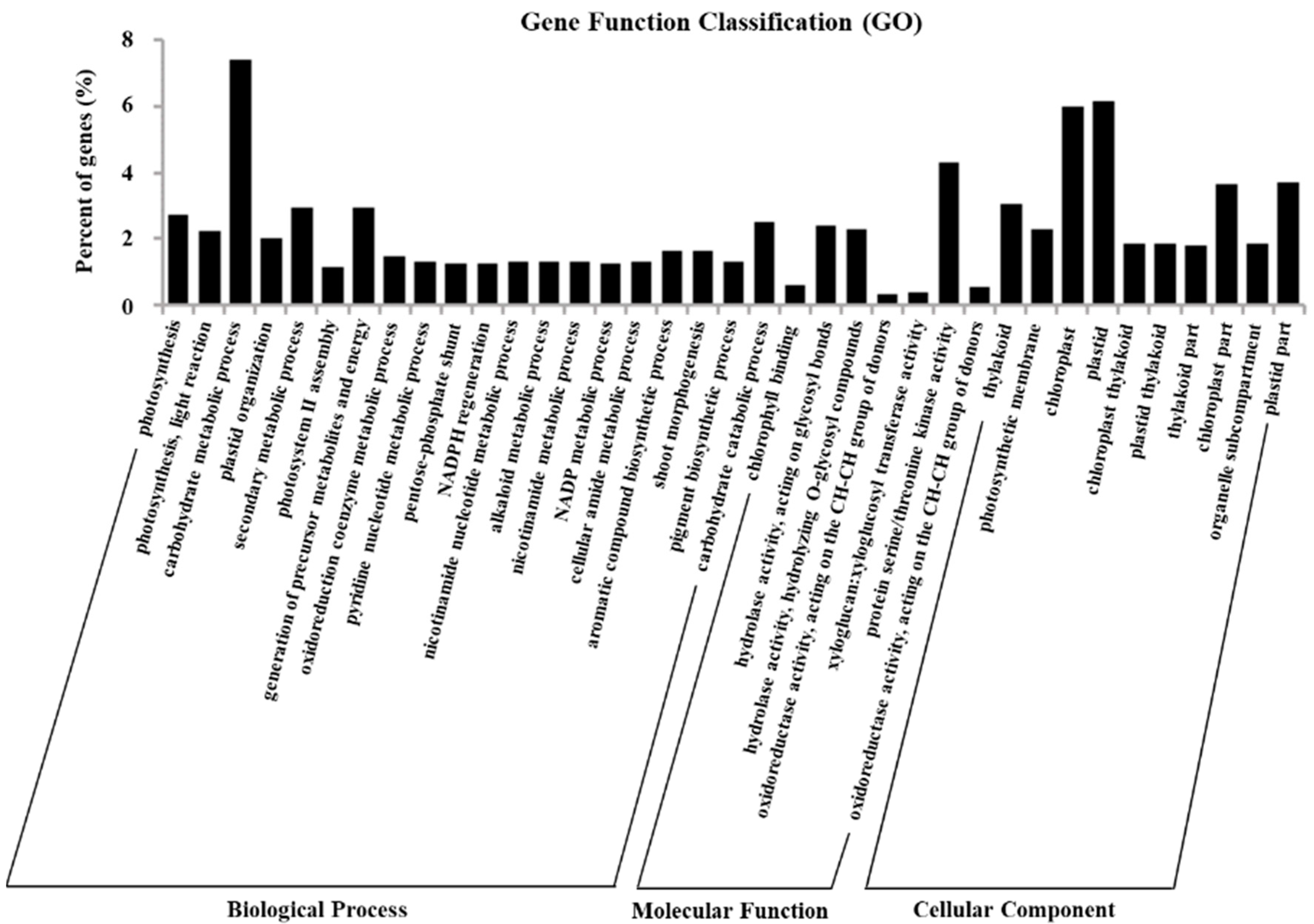
2.5. Gene Ontology Assignments of DEGs
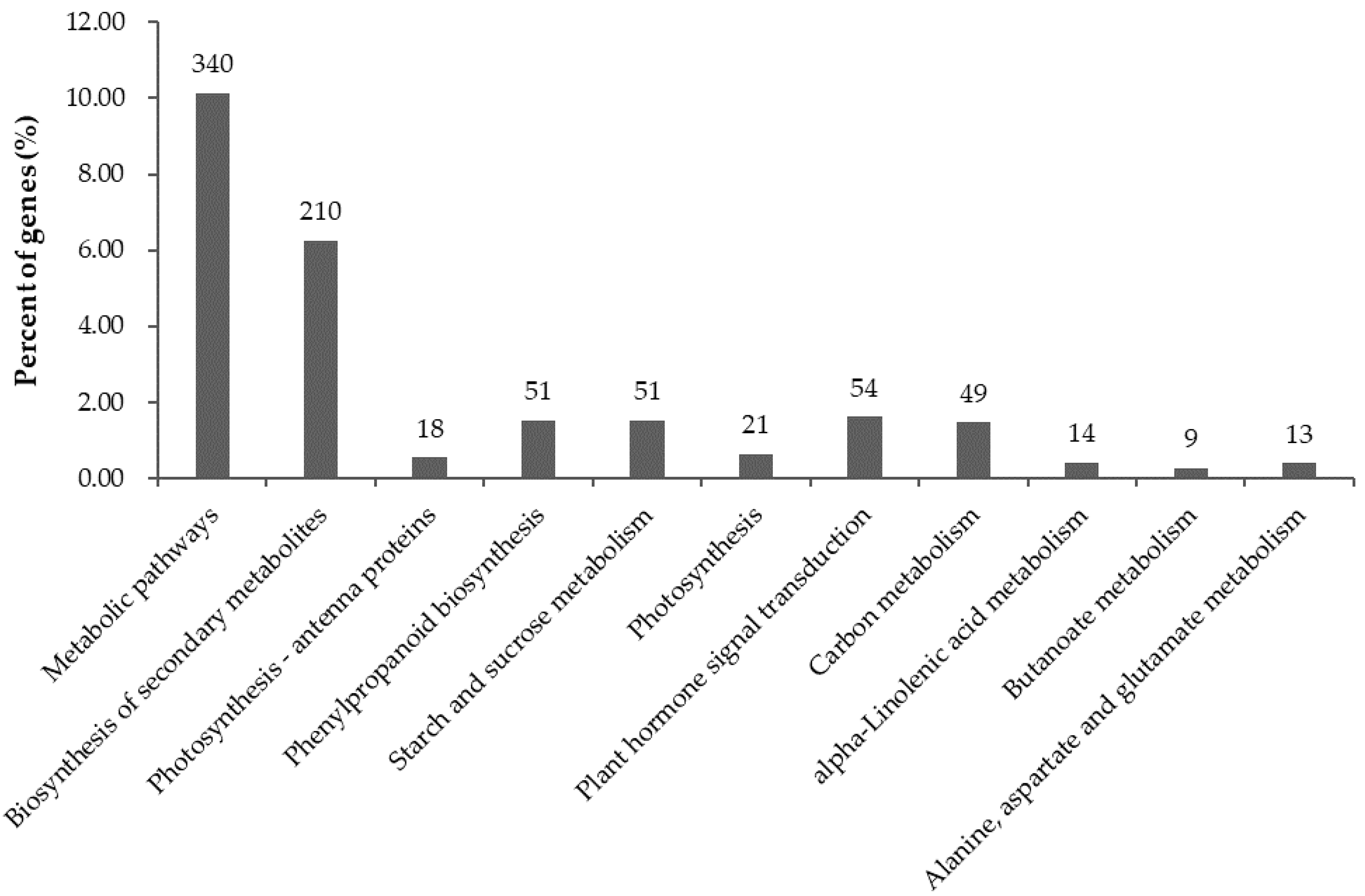
2.6. Significantly Enriched KEGG Pathways in DEGs
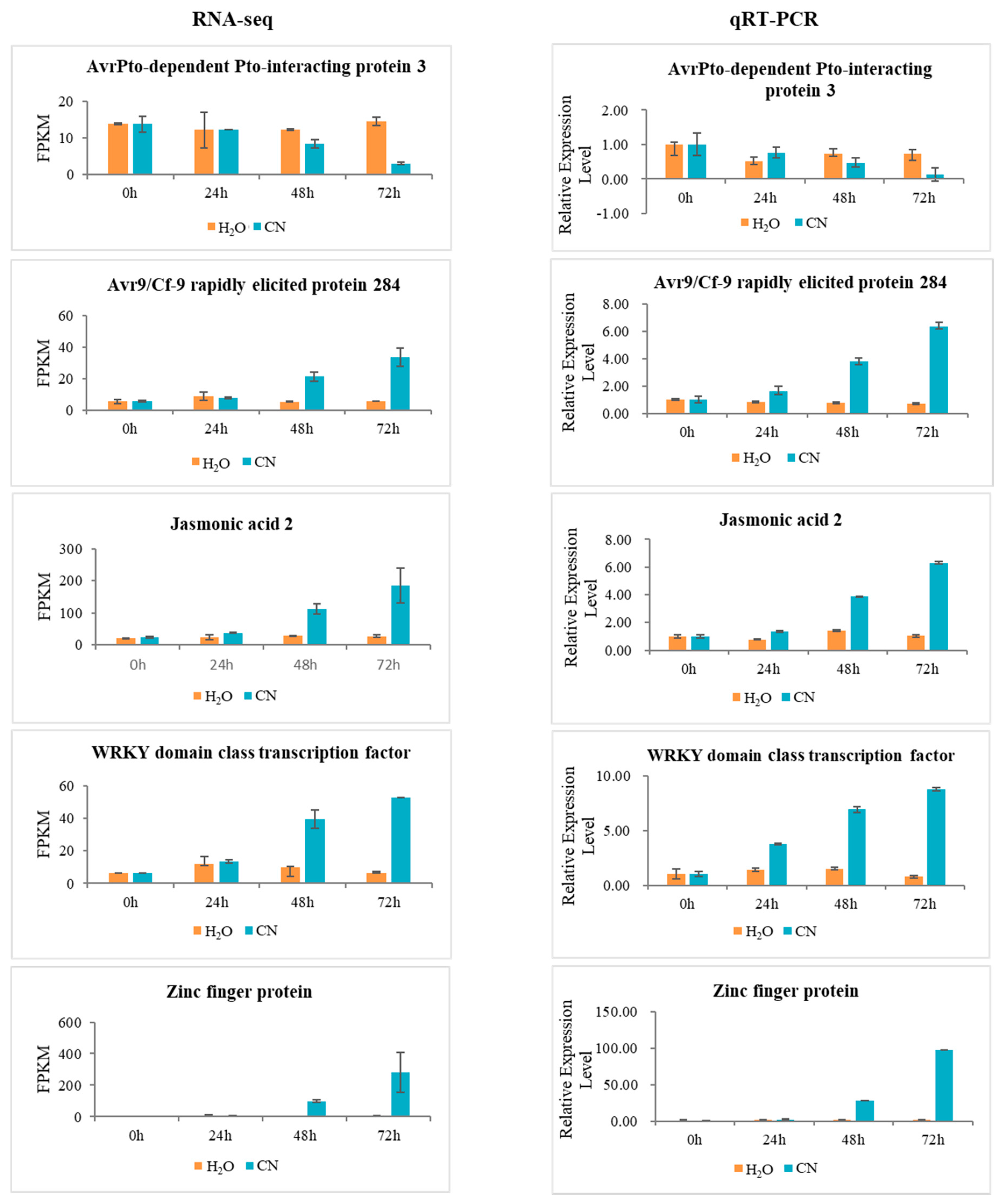
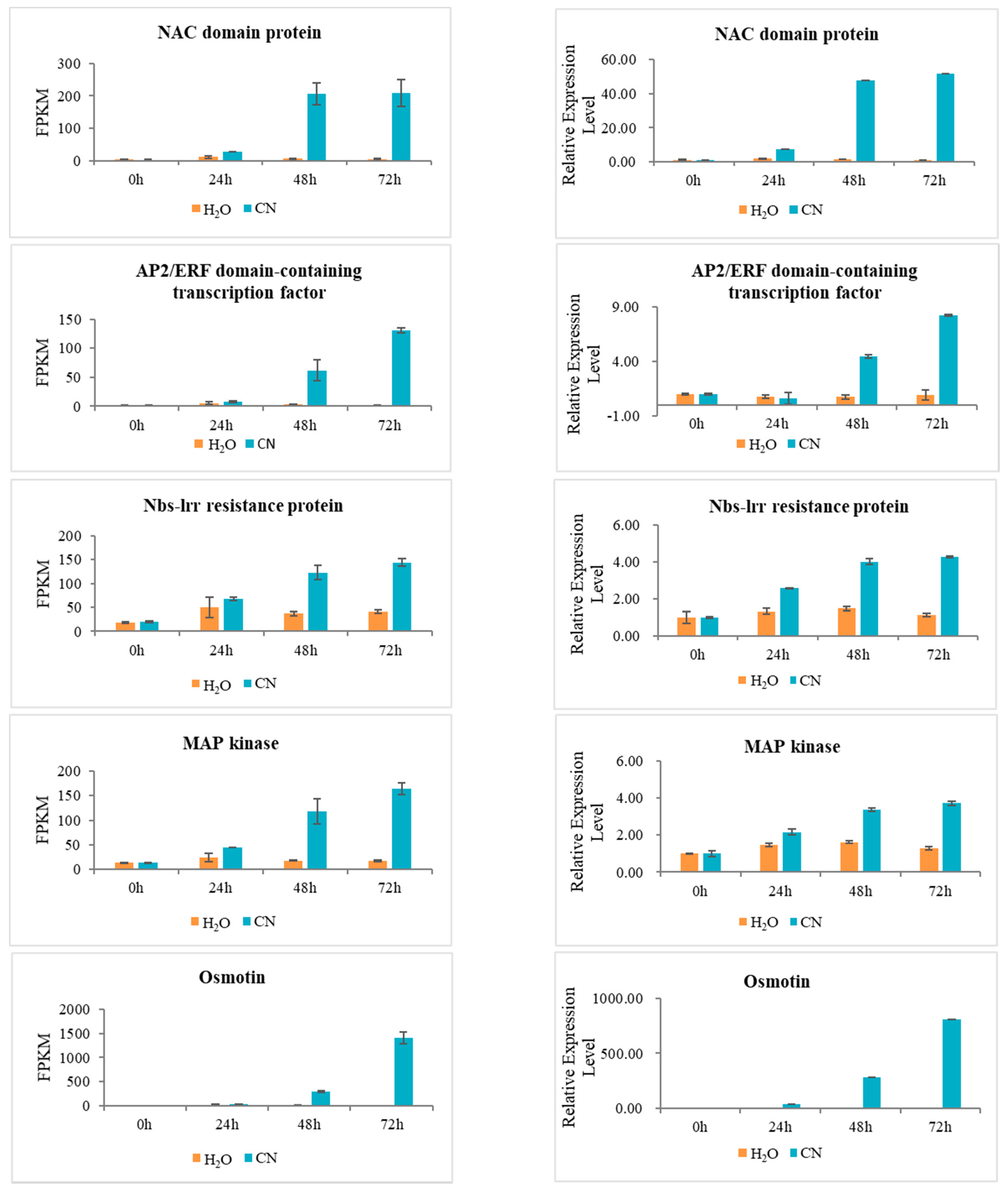
2.7. Verification of RNA-Seq Data Using Quantitative Real-Time PCR
3. Discussion
3.1. Tetraploid Potato Genotype SD20 Is an Ideal Antigen for Late Blight
3.2. RNA-Seq and Differentially Expressed Genes (DEGs) Analysis
3.3. Resistance Gene Expression Profiling in Potato Genotype SD20
3.4. Multi-Signaling Pathways Participated in Resistance and Defense against Pi in SD20
4. Materials and Methods
4.1. Plant Materials
4.2. Inoculation with Phytophthora infestans Isolate
4.3. RNA-Seq Experiments
4.4. Analysis of RNA-Seq Datasets
4.5. Identification of Differentially Expressed Genes
4.6. Clustering Analysis
4.7. GO and KEGG Enrichment Analysis of Differentially Expressed Genes
4.8. Validation of RNA-Seq Data by Real-Time Quantitative PCR (qRT-PCR)
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- International Potato Center (CIP). Potato Facts and Figures. 2016. Available online: http://cipotato.org/potato/facts/ (accessed on 5 June 2018).
