VEGF Upregulation in Viral Infections and Its Possible Therapeutic Implications


Abstract
1. Introduction
2. Upregulation of VEGF Expression in Viral Oncogenesis
2.1. Epstein-Barr Virus (EBV)
2.2. Kaposi’s Sarcoma-Associated Herpesvirus (KSHV)
2.3. Human Papillomavirus (HPV)
2.4. Hepatitis C Viruses (HCV)
2.5. Hepatitis B Viruses (HBV)
3. Upregulation of VEGF Expression in Non-Oncogenic Viral Infections
3.1. Herpes Simplex Virus-1 (HSV-1)
3.2. Dengue Virus (DENV)
3.3. Hantaviruses
4. Viral VEGF Homolog Proteins
5. Therapeutic Applications of Targeting VEGF in Viral Diseases
6. Summary and Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| AP-1 | Activator protein 1 |
| COX-2 | Cyclooxygenase-2 |
| DENV | Dengue virus |
| E5, E6, E7 | HPV early proteins 5, 6 and 7 |
| EBNA1 | EBV nuclear antigen 1 |
| EBV | Epstein-Barr virus |
| EGFR | Epidermal growth factor receptor |
| HBV | Hepatitis B virus |
| HBx | HBV-x protein |
| HCV | Hepatitis C virus |
| HIF-1α | Hypoxia inducible factor-1 alpha |
| HPV | Human papilloma virus |
| HSV-1 | Herpes simples virus-1 |
| hTERT | Human Telomerase reverse transcriptase |
| ICP4 | Infected cell protein 4 |
| JNK | c-Jun N-terminal kinases |
| KSHV | Kaposi’s sarcoma-associated herpesvirus |
| LMP1 | latent membrane protein 1 |
| NF-κB | Nuclear factor kappa B |
| NPC | Nasopharyngeal carcinoma |
| Npr | Neuropilin receptor |
| PI3K | Phosphatidylinositol 3-kinase |
| PlGF | Placental growth factor |
| SP-1 | Specificity protein 1 |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| vFLIP | Viral FLICE inhibitory protein |
| vGPCR | Viral G-protein coupled receptor |
| vIRF3 | Viral interferon regulatory factor 3 |
References
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Evans, I. An Overview of VEGF-Mediated Signal Transduction. Methods Mol. Biol. 2015, 1332, 91–120. [Google Scholar] [PubMed]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R.J. VEGF receptor protein-tyrosine kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2008, 375, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.L.; Thomas, K.A. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L. VEGF receptor signal transduction-A brief update. Vasc. Pharmacol. 2016, 86, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Zarkada, G.; Nurmi, H.; Jakobsson, L.; Heinolainen, K.; Tvorogov, D.; Zheng, W.; Franco, C.A.; Murtomaki, A.; Aranda, E.; et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat. Cell Biol. 2011, 13, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Benedito, R.; Rocha, S.F.; Woeste, M.; Zamykal, M.; Radtke, F.; Casanovas, O.; Duarte, A.; Pytowski, B.; Adams, R.H. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature 2012, 484, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J.; Bates, D.O. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Biselli-Chicote, P.M.; Oliveira, A.R.; Pavarino, E.C.; Goloni-Bertollo, E.M. VEGF gene alternative splicing: Pro- and anti-angiogenic isoforms in cancer. J. Cancer Res. Clin. Oncol. 2012, 138, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein-Barr virus-associated lymphomas. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, C.; Pan, L. Nasopharyngeal carcinoma: A review of current updates. Exp. Ther. Med. 2018, 15, 3687–3692. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.M.; Kim, E.H. Epstein-Barr Virus and Gastric Cancer Risk: A Meta-analysis With Meta-regression of Case-control Studies. J. Prev. Med. Public Health 2016, 49, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.W.; Sunagawa, M.; Li, J.E.; Shimada, S.; Gang, Z.; Tokeshi, Y.; Kosugi, T. The relationship between microvessel density, the expression of vascular endothelial growth factor (VEGF), and the extension of nasopharyngeal carcinoma. Laryngoscope 2000, 110, 2066–2069. [Google Scholar]
- Qian, C.N.; Zhang, C.Q.; Guo, X.; Hong, M.H.; Cao, S.M.; Mai, W.Y.; Min, H.Q.; Zeng, Y.X. Elevation of serum vascular endothelial growth factor in male patients with metastatic nasopharyngeal carcinoma. Cancer 2000, 88, 255–261. [Google Scholar] [CrossRef]
