Connexins and Pannexins in Vascular Function and Disease

 , ,
, ,
Abstract
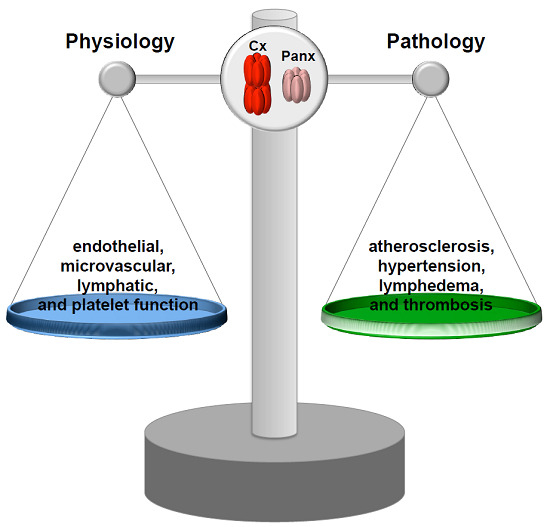
{kind=link}
{kind=link}
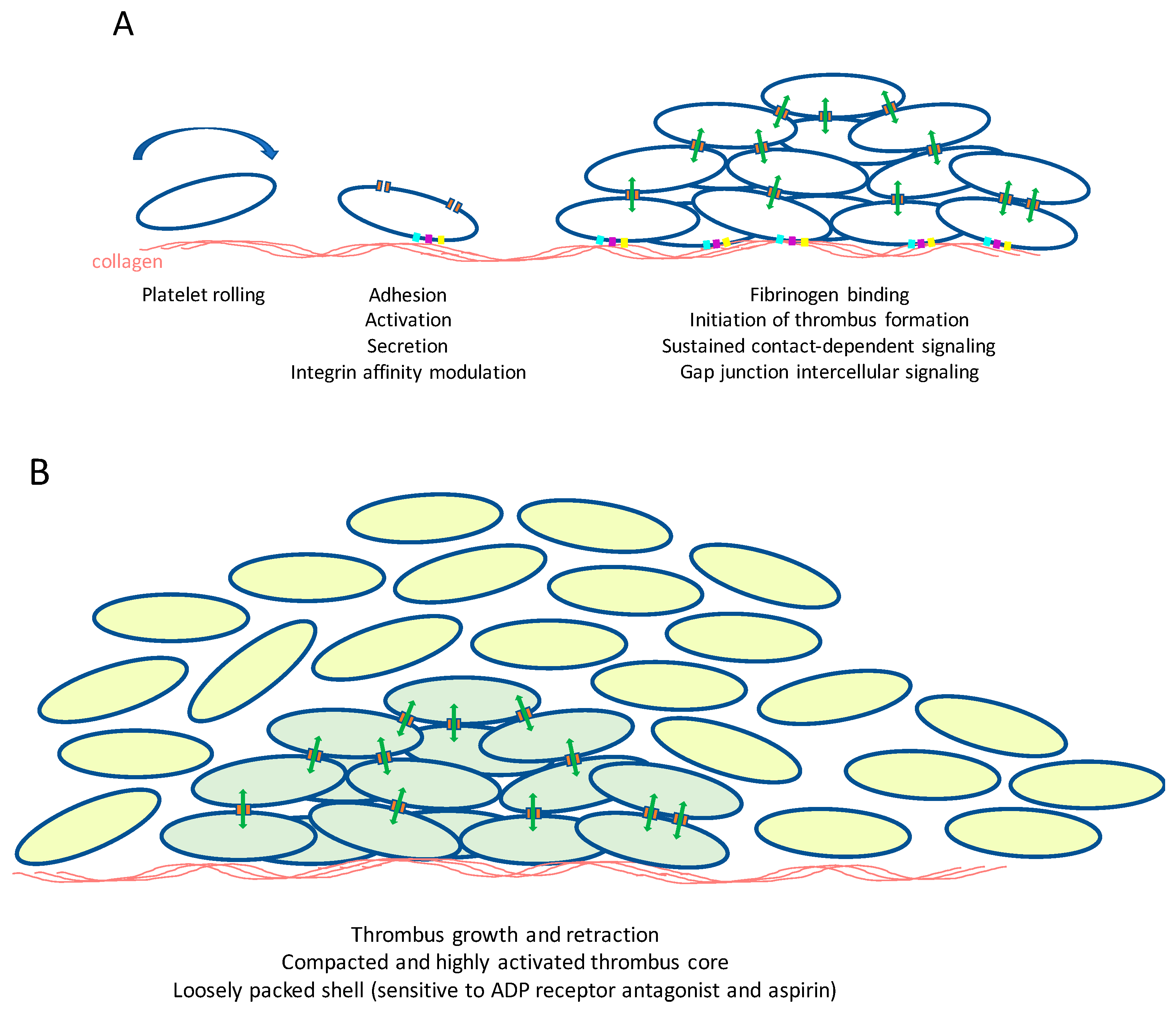{kind=link}
1. Introduction
2. Connexins and Pannexins
3. Role of Cxs and Panxs in Distributing Arteries and Atherosclerosis
3.1. Connexins and Atherosclerotic Disease
3.2. Panx1 and Atherosclerosis
4. Coordination of Microvascular Function by Gap Junctions
4.1. Radial Conduction in the Vascular Wall
4.2. Longitudinal Conduction of Vasomotor Responses
5. Coordination of Microvascular Function by Pannexins
6. Role of Connexins and Pannexins in Venous and Lymphatic Function
7. Connexins and Pannexins in the Control of Platelet Function, Haemostasis and Thrombosis
7.1. Platelets: Mediators of Haemostasis and Thrombosis
7.2. Platelets Possess Connexins
7.3. Platelet Gap Junction Formation—Orchestration of Intercellular Signaling within Arterial Thrombi
7.4. Panx1 Contributes to Platelet Function at Low Agonist Concentrations
7.5. Cx37 and Panx1 Polymorphisms
8. Conclusions and Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| Cx | Connexin |
| Panx | Pannexin |
| TM | Transmembrane |
| EL | Extracellular loop |
| CT | Carboxy-terminus |
| NT | Amino-terminus |
| IL | Intracellular loop |
| ER | Endoplasmic reticulum |
| EC | Endothelial cell |
| LDL | Low-density lipoprotein |
| ECM | Extracellular matrix |
| SMC | Smooth muscle cell |
| TF | Tissue factor |
| POPVC | 1-palmitoyl-2-(5′-oxo-valeroyl)-sn-glycero-3-phosphocholine |
| eNOS | Endothelial NO synthase |
| SNP | Single nucleotide polymorphism |
| TG | Triglycerides |
| FFA | Free fatty acids |
| EDHF | Endothelium-derived hyperpolarizing factor |
| EDH | Endothelium-derived hyperpolarization |
| ACh | Acetylcholine |
| PE | Phenylephrine |
| AR | Adrenergic receptor |
| RBC | Red blood cells |
| VWF | von Willebrand factor |
| GP | Glycoprotein |
| FRAP | Fluorescence recovery after photo-bleaching |
References
- Molica, F.; Meens, M.J.; Morel, S.; Kwak, B.R. Mutations in cardiovascular connexin genes. Biol. Cell 2014, 106, 269–293. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R. Evolutionary analyses of gap junction protein families. Biochim. Biophys. Acta 2013, 1828, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394 Pt 3, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Thevenin, A.F.; Kowal, T.J.; Fong, J.T.; Kells, R.M.; Fisher, C.G.; Falk, M.M. Proteins and mechanisms regulating gap-junction assembly, internalization, and degradation. Physiology 2013, 28, 93–116. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Zhang, X.; Veenstra, R.D. Connexin hemichannel and pannexin channel electrophysiology: How do they differ? FEBS Lett. 2014, 588, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Saez, J.C.; Leybaert, L. Hunting for connexin hemichannels. FEBS Lett. 2014, 588, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.S.; Axelsen, L.N.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.H. Gap junctions. Compr. Physiol. 2012, 2, 1981–2035. [Google Scholar] [PubMed]
- Solan, J.L.; Lampe, P.D. Connexin43 phosphorylation: Structural changes and biological effects. Biochem. J. 2009, 419, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W. The gap junction proteome and its relationship to disease. Trends Cell Biol. 2010, 20, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Agullo-Pascual, E.; Cerrone, M.; Delmar, M. Arrhythmogenic cardiomyopathy and Brugada syndrome: Diseases of the connexome. FEBS Lett. 2014, 588, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavan, R.; Poelzing, S.; Gourdie, R.G. Old cogs, new tricks: A scaffolding role for connexin43 and a junctional role for sodium channels? FEBS Lett. 2014, 588, 1244–1248. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.J.; Simek, J.; Laird, D.W. Mechanisms linking connexin mutations to human diseases. Cell Tissue Res. 2015, 360, 701–721. [Google Scholar] [CrossRef] [PubMed]
- Pfenniger, A.; Wohlwend, A.; Kwak, B.R. Mutations in connexin genes and disease. Eur. J. Clin. Investig. 2011, 41, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Bond, S.R.; Naus, C.C. The pannexins: Past and present. Front. Physiol. 2014, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Dahl, G.; Muller, K.J. Innexin and pannexin channels and their signaling. FEBS Lett. 2014, 588, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Penuela, S.; Harland, L.; Simek, J.; Laird, D.W. Pannexin channels and their links to human disease. Biochem. J. 2014, 461, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Boassa, D.; Ambrosi, C.; Qiu, F.; Dahl, G.; Gaietta, G.; Sosinsky, G. Pannexin1 channels contain a glycosylation site that targets the hexamer to the plasma membrane. J. Biol. Chem. 2007, 282, 31733–31743. [Google Scholar] [CrossRef] [PubMed]
- Scemes, E. Nature of plasmalemmal functional “hemichannels”. Biochim. Biophys. Acta 2012, 1818, 1880–1883. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Leybaert, L.; Giaume, C. Connexin Channels at the Glio-Vascular Interface: Gatekeepers of the Brain. Neurochem. Res. 2017, 42, 2519–2536. [Google Scholar] [CrossRef] [PubMed]