- Haverkort, A.J.; Boonekamp, P.M.; Hutten, R.; Jacobsen, E.; Lotz, L.A.P.; Kessel, G.J.T.; Visser, R.G.F.; van der Vossen, E.A.G. Societal costs of late blight in potato and prospects of durable resistance through cisgenic modification. Potato Res. 2008, 51, 47–57. [Google Scholar] [CrossRef]
- Song, J.; Bradeen, J.M.; Naess, S.K.; Raasch, J.A.; Wielgus, S.M.; Haberlach, G.T.; Liu, J.; Kuang, H.; Austin-Phillips, S.; Buell, C.R.; et al. Gene RB cloned from Solanum bulbocastanum confers broad spectrum resistance to potato late blight. Proc. Natl. Acad. Sci. USA 2003, 100, 9128–9133. [Google Scholar] [CrossRef] [PubMed]
- Van der Vossen, E.A.G.; Gros, J.; Sikkema, A.; Muskens, M.; Wouters, D.; Wolters, P.; Pereira, A.; Allefs, S. The Rpi-blb2 gene from Solanum bulbocastanum is an Mi-1 gene homolog conferring broad-spectrum late blight resistance in potato. Plant J. 2005, 44, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Vossen, J.H.; van Arkel, G.; Bergervoet, M.; Jo, K.; Jacobsen, E.; Visser, R.G.F. The Solanum demissum R8 late blight resistance gene is an Sw-5 homologue that has been deployed worldwide in late blight resistant varieties. Theor. Appl. Genet. 2016, 129, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Li, J.; Tian, Z.; Du, J.; Armstrong, M.; Baker, K.; Lim, J.T.; Vossen, J.H.; He, H.; Portal, L.; et al. Potato late blight field resistance from QTL dPI09c is conferred by the NB-LRR gene R8. J. Exp. Bot. 2018. [Google Scholar] [CrossRef] [PubMed]
- Beketova, M.P.; Drobyazina, P.E.; Khavkin, E.E. The R1 gene for late blight resistance in early and late maturing potato cultivars. Rus. J. Plant Physiol. 2006, 53, 384–389. [Google Scholar] [CrossRef]
- Huang, S.; Vleeshouwers, V.G.; Werij, J.S.; Hutten, R.C.; van Eck, H.J.; Visser, R.G.; Jacobsen, E. The R3 resistance to Phytophthora infestans in potato is conferred by two closely linked R genes with distinct specificities. Mol. Plant-Microbe Interact. 2004, 17, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Li, G.C.; Huang, S.W.; Guo, X.; Li, Y.; Yang, Y.; Guo, Z.; Kuang, H.H.; Rietman, H.; Bergervoet, M.; Vleeshouwers, V.G.; et al. Cloning and characterization of R3b; Members of the R3 super family of late blight resistance genes show sequence and functional divergence. Mol. Plant-Microbe Interact. 2011, 24, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Li, Y.; Vossen, J.H.; Visser, R.G.F.; Jacobsen, E. Functional stacking of three resistance genes against Phytophthora infestans in potato. Transgenic Res. 2012, 21, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Rietman, H.; Bijsterbosch, G.; Cano, L.M.; Lee, H.R.; Vossen, J.H.; Jacobsen, E.; Visser, R.G.; Kamoun, S.; Vleeshouwers, V.G. Qualitative and quantitative late blight resistance in the potato cultivar Sarpo Mira is determined by the perception of five distinct RXLR effectors. Mol. Plant-Microbe Interact. 2012, 25, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.; Handsaker, R.E.; Cano, L.M.; Grabherr, M.; Kodira, C.D.; Raffaele, S.; Torto-Alalibo, T.; et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 2009, 461, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shen, H.; Zhou, Q.; Qian, K.; van der Lee, T.; Huang, S. Changing ploidy as a strategy: The Irish potato famine pathogen shifts ploidy in relation to its sexuality. Mol. Plant-Microbe Interact. 2017, 30, 45–52. [Google Scholar] [CrossRef] [PubMed]
- The Potato Genome Sequencing Consortium (PGSC). Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [CrossRef]
- Chen, C.; Li, A. Transcriptome analysis of differentially expressed genes involved in Proantho cyanidin accumulation in the rhizomes of Fagopyrum dibotrys and an irradiation-induced mutant. Front. Plant Physiol. 2016, 7, 100. [Google Scholar] [CrossRef]