- Wakisaka, N.; Wen, Q.H.; Yoshizaki, T.; Nishimura, T.; Furukawa, M.; Kawahara, E.; Nakanishi, I. Association of vascular endothelial growth factor expression with angiogenesis and lymph node metastasis in nasopharyngeal carcinoma. Laryngoscope 1999, 109, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.M.; James, S.; Balaram, P. Expression of VEGF as prognosticator in primary nasopharyngeal cancer and its relation to EBV status. Virus Res. 2006, 115, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Polz-Dacewicz, M.; Strycharz-Dudziak, M.; Dworzanski, J.; Stec, A.; Kocot, J. Salivary and serum IL-10, TNF-alpha, TGF-beta, VEGF levels in oropharyngeal squamous cell carcinoma and correlation with HPV and EBV infections. Infect. Agent Cancer 2016, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Lu, F.; Lieberman, P.M. Epigenetic regulation of EBV and KSHV latency. Curr. Opin. Virol. 2013, 3, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.; Charalambous, C.; Wilson, J.B. Epstein-Barr virus latent membrane protein 1 (CAO) up-regulates VEGF and TGF alpha concomitant with hyperlasia, with subsequent up-regulation of p16 and MMP9. Cancer Res. 2005, 65, 8826–8835. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, L.; Liu, L.; Xu, Z.; Liao, W.; Feng, D.; Dong, X.; Xu, S.; Xiao, L.; Lu, J.; Luo, X.; et al. EBV-LMP1 targeted DNAzyme enhances radiosensitivity by inhibiting tumor angiogenesis via the JNKs/HIF-1 pathway in nasopharyngeal carcinoma. Oncotarget 2015, 6, 5804–5817. [Google Scholar] [CrossRef] [PubMed]
- Murono, S.; Inoue, H.; Tanabe, T.; Joab, I.; Yoshizaki, T.; Furukawa, M.; Pagano, J.S. Induction of cyclooxygenase-2 by Epstein-Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proc. Natl. Acad. Sci. USA 2001, 98, 6905–6910. [Google Scholar] [CrossRef] [PubMed]
- Paydas, S.; Ergin, M.; Erdogan, S.; Seydaoglu, G. Prognostic significance of EBV-LMP1 and VEGF-A expressions in non-Hodgkin’s lymphomas. Leuk. Res. 2008, 32, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, J.D.; Owen, T.J.; Wood, V.H.; Date, K.L.; Valentine, R.; Chukwuma, M.B.; Arrand, J.R.; Dawson, C.W.; Young, L.S. Epstein-Barr virus-encoded EBNA1 modulates the AP-1 transcription factor pathway in nasopharyngeal carcinoma cells and enhances angiogenesis in vitro. J. Gen. Virol. 2008, 89, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Morgan, D.R.; Meyers, M.O.; Dominguez, R.L.; Martinez, E.; Kakudo, K.; Kuan, P.F.; Banet, N.; Muallem, H.; Woodward, K.; et al. Epstein-barr virus infected gastric adenocarcinoma expresses latent and lytic viral transcripts and has a distinct human gene expression profile. Infect. Agent Cancer 2012, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.W.; Chu, Y.C.; Chen, P.R.; Liao, M.H.; Lee, J.W. Positive regulation of HIF-1A expression by EBV oncoprotein LMP1 in nasopharyngeal carcinoma cells. Cancer Lett. 2016, 382, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Boshoff, C. Kaposi’s sarcoma. Coupling herpesvirus to angiogenesis. Nature 1998, 391, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Ensoli, B.; Sturzl, M. HHV-8 and multistep tumorigenesis. Trends Microbiol. 1999, 7, 310–312. [Google Scholar] [CrossRef]
- Marchio, S.; Primo, L.; Pagano, M.; Palestro, G.; Albini, A.; Veikkola, T.; Cascone, I.; Alitalo, K.; Bussolino, F. Vascular endothelial growth factor-C stimulates the migration and proliferation of Kaposi’s sarcoma cells. J. Biol. Chem. 1999, 274, 27617–27622. [Google Scholar] [CrossRef] [PubMed]
- Arasteh, K.; Hannah, A. The role of vascular endothelial growth factor (VEGF) in AIDS-related Kaposi’s sarcoma. Oncologist 2000, 5, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.F.; Tognazzi, K.; Dvorak, H.F.; Harrist, T.J. Strong expression of kinase insert domain-containing receptor, a vascular permeability factor/vascular endothelial growth factor receptor in AIDS-associated Kaposi’s sarcoma and cutaneous angiosarcoma. Am. J. Pathol. 1996, 148, 1065–1074. [Google Scholar] [PubMed]
- Flore, O.; Rafii, S.; Ely, S.; O’Leary, J.J.; Hyjek, E.M.; Cesarman, E. Transformation of primary human endothelial cells by Kaposi’s sarcoma-associated herpesvirus. Nature 1998, 394, 588–592. [Google Scholar] [CrossRef] [PubMed]
- An, F.Q.; Folarin, H.M.; Compitello, N.; Roth, J.; Gerson, S.L.; McCrae, K.R.; Fakhari, F.D.; Dittmer, D.P.; Renne, R. Long-term-infected telomerase-immortalized endothelial cells: A model for Kaposi’s sarcoma-associated herpesvirus latency in vitro and in vivo. J. Virol. 2006, 80, 4833–4846. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, A.D.; Cavallin, L.E.; Vincent, L.; Chiozzini, C.; Eroles, P.; Duran, E.M.; Asgari, Z.; Hooper, A.T.; La Perle, K.M.; Hilsher, C.; et al. In vivo-restricted and reversible malignancy induced by human herpesvirus-8 KSHV: A cell and animal model of virally induced Kaposi’s sarcoma. Cancer Cell 2007, 11, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Alkharsah, K.R.; Singh, V.V.; Bosco, R.; Santag, S.; Grundhoff, A.; Konrad, A.; Sturzl, M.; Wirth, D.; Dittrich-Breiholz, O.; Kracht, M.; et al. Deletion of Kaposi’s sarcoma-associated herpesvirus FLICE inhibitory protein, vFLIP, from the viral genome compromises the activation of STAT1-responsive cellular genes and spindle cell formation in endothelial cells. J. Virol. 2011, 85, 10375–10388. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Podgrabinska, S.; Skobe, M.; Ganem, D. Activation of NF-kappaB by the latent vFLIP gene of Kaposi’s sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J. Virol. 2006, 80, 7179–7185. [Google Scholar] [CrossRef] [PubMed]