- Danesh-Meyer, H.V.; Zhang, J.; Acosta, M.L.; Rupenthal, I.D.; Green, C.R. Connexin43 in retinal injury and disease. Prog. Retin. Eye Res. 2016, 51, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Davidson, J.O.; Gunn, K.C.; Phillips, A.R.; Green, C.R.; Gunn, A.J. Role of Hemichannels in CNS Inflammation and the Inflammasome Pathway. Adv. Protein Chem. Struct. Biol. 2016, 104, 1–37. [Google Scholar] [PubMed]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Lichtman, A.H.; Hansson, G.K. Immune effector mechanisms implicated in atherosclerosis: From mice to humans. Immunity 2013, 38, 1092–1104. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C. Metalloproteinases promote plaque rupture and myocardial infarction: A persuasive concept waiting for clinical translation. Matrix Biol. 2015, 44–46, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Owens, A.P., 3rd; Mackman, N. Role of tissue factor in atherothrombosis. Curr. Atheroscler. Rep. 2012, 14, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G.; Dejana, E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef] [PubMed]
- Van Rijen, H.V.; van Kempen, M.J.; Postma, S.; Jongsma, H.J. Tumour necrosis factor alpha alters the expression of connexin43, connexin40, and connexin37 in human umbilical vein endothelial cells. Cytokine 1998, 10, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Danesh-Meyer, H.V.; Huang, R.; Nicholson, L.F.; Green, C.R. Connexin43 antisense oligodeoxynucleotide treatment down-regulates the inflammatory response in an in vitro interphase organotypic culture model of optic nerve ischaemia. J. Clin. Neurosci. 2008, 15, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Culot, M.; Wang, N.; Bol, M.; Decrock, E.; De Vuyst, E.; da Costa, A.; Dauwe, I.; Vinken, M.; Simon, A.M.; et al. Connexin channels provide a target to manipulate brain endothelial calcium dynamics and blood-brain barrier permeability. J. Cereb. Blood Flow Metab. 2011, 31, 1942–1957. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.R.; Mulhaupt, F.; Veillard, N.; Gros, D.B.; Mach, F. Altered pattern of vascular connexin expression in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, J.P.; Peters, N.S.; Yeh, H.I.; Rothery, S.; Green, C.R.; Severs, N.J. Upregulation of connexin43 gap junctions during early stages of human coronary atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Polacek, D.; Bech, F.; McKinsey, J.F.; Davies, P.F. Connexin43 gene expression in the rabbit arterial wall: Effects of hypercholesterolemia, balloon injury and their combination. J. Vasc. Res. 1997, 34, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Polacek, D.; Lal, R.; Volin, M.V.; Davies, P.F. Gap junctional communication between vascular cells. Induction of connexin43 messenger RNA in macrophage foam cells of atherosclerotic lesions. Am. J. Pathol. 1993, 142, 593–606. [Google Scholar] [PubMed]
- Johnstone, S.R.; Ross, J.; Rizzo, M.J.; Straub, A.C.; Lampe, P.D.; Leitinger, N.; Isakson, B.E. Oxidized phospholipid species promote in vivo differential cx43 phosphorylation and vascular smooth muscle cell proliferation. Am. J. Pathol. 2009, 175, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.I.; Lu, C.S.; Wu, Y.J.; Chen, C.C.; Hong, R.C.; Ko, Y.S.; Shiao, M.S.; Severs, N.J.; Tsai, C.H. Reduced expression of endothelial connexin37 and connexin40 in hyperlipidemic mice: Recovery of connexin37 after 7-day simvastatin treatment. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Reaume, A.G.; de Sousa, P.A.; Kulkarni, S.; Langille, B.L.; Zhu, D.; Davies, T.C.; Juneja, S.C.; Kidder, G.M.; Rossant, J. Cardiac malformation in neonatal mice lacking connexin43. Science 1995, 267, 1831–1834. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.R.; Veillard, N.; Pelli, G.; Mulhaupt, F.; James, R.W.; Chanson, M.; Mach, F. Reduced connexin43 expression inhibits atherosclerotic lesion formation in low-density lipoprotein receptor-deficient mice. Circulation 2003, 107, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Morel, S.; Chanson, M.; Nguyen, T.D.; Glass, A.M.; Richani Sarieddine, M.Z.; Meens, M.J.; Burnier, L.; Kwak, B.R.; Taffet, S.M. Titration of the gap junction protein Connexin43 reduces atherogenesis. Thromb. Haemost. 2014, 112, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Sun, G.; Zhang, R.; Luo, C.; Ge, M.; Luo, G.; Hei, Z. Connexin 43 expressed in endothelial cells modulates monocyteendothelial adhesion by regulating cell adhesion proteins. Mol. Med. Rep. 2015, 12, 7146–7152. [Google Scholar] [CrossRef] [PubMed]
- Chadjichristos, C.E.; Scheckenbach, K.E.; van Veen, T.A.; Richani Sarieddine, M.Z.; de Wit, C.; Yang, Z.; Roth, I.; Bacchetta, M.; Viswambharan, H.; Foglia, B.; et al. Endothelial-specific deletion of connexin40 promotes atherosclerosis by increasing CD73-dependent leukocyte adhesion. Circulation 2010, 121, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Denis, J.F.; Scheckenbach, K.E.L.; Pfenniger, A.; Meens, M.J.; Krams, R.; Miquerol, L.; Taffet, S.; Chanson, M.; Delmar, M.; Kwak, B.R. Connexin40 controls endothelial activation by dampening NFkappaB activation. Oncotarget 2017, 8, 50972–50986. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Christen, T.; Roth, I.; Chadjichristos, C.E.; Derouette, J.P.; Foglia, B.F.; Chanson, M.; Goodenough, D.A.; Kwak, B.R. Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 2006, 12, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Pfenniger, A.; Meens, M.J.; Pedrigi, R.M.; Foglia, B.; Sutter, E.; Pelli, G.; Rochemont, V.; Petrova, T.V.; Krams, R.; Kwak, B.R. Shear stress-induced atherosclerotic plaque composition in ApoE(−/−) mice is modulated by connexin37. Atherosclerosis 2015, 243, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pfenniger, A.; Derouette, J.P.; Verma, V.; Lin, X.; Foglia, B.; Coombs, W.; Roth, I.; Satta, N.; Dunoyer-Geindre, S.; Sorgen, P.; et al. Gap junction protein Cx37 interacts with endothelial nitric oxide synthase in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Meens, M.J.; Pfenniger, A.; Kwak, B.R. Risky communication in atherosclerosis and thrombus formation. Swiss Med. Wkly. 2012, 142, w13553. [Google Scholar] [CrossRef] [PubMed]