- Muthusamy, M.; Uma, S.; Backiyarani, S.; Saraswathi, M.S.; Chandrasekar, A. Transcriptomic changes of drought-tolerant and sensitive banana cultivars exposed to drought stress. Front. Plant Sci. 2016, 7, 1609. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Meng, J.; Yang, S.; Guo, F.; Zhang, J.; Geng, Y.; Cui, L.; Wan, S.; Li, X. Transcriptome analysis of calcium and hormone-related gene expressions during different stages of peanut pod development. Front. Plant Sci. 2017, 8, 1241. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.L.; Tu, Z.J.; Millett, B.P.; Bradeen, J.M. Insights into organ-specific pathogen defense responses in plants: RNA-seq analysis of potato tuber-Phytophthora infestans interactions. BMC Genom. 2013, 14, 340. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Bradeen, J.M. Contrasting potato foliage and tuber defense mechanisms against the late blight pathogen Phytophthora infestans. PLoS ONE 2016, 11, e0159969. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Alexandersson, E.; Sandin, M.; Resjö, S.; Lenman, M.; Hedley, P.; Levander, F.; Andreasson, E. Quantitative proteomics and transcriptomics of potato in response to Phytophthora infestans in compatible and incompatible interactions. BMC Genom. 2014, 15, 497. [Google Scholar] [CrossRef] [PubMed]
- Frades, I.; Abreha, K.B.; Proux-Wéra, E.; Lankinen, A.; Andreasson, E.; Andreasson, E. A novel workflow correlating RNA-seq data to Phythophthora infestans resistance levels in wild Solanum species and potato clones. Front. Plant Sci. 2015, 8, 718. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Gyetvai, G.; Sonderkaer, M.; Gobel, U.; Basekow, R.; Ballvora, A.; Imhoff, M.; Kersten, B.; Nielsen, K.L.; Gebhardt, C. The transcriptome of compatible and incompatible interactions of potato (Solanum tuberosum) with Phytophthora infestans revealed by DeepSAGE analysis. PLoS ONE 2012, 7, e31526. [Google Scholar] [CrossRef] [PubMed]
- Massa, A.N.; Childs, K.L.; Lin, H.; Bryan, G.J.; Giuliano, G.; Buell, C.R. The transcriptome of the reference potato genome Solanum tuberosum group Phureja clone DM1-3 516R44. PLoS ONE 2011, 6, e26801. [Google Scholar] [CrossRef] [PubMed]
- Rejeb, I.B.; Pastor, V.; Mauch-Mani, B. Plant responses to simultaneous biotic and abiotic stress: Molecular mechanisms. Plants 2014, 3, 458–475. [Google Scholar] [CrossRef] [PubMed]
- Kushalappa, A.C.; Gunnaiah, R. Metabolo-proteomics to discover plant biotic stress resistance genes. Trends Plant Sci. 2013, 18, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, I.; Kissen, R.; Bones, A.M. Phytoalexins in defense against pathogens. Trends Plant Sci. 2012, 17, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Senthil-Kumar, M.; Mysore, K.S. Ornithine-delta-aminotransferase and proline dehydrogenase genes play a role in non-host disease resistance by regulating pyrroline-5-carboxylate metabolism-induced hypersensitive response. Plant Cell Environ. 2012, 35, 1329–1343. [Google Scholar] [CrossRef] [PubMed]
- Li, W.T.; Zhu, Z.W.; Chern, M.S.; Yin, J.J.; Yang, C.; Ran, L.; Cheng, M.; He, M.; Wang, K.; Wang, J.; et al. A natural allele of a transcription factor in rice confers broad-spectrum blast resistance. Cell 2017, 70, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.S.; Dadalto, S.P.; Goncalves, A.B.; de Souza, G.B.; Barros, V.A.; Fietto, L.G. Transcription factor functional protein-protein interactions in plant defense responses. Proteomes 2014, 2, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, K.; Somssich, I. Transcriptional networks in plant immunity. New Phytol. 2015, 206, 932–947. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Liu, S.; Meng, C.; Qin, L.; Kong, L.; Xi, G. WRKY transcription factors in wheat and their induction by biotic and abiotic stress. Plant Mol. Biol. Rep. 2013, 31, 1053–1067. [Google Scholar] [CrossRef]