- Field, N.; Low, W.; Daniels, M.; Howell, S.; Daviet, L.; Boshoff, C.; Collins, M. KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell Sci. 2003, 116, 3721–3728. [Google Scholar] [CrossRef] [PubMed]
- Matta, H.; Mazzacurati, L.; Schamus, S.; Yang, T.; Sun, Q.; Chaudhary, P.M. Kaposi’s sarcoma-associated herpesvirus (KSHV) oncoprotein K13 bypasses TRAFs and directly interacts with the IkappaB kinase complex to selectively activate NF-kappaB without JNK activation. J. Biol. Chem. 2007, 282, 24858–24865. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, S.; Pise-Masison, C.A.; Brady, J.N.; Tosato, G. Gene regulation and functional alterations induced by Kaposi’s sarcoma-associated herpesvirus-encoded ORFK13/vFLIP in endothelial cells. J. Virol. 2009, 83, 2140–2153. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Tosato, G. Role of vascular endothelial growth factor/vascular permeability factor in the pathogenesis of Kaposi’s sarcoma-associated herpesvirus-infected primary effusion lymphomas. Blood 1999, 94, 4247–4254. [Google Scholar] [PubMed]
- Wang, L.; Wakisaka, N.; Tomlinson, C.C.; DeWire, S.M.; Krall, S.; Pagano, J.S.; Damania, B. The Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) K1 protein induces expression of angiogenic and invasion factors. Cancer Res. 2004, 64, 2774–2781. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.Y.; Chen, S.C.; Leach, M.W.; Manfra, D.; Homey, B.; Wiekowski, M.; Sullivan, L.; Jenh, C.H.; Narula, S.K.; Chensue, S.W.; et al. Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi’s sarcoma. J. Exp. Med. 2000, 191, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.C.; Joo, C.H.; Gack, M.U.; Lee, H.R.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1 alpha to induce vascular endothelial growth factor expression. Cancer Res. 2008, 68, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.; O’Hearn, P.M. Use of the red fluorescent protein as a marker of Kaposi’s sarcoma-associated herpesvirus lytic gene expression. Virology 2004, 325, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Spence, T.; Bruce, J.; Yip, K.W.; Liu, F.F. HPV Associated Head and Neck Cancer. Cancers 2016, 8, 75. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.L. Cancer Prevention: HPV Vaccination. Semin. Oncol. Nurs. 2016, 32, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Bratman, S.V.; Bruce, J.P.; O’Sullivan, B.; Pugh, T.J.; Xu, W.; Yip, K.W.; Liu, F.F. Human Papillomavirus Genotype Association With Survival in Head and Neck Squamous Cell Carcinoma. JAMA Oncol. 2016, 2, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, C.; Mena, M.; Alemany, L.; Arbyn, M.; Castellsague, X.; Laporte, L.; Bosch, F.X.; de Sanjose, S.; Trottier, H. HPV DNA, E6/E7 mRNA, and p16INK4a detection in head and neck cancers: A systematic review and meta-analysis. Lancet Oncol. 2014, 15, 1319–1331. [Google Scholar] [CrossRef]
- Anderson, L.A.; O’Rorke, M.A.; Wilson, R.; Jamison, J.; Gavin, A.T.; Northern Ireland HPV Working Group. HPV prevalence and type-distribution in cervical cancer and premalignant lesions of the cervix: A population-based study from Northern Ireland. J. Med. Virol. 2016, 88, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, D.; Petti, L.M. The E5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Song, S.; Liem, A.; Miller, J.A.; Lambert, P.F. Human papillomavirus types 16 E6 and E7 contribute differently to carcinogenesis. Virology 2000, 267, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; He, L.; Zhang, E.; Shi, J.; Zhang, Q.; Le, A.D.; Zhou, K.; Tang, X. Overexpression of human papillomavirus (HPV) type 16 oncoproteins promotes angiogenesis via enhancing HIF-1alpha and VEGF expression in non-small cell lung cancer cells. Cancer Lett. 2011, 311, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, Q.; Nishitani, J.; Brown, J.; Shi, S.; Le, A.D. Overexpression of human papillomavirus type 16 oncoproteins enhances hypoxia-inducible factor 1 alpha protein accumulation and vascular endothelial growth factor expression in human cervical carcinoma cells. Clin. Cancer Res. 2007, 13, 2568–2576. [Google Scholar] [CrossRef] [PubMed]
- Toussaint-Smith, E.; Donner, D.B.; Roman, A. Expression of human papillomavirus type 16 E6 and E7 oncoproteins in primary foreskin keratinocytes is sufficient to alter the expression of angiogenic factors. Oncogene 2004, 23, 2988–2995. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ocejo, O.; Viloria-Petit, A.; Bequet-Romero, M.; Mukhopadhyay, D.; Rak, J.; Kerbel, R.S. Oncogenes and tumor angiogenesis: The HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene 2000, 19, 4611–4620. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Cui, J. Human telomerase reverse transcriptase regulates vascular endothelial growth factor expression via human papillomavirus oncogene E7 in HPV-18-positive cervical cancer cells. Med. Oncol. 2015, 32, 199. [Google Scholar] [CrossRef] [PubMed]