- Derouette, J.P.; Desplantez, T.; Wong, C.W.; Roth, I.; Kwak, B.R.; Weingart, R. Functional differences between human Cx37 polymorphic hemichannels. J. Mol. Cell. Cardiol. 2009, 46, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Morel, S.; Burnier, L.; Roatti, A.; Chassot, A.; Roth, I.; Sutter, E.; Galan, K.; Pfenniger, A.; Chanson, M.; Kwak, B.R. Unexpected role for the human Cx37 C1019T polymorphism in tumour cell proliferation. Carcinogenesis 2010, 31, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Isakson, B.E.; Thompson, R.J. Pannexin-1 as a potentiator of ligand-gated receptor signaling. Channels 2014, 8, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Meens, M.J.; Kwak, B.R.; Duffy, H.S. Role of connexins and pannexins in cardiovascular physiology. Cell. Mol. Life Sci. 2015, 72, 2779–2792. [Google Scholar] [CrossRef] [PubMed]
- Adamson, S.E.; Leitinger, N. The role of pannexin1 in the induction and resolution of inflammation. FEBS Lett. 2014, 588, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Sandilos, J.K.; Chiu, Y.H.; Chekeni, F.B.; Armstrong, A.J.; Walk, S.F.; Ravichandran, K.S.; Bayliss, D.A. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J. Biol. Chem. 2012, 287, 11303–11311. [Google Scholar] [CrossRef] [PubMed]
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, J.M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, D.W.; Salvesen, G.S.; et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 2010, 467, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Misaghi, S.; Newton, K.; Gilmour, L.L.; Louie, S.; Cupp, J.E.; Dubyak, G.R.; Hackos, D.; Dixit, V.M. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J. Immunol. 2011, 186, 6553–6561. [Google Scholar] [CrossRef] [PubMed]
- Molica, F.; Meens, M.J.; Dubrot, J.; Ehrlich, A.; Roth, C.L.; Morel, S.; Pelli, G.; Vinet, L.; Braunersreuther, V.; Ratib, O.; et al. Pannexin1 links lymphatic function to lipid metabolism and atherosclerosis. Sci. Rep. 2017, 7, 13706. [Google Scholar] [CrossRef] [PubMed]
- Segal, S.S. Integration and Modulation of Intercellular Signaling Underlying Blood Flow Control. J. Vasc. Res. 2015, 52, 136–157. [Google Scholar] [CrossRef] [PubMed]
- Feletou, M.; Huang, Y.; Vanhoutte, P.M. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br. J. Pharmacol. 2011, 164, 894–912. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Higgs, E.A. The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S193–S201. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M. COX-1 and vascular disease. Clin. Pharmacol. Ther. 2009, 86, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Busse, R.; Edwards, G.; Feletou, M.; Fleming, I.; Vanhoutte, P.M.; Weston, A.H. EDHF: Bringing the concepts together. Trends Pharmacol. Sci. 2002, 23, 374–380. [Google Scholar] [CrossRef]
- Figueroa, X.F.; Duling, B.R. Gap junctions in the control of vascular function. Antioxid. Redox Signal. 2009, 11, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Gaete, P.S.; Lillo, M.A.; Ardiles, N.M.; Perez, F.R.; Figueroa, X.F. Ca2+-activated K+ channels of small and intermediate conductance control eNOS activation through NAD(P)H oxidase. Free Radic. Biol. Med. 2012, 52, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, X.F.; Isakson, B.E.; Duling, B.R. Connexins: Gaps in our knowledge of vascular function. Physiology 2004, 19, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, V.J.; Wolfle, S.E.; Boettcher, M.; de Wit, C. Gap junctions synchronize vascular tone within the microcirculation. Pharmacol. Rep. 2008, 60, 68–74. [Google Scholar] [PubMed]
- Segal, S.S. Regulation of blood flow in the microcirculation. Microcirculation 2005, 12, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Beny, J.L.; Koenigsberger, M.; Sauser, R. Role of myoendothelial communication on arterial vasomotion. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2036–H2038. [Google Scholar] [CrossRef] [PubMed]
- Emerson, G.G.; Segal, S.S. Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: Role in vasomotor control. Circ. Res. 2000, 87, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Looft-Wilson, R.; Doran, B.; Grayson, T.H.; Segal, S.S.; Hill, C.E. Expression of homocellular and heterocellular gap junctions in hamster arterioles and feed arteries. Cardiovasc. Res. 2003, 60, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Dora, K.A.; Doyle, M.P.; Duling, B.R. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc. Natl. Acad. Sci. USA 1997, 94, 6529–6534. [Google Scholar] [CrossRef] [PubMed]