- Wang, H.Y.; Wang, H.L.; Shao, H.B.; Tang, X.L. Recent advances in utilizing transcription factors to improve plant abiotic stress tolerance by transgenic technology. Front. Plant Sci. 2016, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Machinandiarena, M.F.; Lobato, M.C.; Feldman, M.L.; Daleo, G.R.; Andreu, A.B. Potassium phosphite primes defense responses in potato against Phytophthora infestans. J. Plant Physiol. 2012, 169, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Yogendra, K.N.; Kumar, A.; Sarkar, K.; Li, Y.; Pushpa, D.; Mosa, K.A.; Duggavathi, R.; Kushalappa, A.C. Transcription factor StWRKY1 regulates phenylpropanoid metabolites conferring late blight resistance in potato. J. Exp. Bot. 2015, 66, 7377–7389. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wang, H.; Li, Y.; Chen, Y.; Wang, J.; Tian, Z. An early β-aminobutyric acid responsive gene StWRKY5 confers resistance to late blight in potato. Mol. Plant Breed. 2015, 13, 1207–1213. [Google Scholar] [CrossRef]
- Yogendra, K.N.; Sarkar, K.; Kage, U.; Kushalappa, A.C. Potato NAC43 and MYB8 mediated transcriptional regulation of secondary cell wall biosynthesis to contain Phytophthora infestans infection. Plant Mol. Biol. Rep. 2017, 35, 519–533. [Google Scholar] [CrossRef]
- Shan, T.; Hong, Y.; Du, L.; Xu, H.; Wei, X.; Zhang, Z. Development and characterization of TaMYB86-overexpressing transgenic wheat lines with resistance to common root rot. Acta Agron. Sin. 2016, 42, 1429–1436. [Google Scholar] [CrossRef]
- Tian, Z.; He, Q.; Wang, H.; Liu, Y.; Zhang, Y.; Shao, F.; Xie, C. The potato ERF transcription factor StERF3 negatively regulates resistance to Phytophthora infestans and salt tolerance in potato. Plant Cell Physiol. 2015, 56, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, D.; Chen, J.; Yang, Y.; Huang, Z.; Huang, D.; Wang, X.C.; Huang, R. Tomato stress-responsive factor TSRF1 interacts with ethylene responsive element GCC box and regulates pathogen resistance to Ralstonia solanacearum. Plant Mol. Biol. 2004, 55, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yao, W.; Dong, N.; Liang, H.; Liu, H.; Huang, R. A novel ERF transcription activator in wheat and its induction kinetics after pathogen and hormone treatments. J. Exp. Bot. 2007, 58, 2993–3003. [Google Scholar] [CrossRef] [PubMed]
- Quan, R.; Hu, S.; Zhang, Z.; Zhang, H.; Zhang, Z.; Huang, R. Overexpression of an ERF transcription factor TSRF1 improves rice drought tolerance. Plant Biotechnol. J. 2010, 8, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Oñate-Sánchez, L.; Anderson, J.P.; Young, J.; Singh, K.B. AtERF14, a member of the ERF family of transcription factors, plays a nonredundant role in plant defense. Plant Physiol. 2007, 143, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Sheng, P.; Tan, J.; Chen, X.; Lu, G.; Ma, W.; Heng, Y.; Lin, Q.; Zhu, S.; Wang, J.; et al. Plasma membrane receptor-like kinase leaf panicle 2 acts downstream of the DROUGHT AND SALT TOLERANCE transcription factor to regulate drought sensitivity in rice. J. Exp. Bot. 2015, 66, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Ding, Y.; Jiang, Q.; Wang, F.; Sun, J.; Zhu, C. The role of receptor-like protein kinases (RLKs) in abiotic stress response in plants. Plant Cell Rep. 2017, 36, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Bar, M.; Sharfman, M.; Ron, M.; Avni, A. BAK1 is required for the attenuation of ethylene-inducing xylanase (Eix)-induced defense responses by the decoy receptor LeEix1. Plant J. 2010, 63, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Chaparro-Garcia, A.; Wilkinson, R.C.; Gimenez-Ibanez, S.; Findlay, K.; Coffey, M.D.; Zipfel, C.; Rathjen, J.P.; Kamoun, S.; Schornack, S. The receptor-like kinase SERK3/BAK1 is required for basal resistance against the late blight pathogen Phytophthora infestans in Nicotianaben thamiana. PLoS ONE 2011, 6, e16608. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Tian, Z.; Liu, J.; Xie, C. A novel leucine-rich repeat receptor-like kinase gene in potato, StLRPK1, is involved in response to diverse stresses. Mol. Biol. Rep. 2009, 36, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.B.; Han, L.B.; Wang, H.Y.; Zhang, J.; Sun, S.T.; Feng, D.Q.; Yang, C.L.; Sun, Y.D.; Zhong, N.Q.; Xia, G.X. The thioredoxin GbNRX1 plays a crucial role in homeostasis of apoplastic reactive oxygen species in response to Verticillium dahliae infection in cotton. Plant Physiol. 2016, 170, 2392–2406. [Google Scholar] [CrossRef] [PubMed]