- Tilborghs, S.; Corthouts, J.; Verhoeven, Y.; Arias, D.; Rolfo, C.; Trinh, X.B.; van Dam, P.A. The role of Nuclear Factor-kappa B signaling in human cervical cancer. Crit. Rev. Oncol. Hematol. 2017, 120, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Vescovo, T.; Refolo, G.; Vitagliano, G.; Fimia, G.M.; Piacentini, M. Molecular mechanisms of hepatitis C virus-induced hepatocellular carcinoma. Clin. Microbiol. Infect. 2016, 22, 853–861. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Kramer, J.R.; Chen, G.J.; Duan, Z.; Richardson, P.A.; Davila, J.A. Effectiveness of AFP and ultrasound tests on hepatocellular carcinoma mortality in HCV-infected patients in the USA. Gut 2011, 60, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Messerini, L.; Novelli, L.; Comin, C.E. Microvessel density and clinicopathological characteristics in hepatitis C virus and hepatitis B virus related hepatocellular carcinoma. J. Clin. Pathol. 2004, 57, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Pena, C.E.; Lathia, C.D.; Shan, M.; Meinhardt, G.; Bruix, J. Plasma biomarkers as predictors of outcome in patients with advanced hepatocellular carcinoma. Clin. Cancer Res. 2012, 18, 2290–2300. [Google Scholar] [CrossRef] [PubMed]
- Mukozu, T.; Nagai, H.; Matsui, D.; Kanekawa, T.; Sumino, Y. Serum VEGF as a tumor marker in patients with HCV-related liver cirrhosis and hepatocellular carcinoma. Anticancer Res. 2013, 33, 1013–1021. [Google Scholar] [CrossRef]
- Yvamoto, E.Y.; Ferreira, R.F.; Nogueira, V.; Pinhe, M.A.; Tenani, G.D.; Andrade, J.G.; Baitello, M.E.; Gregorio, M.L.; Fucuta, P.S.; Silva, R.F.; Souza, D.R.; et al. Influence of vascular endothelial growth factor and alpha-fetoprotein on hepatocellular carcinoma. Genet. Mol. Res. 2015, 14, 17453–17462. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, K.; Mori, M.; Shibuta, K.; Banner, B.F.; Barnard, G.F. Vascular endothelial growth factor/vascular permeability factor mRNA expression in patients with chronic hepatitis C and hepatocellular carcinoma. Int. J. Oncol. 1999, 14, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Koga, H.; Yoshida, T.; Masuda, H.; Iwamoto, H.; Sakata, M.; Hanada, S.; Nakamura, T.; Taniguchi, E.; Kawaguchi, T.; et al. Hepatitis C virus core protein upregulates the expression of vascular endothelial growth factor via the nuclear factor-kappaB/hypoxia-inducible factor-1alpha axis under hypoxic conditions. Hepatol. Res. 2012, 42, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Liu, X.; Wang, S.; Yan, X.; Tang, Z.; Wu, K.; Li, Y.; Liu, F. Hepatitis C virus core protein induces hypoxia-inducible factor 1alpha-mediated vascular endothelial growth factor expression in Huh7.5.1 cells. Mol. Med. Rep. 2014, 9, 2010–2014. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus core protein augments androgen receptor-mediated signaling. J. Virol. 2008, 82, 11066–11072. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.Y.; Hsieh, M.S.; Wang, H.Y.; Li, Y.S.; Lin, H.; Hsu, H.W.; Huang, C.Y.; Hsu, C.H.; Cheng, A.L. Hepatitis C virus core protein potentiates proangiogenic activity of hepatocellular carcinoma cells. Oncotarget 2017, 8, 86681–86692. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ringelhan, M.; O’Connor, T.; Protzer, U.; Heikenwalder, M. The direct and indirect roles of HBV in liver cancer: Prospective markers for HCC screening and potential therapeutic targets. J. Pathol. 2015, 235, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Ringelhan, M.; Protzer, U. Oncogenic potential of hepatitis B virus encoded proteins. Curr. Opin. Virol. 2015, 14, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Bonilla Guerrero, R.; Roberts, L.R. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J. Hepatol. 2005, 42, 760–777. [Google Scholar] [CrossRef] [PubMed]