- Isakson, B.E.; Ramos, S.I.; Duling, B.R. Ca2+ and inositol 1,4,5-trisphosphate-mediated signaling across the myoendothelial junction. Circ. Res. 2007, 100, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Nausch, L.W.; Bonev, A.D.; Heppner, T.J.; Tallini, Y.; Kotlikoff, M.I.; Nelson, M.T. Sympathetic nerve stimulation induces local endothelial Ca2+ signals to oppose vasoconstriction of mouse mesenteric arteries. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H594–H602. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.; Dora, K.A.; Gardener, M.J.; Garland, C.J.; Weston, A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 1998, 396, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Gragasin, F.S.; Wu, X.; Wang, S.; McMurtry, S.; Kim, D.H.; Platonov, M.; Koshal, A.; Hashimoto, K.; Campbell, W.B.; et al. Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BK(Ca) channels. Circulation 2003, 107, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I. Cytochrome P450 epoxygenases as EDHF synthase(s). Pharmacol. Res. 2004, 49, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H.; Morikawa, K. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J. Mol. Cell. Cardiol. 2005, 39, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, A.; Hobbs, A.J. Endothelium-derived C-type natriuretic peptide: More than just a hyperpolarizing factor. Trends Pharmacol. Sci. 2005, 26, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.D.; Nilsson, H.; Ahluwalia, A.; Hobbs, A.J. Release of C-type natriuretic peptide accounts for the biological activity of endothelium-derived hyperpolarizing factor. Proc. Natl. Acad. Sci. USA 2003, 100, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.J.; Dora, K.A. EDH: Endothelium-dependent hyperpolarization and microvascular signalling. Acta Physiol. 2017, 219, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, M.; de Wit, C. Distinct endothelium-derived hyperpolarizing factors emerge in vitro and in vivo and are mediated in part via connexin 40-dependent myoendothelial coupling. Hypertension 2011, 57, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Eichler, I.; Wibawa, J.; Grgic, I.; Knorr, A.; Brakemeier, S.; Pries, A.R.; Hoyer, J.; Kohler, R. Selective blockade of endothelial Ca2+-activated small- and intermediate-conductance K+-channels suppresses EDHF-mediated vasodilation. Br. J. Pharmacol. 2003, 138, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Doughty, J.M.; Plane, F.; Langton, P.D. Charybdotoxin and apamin block EDHF in rat mesenteric artery if selectively applied to the endothelium. Am. J. Physiol. 1999, 276 Pt 2, H1107–H1112. [Google Scholar] [CrossRef] [PubMed]
- Dora, K.A.; Sandow, S.L.; Gallagher, N.T.; Takano, H.; Rummery, N.M.; Hill, C.E.; Garland, C.J. Myoendothelial gap junctions may provide the pathway for EDHF in mouse mesenteric artery. J. Vasc. Res. 2003, 40, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Feletou, M.; Vanhoutte, P.M.; Weston, A.H.; Edwards, G. EDHF and endothelial potassiun channels: IKCa and SKCa. Br. J. Pharmacol. 2003, 140, 225. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.M. Endothelium-dependent smooth muscle hyperpolarization: Do gap junctions provide a unifying hypothesis? Br. J. Pharmacol. 2004, 141, 881–903. [Google Scholar] [CrossRef] [PubMed]
- Feletou, M.; Vanhoutte, P.M. Endothelium-dependent hyperpolarization: No longer an f-word! J. Cardiovasc. Pharmacol. 2013, 61, 91–92. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Hill, C.E. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ. Res. 2000, 86, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H.; Yasutake, H.; Fujii, K.; Owada, M.K.; Nakaike, R.; Fukumoto, Y.; Takayanagi, T.; Nagao, T.; Egashira, K.; Fujishima, M.; et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J. Cardiovasc. Pharmacol. 1996, 28, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Chaytor, A.T.; Bakker, L.M.; Edwards, D.H.; Griffith, T.M. Connexin-mimetic peptides dissociate electrotonic EDHF-type signalling via myoendothelial and smooth muscle gap junctions in the rabbit iliac artery. Br. J. Pharmacol. 2005, 144, 108–114. [Google Scholar] [CrossRef] [PubMed]
- De Vriese, A.S.; Van de Voorde, J.; Lameire, N.H. Effects of connexin-mimetic peptides on nitric oxide synthase- and cyclooxygenase-independent renal vasodilation. Kidney Int. 2002, 61, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Mather, S.; Dora, K.A.; Sandow, S.L.; Winter, P.; Garland, C.J. Rapid endothelial cell-selective loading of connexin 40 antibody blocks endothelium-derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ. Res. 2005, 97, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Straub, A.C.; Lohman, A.W.; Billaud, M.; Johnstone, S.R.; Dwyer, S.T.; Lee, M.Y.; Bortz, P.S.; Best, A.K.; Columbus, L.; Gaston, B.; et al. Endothelial cell expression of haemoglobin alpha regulates nitric oxide signalling. Nature 2012, 491, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, X.F.; Lillo, M.A.; Gaete, P.S.; Riquelme, M.A.; Saez, J.C. Diffusion of nitric oxide across cell membranes of the vascular wall requires specific connexin-based channels. Neuropharmacology 2013, 75, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Bolz, S.S.; de Wit, C.; Pohl, U. Endothelium-derived hyperpolarizing factor but not NO reduces smooth muscle Ca2+ during acetylcholine-induced dilation of microvessels. Br. J. Pharmacol. 1999, 128, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Bolz, S.S.; Vogel, L.; Sollinger, D.; Derwand, R.; de Wit, C.; Loirand, G.; Pohl, U. Nitric oxide-induced decrease in calcium sensitivity of resistance arteries is attributable to activation of the myosin light chain phosphatase and antagonized by the RhoA/Rho kinase pathway. Circulation 2003, 107, 3081–3087. [Google Scholar] [CrossRef] [PubMed]