- Derksen, H.; Rampitsch, C.; Daayf, F. Signaling cross-talk in plant disease resistance. Plant Sci. 2013, 207, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Wiesel, L.; Davis, J.L.; Milne, L.; Fernandez, V.R.; Herold, M.B.; Middlefell Williams, J.; Morris, J.; Hedley, P.E.; Harrower, B.; Newton, A.C.; et al. A transcriptional reference map of defence hormone responses in potato. Sci. Rep. 2015, 5, 15229. [Google Scholar] [CrossRef] [PubMed]
- Louis, J.; Gobbato, E.; Mondal, H.A.; Feys, B.J.; Parker, J.E.; Shah, J. Discrimination of Arabidopsis PAD4 activities in defense against green peach aphid and pathogens. Plant Physiol. 2012, 158, 1860–1872. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Yang, K.Y.; Li, G.; Liu, Y.; Zhang, S. Activation of Ntf4, a tobacco mitogen-activated protein kinase, during plant defense response and its involvement in hypersensitive response-like cell death. Plant Physiol. 2006, 141, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Yamamizo, C.; Kuchimura, K.; Kobayashi, A.; Katou, S.; Kawakita, K.; Jones, J.D.G.; Doke, N.; Yoshioka, H. Rewiring mitogen-activated protein kinase cascade by positive feedback confers potato blight resistance. Plant Physiol. 2006, 140, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Kishi-Kaboshi, M.; Kurimoto, L.; Murakami, S.; Umezawa, T.; Shibuya, N.; Yamane, H.; Miyao, A.; Takatsuji, H.; Takahashi, A.; Hirochika, H.; et al. A rice fungal MAMP-responsive MAPK cascade regulates metabolic flow to antimicrobial metabolite synthesis. Plant J. 2010, 63, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, N.; Yamada, R.; Yoshioka, M.; Katou, S.; Yoshioka, H. Phosphorylation of the Nicotiana benthamiana WRKY8 transcription factor by MAPK functions in the defense response. Plant Cell 2011, 23, 1153–1170. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhang, C.; Albrecht, U.; Shimizu, R.; Wang, G.; Bowman, K.D. Overexpression of a citrus NDR1 ortholog increases disease resistance in Arabidopsis. Front. Plant Sci. 2013, 4, 157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, F.; Na, R.; Zhang, X.; Yang, S.; Gao, J.; Fan, M.; Zhao, Y.; Zhao, J. AtROP1 negatively regulates potato resistance to Phytophthora infestans via NADPH oxidase-mediated accumulation of H2O2. BMC Plant Biol. 2014, 14, 392. [Google Scholar] [CrossRef] [PubMed]
- Bolser, D.M.; Staines, D.M.; Perry, E.; Kersey, P.J. Ensemble Plants: Integrating tools for visualizing, mining, and analyzing plant genomic data. In Plant Genomics Databases, Methods in Molecular Biology; van Dijk, A., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1533, pp. 1–31. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Reads | Total Bases (G) | Mapped Reads | Uniquely Mapped Reads |
|---|---|---|---|---|
| H2O_0h_rep1 | 100,379,690 | 14.73 | 85,471,167 (85.15%) | 61,107,575 (60.88%) |
| H2O_0h_rep2 | 69,333,843 | 10.33 | 58,488,919 (84.36%) | 40,952,418 (59.07%) |
| H2O_24h_rep1 | 72,144,160 | 10.70 | 58,778,306 (81.47%) | 52,037,875 (72.13%) |
| H2O_24h_rep2 | 89,027,098 | 13.23 | 75,360,165 (84.65%) | 54,980,812 (61.76%) |
| H2O_48h_rep1 | 70,254,604 | 11.67 | 59,645,988 (84.90%) | 48,692,495 (69.31%) |
| H2O_48h_rep2 | 101,393,166 | 15.09 | 86,146,832 (84.96%) | 59,650,502 (58.83%) |
| H2O_72h_rep1 | 88,635,452 | 13.11 | 72,436,689 (81.72%) | 63,188,455 (71.29%) |
| H2O_72h_rep2 | 58,767,684 | 8.72 | 49,143,727 (83.62%) | 37,364,283 (63.58%) |
| CN_0h_rep1 | 74,384,150 | 11.03 | 61,061,372 (82.09%) | 51,846,675 (69.70%) |
| CN_0h_rep2 | 90,097,961 | 13.35 | 76,294,952 (84.68%) | 53,857,487 (59.78%) |
| CN_24h_rep1 | 62,023,469 | 9.21 | 50,950,700 (82.15%) | 42,735,432 (68.90%) |
| CN_24h_rep2 | 94,069,938 | 13.94 | 79,439,329 (84.45%) | 55,514,195 (59.01%) |
| CN_48h_rep1 | 65,892,845 | 9.69 | 52,862,524 (80.22%) | 44,911,407 (68.16%) |
| CN_48h_rep2 | 73,755,259 | 10.94 | 59,183,527 (80.24%) | 43,261,168 (58.66%) |
| CN_72h_rep1 | 58,635,682 | 8.68 | 43,264,784 (73.79%) | 36,679,517 (62.55%) |