- Nosaka, T.; Naito, T.; Hiramatsu, K.; Ohtani, M.; Nemoto, T.; Marusawa, H.; Ma, N.; Hiraku, Y.; Kawanishi, S.; Yamashita, T.; et al. Gene expression profiling of hepatocarcinogenesis in a mouse model of chronic hepatitis B. PLoS ONE 2017, 12, e0185442. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.B.; Shen, S.Q.; Ding, Y.M.; Wang, W.X.; Tao, J.P.; Liang, L.J.; Hu, W.J. The angiogenic and prognostic implications of VEGF, Ang-1, Ang-2, and MMP-9 for hepatocellular carcinoma with background of hepatitis B virus. Med. Oncol. 2009, 26, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.S.; Chan, H.L.; To, K.F.; Leung, W.K.; Chan, K.K.; Liew, C.T.; Sung, J.J. Cyclooxygenase-2 pathway correlates with vascular endothelial growth factor expression and tumor angiogenesis in hepatitis B virus-associated hepatocellular carcinoma. Int. J. Oncol. 2004, 24, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Lee, Y.M.; Bae, S.K.; Murakami, S.; Yun, Y.; Kim, K.W. Human hepatitis B virus X protein is a possible mediator of hypoxia-induced angiogenesis in hepatocarcinogenesis. Biochem. Biophys. Res. Commun. 2000, 268, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.G.; Na, T.Y.; Seo, H.W.; Seong, J.K.; Park, C.K.; Shin, Y.K.; Lee, M.O. Hepatitis B virus X protein induces the expression of MTA1 and HDAC1, which enhances hypoxia signaling in hepatocellular carcinoma cells. Oncogene 2008, 27, 3405–3413. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.G.; Oh, S.H.; Park, E.S.; Cho, H.; Lee, N.; Park, H.; Kim, D.K.; Yu, D.Y.; Seong, J.K.; Lee, M.O. Hepatitis B virus X protein enhances transcriptional activity of hypoxia-inducible factor-1alpha through activation of mitogen-activated protein kinase pathway. J. Biol. Chem. 2003, 278, 39076–39084. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.P.; Hu, B.G.; Ye, C.; Ho, R.L.; Chen, G.G.; Lai, P.B. HBx mutants differentially affect the activation of hypoxia-inducible factor-1alpha in hepatocellular carcinoma. Br. J. Cancer 2014, 110, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.J.; Lin, Y.J.; Yen, C.S.; Tsai, H.W.; Tsai, T.F.; Chang, K.Y.; Huang, W.C.; Lin, P.W.; Chiang, C.W.; Chang, T.T. Hepatitis B virus X protein upregulates mTOR signaling through IKKbeta to increase cell proliferation and VEGF production in hepatocellular carcinoma. PLoS ONE 2012, 7, e41931. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Teng, C.F.; Wu, H.C.; Tsai, H.W.; Chuang, H.C.; Tsai, T.F.; Hsu, Y.H.; Huang, W.; Wu, L.W.; Su, I.J. Enhanced expression of vascular endothelial growth factor-A in ground glass hepatocytes and its implication in hepatitis B virus hepatocarcinogenesis. Hepatology 2009, 49, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- World Health, O. Hepatitis B vaccines: WHO position paper, July 2017-Recommendations. Vaccine 2017. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Robinson, N.J. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: A global review. J. Infect. Dis. 2002, 186, S3–S28. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J.; Chang, M.; Garg, N.; Zhu, J.; Chang, J.H.; Shukla, D. Corneal lymphangiogenesis in herpetic stromal keratitis. Surv. Ophthalmol. 2015, 60, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Wuest, T.R.; Carr, D.J. VEGF-A expression by HSV-1-infected cells drives corneal lymphangiogenesis. J. Exp. Med. 2010, 207, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Deshpande, S.; Lee, S.; Ferrara, N.; Rouse, B.T. Contribution of vascular endothelial growth factor in the neovascularization process during the pathogenesis of herpetic stromal keratitis. J. Virol. 2001, 75, 9828–9835. [Google Scholar] [CrossRef] [PubMed]
- Biswas, P.S.; Banerjee, K.; Kinchington, P.R.; Rouse, B.T. Involvement of IL-6 in the paracrine production of VEGF in ocular HSV-1 infection. Exp. Eye Res. 2006, 82, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Wuest, T.; Zheng, M.; Efstathiou, S.; Halford, W.P.; Carr, D.J. The herpes simplex virus-1 transactivator infected cell protein-4 drives VEGF-A dependent neovascularization. PLoS Pathog. 2011, 7, e1002278. [Google Scholar] [CrossRef] [PubMed]
- Suryawanshi, A.; Mulik, S.; Sharma, S.; Reddy, P.B.; Sehrawat, S.; Rouse, B.T. Ocular neovascularization caused by herpes simplex virus type 1 infection results from breakdown of binding between vascular endothelial growth factor A and its soluble receptor. J. Immunol. 2011, 186, 3653–3665. [Google Scholar] [CrossRef] [PubMed]
- Suryawanshi, A.; Veiga-Parga, T.; Reddy, P.B.; Rajasagi, N.K.; Rouse, B.T. IL-17A differentially regulates corneal vascular endothelial growth factor (VEGF)-A and soluble VEGF receptor 1 expression and promotes corneal angiogenesis after herpes simplex virus infection. J. Immunol. 2012, 188, 3434–3446. [Google Scholar] [CrossRef] [PubMed]
- Mulik, S.; Xu, J.; Reddy, P.B.; Rajasagi, N.K.; Gimenez, F.; Sharma, S.; Lu, P.Y.; Rouse, B.T. Role of miR-132 in angiogenesis after ocular infection with herpes simplex virus. Am. J. Pathol. 2012, 181, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Gurung, H.R.; Carr, M.M.; Bryant, K.; Chucair-Elliott, A.J.; Carr, D.J. Fibroblast growth factor-2 drives and maintains progressive corneal neovascularization following HSV-1 infection. Mucosal Immunol. 2018, 11, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Screaton, G.; Mongkolsapaya, J.; Yacoub, S.; Roberts, C. New insights into the immunopathology and control of dengue virus infection. Nat. Rev. Immunol. 2015, 15, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Discovery of vascular permeability factor (VPF). Exp. Cell Res. 2006, 312, 522–556. [Google Scholar] [CrossRef] [PubMed]