- Bagher, P.; Segal, S.S. Regulation of blood flow in the microcirculation: Role of conducted vasodilation. Acta Physiol. 2011, 202, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Emerson, G.G.; Segal, S.S. Endothelial cell pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circ. Res. 2000, 86, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, X.F.; Duling, B.R. Dissection of two Cx37-independent conducted vasodilator mechanisms by deletion of Cx40: Electrotonic versus regenerative conduction. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2001–H2007. [Google Scholar] [CrossRef] [PubMed]
- De Wit, C.; Griffith, T.M. Connexins and gap junctions in the EDHF phenomenon and conducted vasomotor responses. Pflugers Arch. 2010, 459, 897–914. [Google Scholar] [CrossRef] [PubMed]
- Haefliger, J.A.; Nicod, P.; Meda, P. Contribution of connexins to the function of the vascular wall. Cardiovasc. Res. 2004, 62, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Duling, B.R. Electromechanical coupling and the conducted vasomotor response. Am. J. Physiol. 1995, 269 Pt 2, H2022–H2030. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Little, T.L.; Duling, B.R. Cellular pathways of the conducted electrical response in arterioles of hamster cheek pouch in vitro. Am. J. Physiol. 1995, 269 Pt 2, H2031–H2038. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, F.; Holstein-Rathlou, N. Conducted vasomotor responses in arterioles: Characteristics, mechanisms and physiological significance. Acta Physiol. Scand. 1999, 167, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Welsh, D.G.; Segal, S.S. Endothelial and smooth muscle cell conduction in arterioles controlling blood flow. Am. J. Physiol. 1998, 274 Pt 2, H178–H186. [Google Scholar] [CrossRef] [PubMed]
- Welsh, D.G.; Segal, S.S. Role of EDHF in conduction of vasodilation along hamster cheek pouch arterioles in vivo. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1832–H1839. [Google Scholar] [CrossRef] [PubMed]
- Segal, S.S.; Welsh, D.G.; Kurjiaka, D.T. Spread of vasodilatation and vasoconstriction along feed arteries and arterioles of hamster skeletal muscle. J. Physiol. 1999, 516 Pt 1, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, I.S.; Segal, S.S. Resolution of smooth muscle and endothelial pathways for conduction along hamster cheek pouch arterioles. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H604–H612. [Google Scholar] [CrossRef] [PubMed]
- Budel, S.; Bartlett, I.S.; Segal, S.S. Homocellular conduction along endothelium and smooth muscle of arterioles in hamster cheek pouch: Unmasking an NO wave. Circ. Res. 2003, 93, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Segal, S.S.; Jacobs, T.L. Role for endothelial cell conduction in ascending vasodilatation and exercise hyperaemia in hamster skeletal muscle. J. Physiol. 2001, 536 Pt 3, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akiyama, M.; Takeda, M.; Gabazza, E.C.; Hayashi, T.; Suzuki, K. Connexin32 is expressed in vascular endothelial cells and participates in gap-junction intercellular communication. Biochem. Biophys. Res. Commun. 2009, 382, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Severs, N.J.; Rothery, S.; Dupont, E.; Coppen, S.R.; Yeh, H.I.; Ko, Y.S.; Matsushita, T.; Kaba, R.; Halliday, D. Immunocytochemical analysis of connexin expression in the healthy and diseased cardiovascular system. Microsc. Res. Tech. 2001, 52, 301–322. [Google Scholar] [CrossRef]
- Van Kempen, M.J.; Jongsma, H.J. Distribution of connexin37, connexin40 and connexin43 in the aorta and coronary artery of several mammals. Histochem. Cell Biol. 1999, 112, 479–486. [Google Scholar] [CrossRef] [PubMed]
- De Wit, C.; Roos, F.; Bolz, S.S.; Kirchhoff, S.; Kruger, O.; Willecke, K.; Pohl, U. Impaired conduction of vasodilation along arterioles in connexin40-deficient mice. Circ. Res. 2000, 86, 649–655. [Google Scholar] [CrossRef] [PubMed]
- De Wit, C.; Roos, F.; Bolz, S.S.; Pohl, U. Lack of vascular connexin 40 is associated with hypertension and irregular arteriolar vasomotion. Physiol. Genom. 2003, 13, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, X.F.; Paul, D.L.; Simon, A.M.; Goodenough, D.A.; Day, K.H.; Damon, D.N.; Duling, B.R. Central role of connexin40 in the propagation of electrically activated vasodilation in mouse cremasteric arterioles in vivo. Circ. Res. 2003, 92, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Jobs, A.; Schmidt, K.; Schmidt, V.J.; Lubkemeier, I.; van Veen, T.A.; Kurtz, A.; Willecke, K.; de Wit, C. Defective Cx40 maintains Cx37 expression but intact Cx40 is crucial for conducted dilations irrespective of hypertension. Hypertension 2012, 60, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; de Wit, C.; Kurtz, L.; Grunberger, C.; Kurtz, A.; Schweda, F. Connexin40 is essential for the pressure control of renin synthesis and secretion. Circ. Res. 2007, 100, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Gollob, M.H.; Jones, D.L.; Krahn, A.D.; Danis, L.; Gong, X.Q.; Shao, Q.; Liu, X.; Veinot, J.P.; Tang, A.S.; Stewart, A.F.; et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 2006, 354, 2677–2688. [Google Scholar] [CrossRef] [PubMed]