| CN_72h_rep2 | 94,447,608 | 14.00 | 76,553,151 (81.05%) | 54,108,598 (57.29%) |
| Gene ID | Description | log2FC_24 h | log2FC_48 h | log2FC_72 h |
|---|---|---|---|---|
| PGSC0003DMG400005492 | Phenylalanine ammonia-lyase, PAL | 0.56 | 2.72 | 3.26 |
| PGSC0003DMG400023458 | Phenylalanine ammonia-lyase, PAL | 0.36 | 1.70 | 1.95 |
| PGSC0003DMG402021564 | Phenylalanine ammonia-lyase, PAL | 0.93 | 1.99 | 2.24 |
| PGSC0003DMG400019386 | Phenylalanine ammonia-lyase, PAL | −0.37 | 1.03 | 1.22 |
| PGSC0003DMG400005115 | PR1 protein | 0.00 | 1.94 | 0.71 |
| PGSC0003DMG400005116 | PR1 protein | −0.05 | 1.96 | 1.27 |
| PGSC0003DMG400010635 | BOP/NPR1/NIM1-like regulatory protein | −0.21 | −0.54 | −1.26 |
| PGSC0003DMG400032231 | NDR1 | 1.76 | 0.73 | 0.81 |
| PGSC0003DMG400000519 | Glucan endo-1,3-beta-glucosidase, acidic isoform GI9 | 4.47 | 3.49 | 4.23 |
| PGSC0003DMG400029830 | Glucan endo-1,3-beta-d-glucosidase | 4.35 | 7.36 | 7.31 |
| PGSC0003DMG400010491 | Glucan endo-1,3-beta-d-glucosidase | 2.99 | 3.89 | 5.50 |
| PGSC0003DMG400018523 | Glucan endo-1,3-beta-glucosidase | 0.34 | 0.50 | 1.59 |
| PGSC0003DMG400021848 | Glucan endo-1,3-beta-glucosidase, basic vacuolar isoform GLB | 0.03 | 1.81 | 1.81 |
| PGSC0003DMG400012702 | Glucan endo-1,3-beta-d-glucosidase | 0.08 | 0.41 | 1.96 |
| PGSC0003DMG400000689 | Glucan endo-1,3-beta-glucosidase | −0.20 | −0.73 | −3.78 |
| PGSC0003DMG400032133 | Glucan endo-1,3-beta-glucosidase 4 | −0.19 | −0.65 | −1.78 |
| PGSC0003DMG400005021 | Glucan endo-1,3-beta-glucosidase | −0.06 | −0.73 | −2.48 |
| PGSC0003DMG402016475 | Glucan endo-1,3-beta-glucosidase | −0.10 | −0.61 | −2.22 |
| PGSC0003DMG400004264 | Thaumatin | 1.34 | 1.79 | 2.54 |
| PGSC0003DMG400004262 | Thaumatin | 1.70 | 1.90 | 2.79 |
| PGSC0003DMG400019873 | Phytoalexin-deficient 4-2 protein, PDA4 | 1.63 | 2.46 | 2.93 |
| PGSC0003DMG400010859 | Lipoxygenase, LOX | 4.15 | 7.88 | 9.08 |
| PGSC0003DMG400024693 | Lipoxygenase, LOX | 1.65 | 0.42 | 0.45 |
| PGSC0003DMG400032155 | Lipoxygenase, LOX | 1.45 | 0.71 | −2.16 |
| PGSC0003DMG400022894 | Lipoxygenase, LOX | 0.40 | 1.77 | 2.51 |
| PGSC0003DMG400001149 | Allene oxide synthase 2, AOS | 1.30 | 1.46 | 1.25 |
| PGSC0003DMG400010283 | Class I chitinase | 0.86 | 5.10 | 7.33 |
| PGSC0003DMG400004593 | Chitinase | −0.54 | −1.03 | −2.05 |
| PGSC0003DMG400008673 | Endochitinase (Chitinase) | 4.47 | 4.92 | 5.83 |
| PGSC0003DMG400001528 | Class II chitinase | 6.18 | 7.67 | 9.09 |
| PGSC0003DMG400001529 | Acidic 27 kDa endochitinase | 1.71 | 1.86 | 1.72 |
| PGSC0003DMG402001531 | Chitinase 134 | 2.07 | 3.70 | 6.24 |
| PGSC0003DMG400013894 | 1-aminocyclopropane-1-carboxylate oxidase 2, ACC | 2.85 | 4.68 | 4.82 |
| PGSC0003DMG400007999 | Nbs-lrr resistance protein | 1.71 | 2.37 | 2.70 |
| PGSC0003DMG400027797 | TIR-NBS-LRR disease resistance | 0.72 | 0.80 | 1.10 |
| PGSC0003DMG402004425 | Cc-nbs-lrr resistance protein | −0.17 | −0.59 | −1.35 |
| PGSC0003DMG400006800 | NBS-LRR protein | −0.02 | −0.79 | −1.37 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Guo, X.; Yang, Y.; Ye, P.; Xiong, X.; Liu, J.; Dong, D.; Li, G. Gene Profiling in Late Blight Resistance in Potato Genotype SD20. Int. J. Mol. Sci. 2018, 19, 1728. https://doi.org/10.3390/ijms19061728
Yang X, Guo X, Yang Y, Ye P, Xiong X, Liu J, Dong D, Li G. Gene Profiling in Late Blight Resistance in Potato Genotype SD20. International Journal of Molecular Sciences. 2018; 19(6):1728. https://doi.org/10.3390/ijms19061728
Chicago/Turabian StyleYang, Xiaohui, Xiao Guo, Yu Yang, Pei Ye, Xingyao Xiong, Jun Liu, Daofeng Dong, and Guangcun Li. 2018. "Gene Profiling in Late Blight Resistance in Potato Genotype SD20" International Journal of Molecular Sciences 19, no. 6: 1728. https://doi.org/10.3390/ijms19061728
APA StyleYang, X., Guo, X., Yang, Y., Ye, P., Xiong, X., Liu, J., Dong, D., & Li, G. (2018). Gene Profiling in Late Blight Resistance in Potato Genotype SD20. International Journal of Molecular Sciences, 19(6), 1728. https://doi.org/10.3390/ijms19061728