- Del Moral-Hernandez, O.; Martinez-Hernandez, N.E.; Mosso-Pani, M.A.; Hernandez-Sotelo, D.; Illades-Aguiar, B.; Flores-Alfaro, E.; Antonio-Vejar, V.; Leyva-Vazquez, M.A. Association DENV1 and DENV2 infection with high serum levels of soluble thrombomodulin and VEGF in patients with dengue fever and dengue hemorrhagic fever. Int. J. Clin. Exp. Med. 2014, 7, 370–378. [Google Scholar] [PubMed]
- Furuta, T.; Murao, L.A.; Lan, N.T.; Huy, N.T.; Huong, V.T.; Thuy, T.T.; Tham, V.D.; Nga, C.T.; Ha, T.T.; Ohmoto, Y.; et al. Association of mast cell-derived VEGF and proteases in Dengue shock syndrome. PLoS Negl. Trop. Dis. 2012, 6, e1505. [Google Scholar] [CrossRef] [PubMed]
- Srikiatkhachorn, A.; Ajariyakhajorn, C.; Endy, T.P.; Kalayanarooj, S.; Libraty, D.H.; Green, S.; Ennis, F.A.; Rothman, A.L. Virus-induced decline in soluble vascular endothelial growth receptor 2 is associated with plasma leakage in dengue hemorrhagic Fever. J. Virol. 2007, 81, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.S.; Lo, H.W.; Teng, H.C.; Lo, W.C.; Ker, C.G. Elevated levels of plasma VEGF in patients with dengue hemorrhagic fever. FEMS Immunol. Med. Microbiol. 2005, 43, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Van de Weg, C.A.; Pannuti, C.S.; van den Ham, H.J.; de Araujo, E.S.; Boas, L.S.; Felix, A.C.; Carvalho, K.I.; Levi, J.E.; Romano, C.M.; Centrone, C.C.; et al. Serum angiopoietin-2 and soluble VEGF receptor 2 are surrogate markers for plasma leakage in patients with acute dengue virus infection. J. Clin. Virol. 2014, 60, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Azizan, A.; Fitzpatrick, K.; Signorovitz, A.; Tanner, R.; Hernandez, H.; Stark, L.; Sweat, M. Profile of time-dependent VEGF upregulation in human pulmonary endothelial cells, HPMEC-ST1.6R infected with DENV-1, -2, -3, and -4 viruses. Virol. J. 2009, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Azizan, A.; Sweat, J.; Espino, C.; Gemmer, J.; Stark, L.; Kazanis, D. Differential proinflammatory and angiogenesis-specific cytokine production in human pulmonary endothelial cells, HPMEC-ST1.6R infected with dengue-2 and dengue-3 virus. J. Virol. Methods 2006, 138, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C.; Zhang, Y.Z. The evolution and emergence of hantaviruses. Curr. Opin. Virol. 2015, 10, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Kruger, D.H.; Figueiredo, L.T.; Song, J.W.; Klempa, B. Hantaviruses—Globally emerging pathogens. J. Clin. Virol. 2015, 64, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, W.; Wang, J.P.; Pan, L.; Zhang, Y.; Yu, H.T.; Jiang, W.; Wang, P.Z.; Bai, X.F. Elevated vascular endothelial growth factor levels induce hyperpermeability of endothelial cells in hantavirus infection. J. Int. Med. Res. 2012, 40, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovskaya, I.N.; Gorbunova, E.E.; Mackow, N.A.; Mackow, E.R. Hantaviruses direct endothelial cell permeability by sensitizing cells to the vascular permeability factor VEGF, while angiopoietin 1 and sphingosine 1-phosphate inhibit hantavirus-directed permeability. J. Virol. 2008, 82, 5797–5806. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovskaya, I.N.; Gorbunova, E.E.; Mackow, E.R. Hypoxia induces permeability and giant cell responses of Andes virus-infected pulmonary endothelial cells by activating the mTOR-S6K signaling pathway. J. Virol. 2013, 87, 12999–13008. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, K.B.; Nguyen Hoang, A.T.; Gupta, S.; Ahlm, C.; Svensson, M.; Klingstrom, J. Andes Hantavirus-Infection of a 3D Human Lung Tissue Model Reveals a Late Peak in Progeny Virus Production Followed by Increased Levels of Proinflammatory Cytokines and VEGF-A. PLoS ONE 2016, 11, e0149354. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, E.; Gavrilovskaya, I.N.; Mackow, E.R. Pathogenic hantaviruses Andes virus and Hantaan virus induce adherens junction disassembly by directing vascular endothelial cadherin internalization in human endothelial cells. J. Virol. 2010, 84, 7405–7411. [Google Scholar] [CrossRef] [PubMed]