- Lubkemeier, I.; Andrie, R.; Lickfett, L.; Bosen, F.; Stockigt, F.; Dobrowolski, R.; Draffehn, A.M.; Fregeac, J.; Schultze, J.L.; Bukauskas, F.F.; et al. The Connexin40A96S mutation from a patient with atrial fibrillation causes decreased atrial conduction velocities and sustained episodes of induced atrial fibrillation in mice. J. Mol. Cell. Cardiol. 2013, 65, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Santa Cruz, A.; Mese, G.; Valiuniene, L.; Brink, P.R.; White, T.W.; Valiunas, V. Altered conductance and permeability of Cx40 mutations associated with atrial fibrillation. J. Gen. Physiol. 2015, 146, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Lohman, A.W.; Billaud, M.; Straub, A.C.; Johnstone, S.R.; Best, A.K.; Lee, M.; Barr, K.; Penuela, S.; Laird, D.W.; Isakson, B.E. Expression of pannexin isoforms in the systemic murine arterial network. J. Vasc. Res. 2012, 49, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Lohman, A.W.; Isakson, B.E. Differentiating connexin hemichannels and pannexin channels in cellular ATP release. FEBS Lett. 2014, 588, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Penuela, S.; Kelly, J.J.; Churko, J.M.; Barr, K.J.; Berger, A.C.; Laird, D.W. Panx1 regulates cellular properties of keratinocytes and dermal fibroblasts in skin development and wound healing. J. Investig. Dermatol. 2014, 134, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Billaud, M.; Lohman, A.W.; Johnstone, S.R.; Biwer, L.A.; Mutchler, S.; Isakson, B.E. Regulation of cellular communication by signaling microdomains in the blood vessel wall. Pharmacol. Rev. 2014, 66, 513–569. [Google Scholar] [CrossRef] [PubMed]
- Angus, J.A.; Wright, C.E. Novel alpha1-adrenoceptor antagonism by the fluroquinolone antibiotic trovafloxacin. Eur. J. Pharmacol. 2016, 791, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.H.; Jin, X.; Medina, C.B.; Leonhardt, S.A.; Kiessling, V.; Bennett, B.C.; Shu, S.; Tamm, L.K.; Yeager, M.; Ravichandran, K.S.; et al. A quantized mechanism for activation of pannexin channels. Nat. Commun. 2017, 8, 14324. [Google Scholar] [CrossRef] [PubMed]
- Good, M.E.; Chiu, Y.H.; Poon, I.K.H.; Medina, C.B.; Butcher, J.T.; Mendu, S.K.; DeLalio, L.J.; Lohman, A.W.; Leitinger, N.; Barrett, E.; et al. Pannexin 1 Channels as an Unexpected New Target of the Anti-Hypertensive Drug Spironolactone. Circ. Res. 2018, 122, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Kauffenstein, G.; Tamareille, S.; Prunier, F.; Roy, C.; Ayer, A.; Toutain, B.; Billaud, M.; Isakson, B.E.; Grimaud, L.; Loufrani, L.; et al. Central Role of P2Y6 UDP Receptor in Arteriolar Myogenic Tone. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, M.; Piil, P.; Kiehn, O.T.; Maagaard, C.; Jorgensen, T.S.; Egelund, J.; Isakson, B.E.; Nielsen, M.S.; Gliemann, L.; Hellsten, Y. Probenecid Inhibits alpha-Adrenergic Receptor-Mediated Vasoconstriction in the Human Leg Vasculature. Hypertension 2018, 71, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Billaud, M.; Chiu, Y.H.; Lohman, A.W.; Parpaite, T.; Butcher, J.T.; Mutchler, S.M.; DeLalio, L.J.; Artamonov, M.V.; Sandilos, J.K.; Best, A.K.; et al. A molecular signature in the pannexin1 intracellular loop confers channel activation by the alpha1 adrenoreceptor in smooth muscle cells. Sci. Signal. 2015, 8, ra17. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.; Chiu, Y.H.; Armstrong, A.J.; Kinchen, J.M.; Juncadella, I.J.; Bayliss, D.A.; Ravichandran, K.S. Unexpected link between an antibiotic, pannexin channels and apoptosis. Nature 2014, 507, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Lohman, A.W.; Weaver, J.L.; Billaud, M.; Sandilos, J.K.; Griffiths, R.; Straub, A.C.; Penuela, S.; Leitinger, N.; Laird, D.W.; Bayliss, D.A.; et al. S-nitrosylation inhibits pannexin 1 channel function. J. Biol. Chem. 2012, 287, 39602–39612. [Google Scholar] [CrossRef] [PubMed]
- Gaynullina, D.; Tarasova, O.S.; Kiryukhina, O.O.; Shestopalov, V.I.; Panchin, Y. Endothelial function is impaired in conduit arteries of pannexin1 knockout mice. Biol. Direct 2014, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Good, M.E.; Eucker, S.A.; Li, J.; Bacon, H.M.; Lang, S.M.; Butcher, J.T.; Johnson, T.J.; Gaykema, R.P.; Patel, M.K.; Zuo, Z.; et al. Endothelial cell Pannexin1 modulates severity of ischemic stroke by regulating cerebral inflammation and myogenic tone. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Lohman, A.W.; Leskov, I.L.; Butcher, J.T.; Johnstone, S.R.; Stokes, T.A.; Begandt, D.; DeLalio, L.J.; Best, A.K.; Penuela, S.; Leitinger, N.; et al. Pannexin 1 channels regulate leukocyte emigration through the venous endothelium during acute inflammation. Nat. Commun. 2015, 6, 7965. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.S.; Diederich, L.; Panknin, C.; DeLalio, L.J.; Drake, J.C.; Sherman, R.; Jackson, E.K.; Yan, Z.; Kelm, M.; Cortese-Krott, M.M.; et al. Possible roles for ATP release from RBCs exclude the cAMP-mediated Panx1 pathway. Am. J. Physiol. Cell Physiol. 2017, 313, C593–C603. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Charles, E.J.; Zhao, Y.; Narahari, A.K.; Baderdinni, P.K.; Good, M.E.; Lorenz, U.M.; Kron, I.L.; Bayliss, D.A.; Ravichandran, K.S.; et al. Pannexin 1 channels on endothelial cells mediate vascular inflammation during lung ischemia-reperfusion injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.; Perry, H.M.; Medina, C.B.; Huang, L.; Yao, Y.; Bajwa, A.; Lorenz, U.M.; Rosin, D.L.; Ravichandran, K.S.; Isakson, B.E.; et al. Epithelial and Endothelial Pannexin 1 Channels Mediate AKI. J. Am. Soc. Nephrol. 2018, in press. [Google Scholar]
- Sikora, J.; Orlov, S.N.; Furuya, K.; Grygorczyk, R. Hemolysis is a primary ATP-release mechanism in human erythrocytes. Blood 2014, 124, 2150–2157. [Google Scholar] [CrossRef] [PubMed]
- Munger, S.J.; Kanady, J.D.; Simon, A.M. Absence of venous valves in mice lacking Connexin37. Dev. Biol. 2013, 373, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Kanady, J.D.; Dellinger, M.T.; Munger, S.J.; Witte, M.H.; Simon, A.M. Connexin37 and Connexin43 deficiencies in mice disrupt lymphatic valve development and result in lymphatic disorders including lymphedema and chylothorax. Dev. Biol. 2011, 354, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Sabine, A.; Agalarov, Y.; Maby-El Hajjami, H.; Jaquet, M.; Hagerling, R.; Pollmann, C.; Bebber, D.; Pfenniger, A.; Miura, N.; Dormond, O.; et al. Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic-valve formation. Dev. Cell. 2012, 22, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Munger, S.J.; Geng, X.; Srinivasan, R.S.; Witte, M.H.; Paul, D.L.; Simon, A.M. Segregated Foxc2, NFATc1 and Connexin expression at normal developing venous valves, and Connexin-specific differences in the valve phenotypes of Cx37, Cx43, and Cx47 knockout mice. Dev. Biol. 2016, 412, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Lyons, O.; Saha, P.; Seet, C.; Kuchta, A.; Arnold, A.; Grover, S.; Rashbrook, V.; Sabine, A.; Vizcay-Barrena, G.; Patel, A.; et al. Human venous valve disease caused by mutations in FOXC2 and GJC2. J. Exp. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Meens, M.J.; Kutkut, I.; Rochemont, V.; Dubrot, J.; Kaladji, F.R.; Sabine, A.; Lyons, O.; Hendrikx, S.; Bernier-Latmani, J.; Kiefer, F.; et al. Cx47 fine-tunes the handling of serum lipids but is dispensable for lymphatic vascular function. PLoS ONE 2017, 12, e0181476. [Google Scholar] [CrossRef] [PubMed]
- Brice, G.; Ostergaard, P.; Jeffery, S.; Gordon, K.; Mortimer, P.S.; Mansour, S. A novel mutation in GJA1 causing oculodentodigital syndrome and primary lymphoedema in a three generation family. Clin. Genet. 2013, 84, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Brouillard, P.; Boon, L.; Vikkula, M. Genetics of lymphatic anomalies. J. Clin. Investig. 2014, 124, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Ferrell, R.E.; Baty, C.J.; Kimak, M.A.; Karlsson, J.M.; Lawrence, E.C.; Franke-Snyder, M.; Meriney, S.D.; Feingold, E.; Finegold, D.N. GJC2 missense mutations cause human lymphedema. Am. J. Hum. Genet. 2010, 86, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, E.; Rodenburg, R.J.; van den Brand, M.; Thomsen, L.L.; Duno, M.; Batbayli, M.; Wibrand, F.; Nijtmans, L. Respiratory chain complex I deficiency due to NDUFA12 mutations as a new cause of Leigh syndrome. J. Med. Genet. 2011, 48, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Finegold, D.N.; Baty, C.J.; Knickelbein, K.Z.; Perschke, S.; Noon, S.E.; Campbell, D.; Karlsson, J.M.; Huang, D.; Kimak, M.A.; Lawrence, E.C.; et al. Connexin 47 mutations increase risk for secondary lymphedema following breast cancer treatment. Clin. Cancer Res. 2012, 18, 2382–2890. [Google Scholar] [CrossRef] [PubMed]
- Hadizadeh, M.; Mohaddes Ardebili, S.M.; Salehi, M.; Young, C.; Mokarian, F.; McClellan, J.; Xu, Q.; Kazemi, M.; Moazam, E.; Mahaki, B.; et al. GJA4/Connexin 37 Mutations Correlate with Secondary Lymphedema Following Surgery in Breast Cancer Patients. Biomedicines 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Kuczma, M.; Lee, J.R.; Kraj, P. Connexin 43 signaling enhances the generation of Foxp3+ regulatory T cells. J. Immunol. 2011, 187, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Oviedo-Orta, E.; Perreau, M.; Evans, W.H.; Potolicchio, I. Control of the proliferation of activated CD4+ T cells by connexins. J. Leukoc. Biol. 2010, 88, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Pfenniger, A.; Chanson, M.; Kwak, B.R. Connexins in atherosclerosis. Biochim. Biophys. Acta 2013, 1828, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Kanaji, T.; Russell, S.; Kunicki, T.J.; Furihata, K.; Kanaji, S.; Marchese, P.; Reininger, A.; Ruggeri, Z.M.; Ware, J. The contribution of glycoprotein VI to stable platelet adhesion and thrombus formation illustrated by targeted gene deletion. Blood 2003, 102, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Gawaz, M.; Gruner, S.; Schulte, V.; Konrad, I.; Zohlnhofer, D.; Heinzmann, U.; Nieswandt, B. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J. Exp. Med. 2003, 197, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Inoue, O.; Suzuki-Inoue, K.; Dean, W.L.; Frampton, J.; Watson, S.P. Integrin alpha2beta1 mediates outside-in regulation of platelet spreading on collagen through activation of Src kinases and PLCgamma2. J. Cell Biol. 2003, 160, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Gibbins, J.; Asselin, J.; Farndale, R.; Barnes, M.; Law, C.L.; Watson, S.P. Tyrosine phosphorylation of the Fc receptor gamma-chain in collagen-stimulated platelets. J. Biol. Chem. 1996, 271, 18095–18099. [Google Scholar] [CrossRef] [PubMed]
- Gibbins, J.M.; Okuma, M.; Farndale, R.; Barnes, M.; Watson, S.P. Glycoprotein VI is the collagen receptor in platelets which underlies tyrosine phosphorylation of the Fc receptor gamma-chain. FEBS Lett. 1997, 413, 255–259. [Google Scholar] [CrossRef]
- Suzuki-Inoue, K.; Tulasne, D.; Shen, Y.; Bori-Sanz, T.; Inoue, O.; Jung, S.M.; Moroi, M.; Andrews, R.K.; Berndt, M.C.; Watson, S.P. Association of Fyn and Lyn with the proline-rich domain of glycoprotein VI regulates intracellular signaling. J. Biol. Chem. 2002, 277, 21561–21566. [Google Scholar] [CrossRef] [PubMed]