- Easterbrook, J.D.; Klein, S.L. Seoul virus enhances regulatory and reduces proinflammatory responses in male Norway rats. J. Med. Virol. 2008, 80, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, E.; Nusshag, C.; Baumann, A.; Schafer, J.; Hofmann, J.; Schnitzler, P.; Klempa, B.; Witkowski, P.T.; Kruger, D.H.; Zeier, M. Clinical characterization of two severe cases of hemorrhagic fever with renal syndrome (HFRS) caused by hantaviruses Puumala and Dobrava-Belgrade genotype Sochi. BMC Infect. Dis. 2016, 16, 675. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fleming, S.B.; Wise, L.M.; Mercer, A.A. Molecular genetic analysis of orf virus: A poxvirus that has adapted to skin. Viruses 2015, 7, 1505–1539. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Oku, A.; Sawano, A.; Yamaguchi, S.; Yazaki, Y.; Shibuya, M. A novel type of vascular endothelial growth factor, VEGF-E (NZ-7 VEGF), preferentially utilizes KDR/Flk-1 receptor and carries a potent mitotic activity without heparin-binding domain. J. Biol. Chem. 1998, 273, 31273–31282. [Google Scholar] [CrossRef] [PubMed]
- Lyttle, D.J.; Fraser, K.M.; Fleming, S.B.; Mercer, A.A.; Robinson, A.J. Homologs of vascular endothelial growth factor are encoded by the poxvirus orf virus. J. Virol. 1994, 68, 84–92. [Google Scholar] [PubMed]
- Wise, L.M.; Savory, L.J.; Dryden, N.H.; Whelan, E.M.; Fleming, S.B.; Mercer, A.A. Major amino acid sequence variants of viral vascular endothelial growth factor are functionally equivalent during Orf virus infection of sheep skin. Virus Res. 2007, 128, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Savory, L.J.; Stacker, S.A.; Fleming, S.B.; Niven, B.E.; Mercer, A.A. Viral vascular endothelial growth factor plays a critical role in orf virus infection. J. Virol. 2000, 74, 10699–106706. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular endothelial growth factor receptor-2: Its unique signaling and specific ligand, VEGF-E. Cancer Sci. 2003, 94, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Clauss, M.; Lepple-Wienhues, A.; Waltenberger, J.; Augustin, H.G.; Ziche, M.; Lanz, C.; Buttner, M.; Rziha, H.J.; Dehio, C. A novel vascular endothelial growth factor encoded by Orf virus, VEGF-E, mediates angiogenesis via signalling through VEGFR-2 (KDR) but not VEGFR-1 (Flt-1) receptor tyrosine kinases. EMBO J. 1999, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Cebe-Suarez, S.; Grunewald, F.S.; Jaussi, R.; Li, X.; Claesson-Welsh, L.; Spillmann, D.; Mercer, A.A.; Prota, A.E.; Ballmer-Hofer, K. Orf virus VEGF-E NZ2 promotes paracellular NRP-1/VEGFR-2 coreceptor assembly via the peptide RPPR. FASEB J. 2008, 22, 3078–3086. [Google Scholar] [CrossRef] [PubMed]
- Wise, L.M.; Veikkola, T.; Mercer, A.A.; Savory, L.J.; Fleming, S.B.; Caesar, C.; Vitali, A.; Makinen, T.; Alitalo, K.; Stacker, S.A. Vascular endothelial growth factor (VEGF)-like protein from orf virus NZ2 binds to VEGFR2 and neuropilin-1. Proc. Natl. Acad. Sci. USA 1999, 96, 3071–3076. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, Y.; Yamazaki, Y.; Morita, T. Localization of heparin- and neuropilin-1-recognition sites of viral VEGFs. Biochem. Biophys. Res. Commun. 2006, 348, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Wise, L.M.; Stacker, S.A.; Fleming, S.B.; Mercer, A.A. Pseudocowpox virus encodes a homolog of vascular endothelial growth factor. Virology 2003, 305, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Inder, M.K.; Ueda, N.; Mercer, A.A.; Fleming, S.B.; Wise, L.M. Bovine papular stomatitis virus encodes a functionally distinct VEGF that binds both VEGFR-1 and VEGFR-2. J. Gen. Virol. 2007, 88, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Inder, M.K.; Wise, L.M.; Fleming, S.B.; Mercer, A.A. Parapoxvirus of red deer in New Zealand encodes a variant of viral vascular endothelial growth factor. Virus Res. 2007, 124, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Hamden, K.E.; Whitman, A.G.; Ford, P.W.; Shelton, J.G.; McCubrey, J.A.; Akula, S.M. Raf and VEGF: Emerging therapeutic targets in Kaposi’s sarcoma-associated herpesvirus infection and angiogenesis in hematopoietic and nonhematopoietic tumors. Leukemia 2005, 19, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.; Lee, V.H.; Kwong, D.L. Novel therapy for nasopharyngeal carcinoma—Where are we. Oral Oncol. 2014, 50, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Zhang, Q.; Pfister, D.G.; Kim, J.; Garden, A.S.; Mechalakos, J.; Hu, K.; Le, Q.T.; Colevas, A.D.; Glisson, B.S.; et al. Addition of bevacizumab to standard chemoradiation for locoregionally advanced nasopharyngeal carcinoma (RTOG 0615): A phase 2 multi-institutional trial. Lancet Oncol. 2012, 13, 172–180. [Google Scholar] [CrossRef]
- Roy, D.; Sin, S.H.; Lucas, A.; Venkataramanan, R.; Wang, L.; Eason, A.; Chavakula, V.; Hilton, I.B.; Tamburro, K.M.; Damania, B.; et al. mTOR inhibitors block Kaposi sarcoma growth by inhibiting essential autocrine growth factors and tumor angiogenesis. Cancer Res. 2013, 73, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Rader, J.S.; Sill, M.W.; Beumer, J.H.; Lankes, H.A.; Benbrook, D.M.; Garcia, F.; Trimble, C.; Tate Thigpen, J.; Lieberman, R.; Zuna, R.E.; et al. A stratified randomized double-blind phase II trial of celecoxib for treating patients with cervical intraepithelial neoplasia: The potential predictive value of VEGF serum levels: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 145, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Argiris, A.; Bauman, J.E.; Ohr, J.; Gooding, W.E.; Heron, D.E.; Duvvuri, U.; Kubicek, G.J.; Posluszny, D.M.; Vassilakopoulou, M.; Kim, S.; et al. Phase II randomized trial of radiation therapy, cetuximab, and pemetrexed with or without bevacizumab in patients with locally advanced head and neck cancer. Ann. Oncol. 2016, 27, 1594–1600. [Google Scholar] [CrossRef] [PubMed]