- Gibbins, J.M. Platelet adhesion signalling and the regulation of thrombus formation. J. Cell Sci. 2004, 117 Pt 16, 3415–3425. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.P.; Auger, J.M.; McCarty, O.J.; Pearce, A.C. GPVI and integrin alphaIIb beta3 signaling in platelets. J. Thromb. Haemost. 2005, 3, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, Z.M. Platelets in atherothrombosis. Nat. Med. 2002, 8, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Shattil, S.J. Signaling through platelet integrin alpha IIb beta 3: Inside-out, outside-in, and sideways. Thromb. Haemost. 1999, 82, 318–325. [Google Scholar] [PubMed]
- Shattil, S.J.; Kashiwagi, H.; Pampori, N. Integrin signaling: The platelet paradigm. Blood 1998, 91, 2645–2657. [Google Scholar] [PubMed]
- Jones, C.I.; Barrett, N.E.; Moraes, L.A.; Gibbins, J.M.; Jackson, D.E. Endogenous inhibitory mechanisms and the regulation of platelet function. Methods Mol. Biol. 2012, 788, 341–366. [Google Scholar] [PubMed]
- Prevost, N.; Woulfe, D.; Tanaka, T.; Brass, L.F. Interactions between Eph kinases and ephrins provide a mechanism to support platelet aggregation once cell-to-cell contact has occurred. Proc. Natl. Acad. Sci. USA 2002, 99, 9219–9224. [Google Scholar] [CrossRef] [PubMed]
- Prevost, N.; Woulfe, D.S.; Jiang, H.; Stalker, T.J.; Marchese, P.; Ruggeri, Z.M.; Brass, L.F. Eph kinases and ephrins support thrombus growth and stability by regulating integrin outside-in signaling in platelets. Proc. Natl. Acad. Sci. USA 2005, 102, 9820–9825. [Google Scholar] [CrossRef] [PubMed]
- Vaiyapuri, S.; Sage, T.; Rana, R.H.; Schenk, M.P.; Ali, M.S.; Unsworth, A.J.; Jones, C.I.; Stainer, A.R.; Kriek, N.; Moraes, L.A.; et al. EphB2 regulates contact-dependent and contact-independent signaling to control platelet function. Blood 2015, 125, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Prevost, N.; Woulfe, D.; Tognolini, M.; Brass, L.F. Contact-dependent signaling during the late events of platelet activation. J. Thromb. Haemost. 2003, 1, 1613–1627. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, W.S.; Giuliano, S.; Kulkarni, S.; Dopheide, S.M.; Harper, I.S.; Jackson, S.P. Intercellular calcium communication regulates platelet aggregation and thrombus growth. J. Cell Biol. 2003, 160, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; Fontana, P.; Burnier, L.; Roth, I.; Sugamele, R.; Brisset, A.; Morel, S.; Nolli, S.; Sutter, E.; Chassot, A.; et al. Connexin 37 limits thrombus propensity by downregulating platelet reactivity. Circulation 2011, 124, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Vaiyapuri, S.; Jones, C.I.; Sasikumar, P.; Moraes, L.A.; Munger, S.J.; Wright, J.R.; Ali, M.S.; Sage, T.; Kaiser, W.J.; Tucker, K.L.; et al. Gap junctions and connexin hemichannels underpin hemostasis and thrombosis. Circulation 2012, 125, 2479–2491. [Google Scholar] [CrossRef] [PubMed]
- Vaiyapuri, S.; Moraes, L.A.; Sage, T.; Ali, M.S.; Lewis, K.R.; Mahaut-Smith, M.P.; Oviedo-Orta, E.; Simon, A.M.; Gibbins, J.M. Connexin40 regulates platelet function. Nat. Commun. 2013, 4, 2564. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.A.; Wright, J.R.; Vial, C.; Evans, R.J.; Mahaut-Smith, M.P. Amplification of human platelet activation by surface pannexin-1 channels. J. Thromb. Haemost. 2014, 12, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Molica, F.; Stierlin, F.B.; Fontana, P.; Kwak, B.R. Pannexin- and Connexin-Mediated Intercellular Communication in Platelet Function. Int. J. Mol. Sci. 2017, 18, 850. [Google Scholar] [CrossRef] [PubMed]
- Stalker, T.J.; Traxler, E.A.; Wu, J.; Wannemacher, K.M.; Cermignano, S.L.; Voronov, R.; Diamond, S.L.; Brass, L.F. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood 2013, 121, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Morel, S.; Burnier, L.; Kwak, B.R. Connexins participate in the initiation and progression of atherosclerosis. Semin. Immunopathol. 2009, 31, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Burger, F.; Pelli, G.; Mach, F.; Kwak, B.R. Dual benefit of reduced Cx43 on atherosclerosis in LDL receptor-deficient mice. Cell Commun. Adhes. 2003, 10, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.R.; Ferdinandy, P.; Beyer, E.C.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in Cardiovascular and Neurovascular Health and Disease: Pharmacological Implications. Pharmacol. Rev. 2017, 69, 396–478. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molica, F.; Figueroa, X.F.; Kwak, B.R.; Isakson, B.E.; Gibbins, J.M. Connexins and Pannexins in Vascular Function and Disease. Int. J. Mol. Sci. 2018, 19, 1663. https://doi.org/10.3390/ijms19061663
Molica F, Figueroa XF, Kwak BR, Isakson BE, Gibbins JM. Connexins and Pannexins in Vascular Function and Disease. International Journal of Molecular Sciences. 2018; 19(6):1663. https://doi.org/10.3390/ijms19061663
Chicago/Turabian StyleMolica, Filippo, Xavier F. Figueroa, Brenda R. Kwak, Brant E. Isakson, and Jonathan M. Gibbins. 2018. "Connexins and Pannexins in Vascular Function and Disease" International Journal of Molecular Sciences 19, no. 6: 1663. https://doi.org/10.3390/ijms19061663
APA StyleMolica, F., Figueroa, X. F., Kwak, B. R., Isakson, B. E., & Gibbins, J. M. (2018). Connexins and Pannexins in Vascular Function and Disease. International Journal of Molecular Sciences, 19(6), 1663. https://doi.org/10.3390/ijms19061663