- Baghban Rahimi, S.; Mohebbi, A.; Vakilzadeh, G.; Biglari, P.; Razeghi Jahromi, S.; Mohebi, S.R.; Shirian, S.; Gorji, A.; Ghaemi, A. Enhancement of therapeutic DNA vaccine potency by melatonin through inhibiting VEGF expression and induction of antitumor immunity mediated by CD8+ T cells. Arch. Virol. 2018, 163, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Ngo, V.C.; Koong, H.N.; Poon, D.; Choo, S.P.; Toh, H.C.; Thng, C.H.; Chow, P.; Ong, H.S.; Chung, A.; et al. AZD6244 enhances the anti-tumor activity of sorafenib in ectopic and orthotopic models of human hepatocellular carcinoma (HCC). J. Hepatol. 2010, 52, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Sampat, K.R.; O’Neil, B. Antiangiogenic therapies for advanced hepatocellular carcinoma. Oncologist 2013, 18, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Himmelsbach, K.; Sauter, D.; Baumert, T.F.; Ludwig, L.; Blum, H.E.; Hildt, E. New aspects of an anti-tumour drug: Sorafenib efficiently inhibits HCV replication. Gut 2009, 58, 1644–1653. [Google Scholar] [CrossRef] [PubMed]
- Descamps, V.; Helle, F.; Louandre, C.; Martin, E.; Brochot, E.; Izquierdo, L.; Fournier, C.; Hoffmann, T.W.; Castelain, S.; Duverlie, G.; et al. The kinase-inhibitor sorafenib inhibits multiple steps of the Hepatitis C Virus infectious cycle in vitro. Antiviral Res. 2015, 118, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wen, F.; Li, J.; Zhang, P.; Yan, W.; Hao, P.; Xia, F.; Bi, F.; Li, Q. A high baseline HBV load and antiviral therapy affect the survival of patients with advanced HBV-related HCC treated with sorafenib. Liver Int. 2015, 35, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Tang, Q.; Biswas, P.S.; Xu, J.; Schiffelers, R.M.; Xie, F.Y.; Ansari, A.M.; Scaria, P.V.; Woodle, M.C.; Lu, P.; et al. Inhibition of ocular angiogenesis by siRNA targeting vascular endothelial growth factor pathway genes: Therapeutic strategy for herpetic stromal keratitis. Am. J. Pathol. 2004, 165, 2177–2185. [Google Scholar] [CrossRef]
- Kalogeropoulos, D.; Geka, A.; Malamos, K.; Kanari, M.; Kalogeropoulos, C. New Therapeutic Perceptions in a Patient with Complicated Herpes Simplex Virus 1 Keratitis: A Case Report and Review of the Literature. Am. J. Case Rep. 2017, 18, 1382–1389. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mackow, E.R.; Gorbunova, E.E.; Dalrymple, N.A.; Gavrilovskaya, I.N. Role of vascular and lymphatic endothelial cells in hantavirus pulmonary syndrome suggests targeted therapeutic approaches. Lymphat. Res. Biol. 2013, 11, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, C.B.; Hooper, J.; Mertz, G. Treatment of hantavirus pulmonary syndrome. Antiviral Res. 2008, 78, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, E.E.; Gavrilovskaya, I.N.; Pepini, T.; Mackow, E.R. VEGFR2 and Src kinase inhibitors suppress Andes virus-induced endothelial cell permeability. J. Virol. 2011, 85, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Bird, B.H.; Shrivastava-Ranjan, P.; Dodd, K.A.; Erickson, B.R.; Spiropoulou, C.F. Effect of Vandetanib on Andes virus survival in the hamster model of Hantavirus pulmonary syndrome. Antiviral Res. 2016, 132, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Zhang, D.W.; Wen, J.; Cao, Q.; Chen, R.J.; Zhu, J.; Feng, Z.Q. A novel LMP1 antibody synergizes with mitomycin C to inhibit nasopharyngeal carcinoma growth in vivo through inducing apoptosis and downregulating vascular endothelial growth factor. Int. J. Mol. Sci. 2012, 13, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Murakami, M.; Takahashi, H.; Yamauchi, M.; Kiba, A.; Yamaguchi, S.; Yabana, N.; Alitalo, K.; Shibuya, M. Chimeric VEGF-E(NZ7)/PlGF promotes angiogenesis via VEGFR-2 without significant enhancement of vascular permeability and inflammation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2019–2026. [Google Scholar] [CrossRef] [PubMed]
- Inoue, N.; Kondo, T.; Kobayashi, K.; Aoki, M.; Numaguchi, Y.; Shibuya, M.; Murohara, T. Therapeutic angiogenesis using novel vascular endothelial growth factor-E/human placental growth factor chimera genes. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Bodaan, C.J.; Wise, L.M.; Wakelin, K.A.; Stuart, G.S.; Real, N.C.; Mercer, A.A.; Riley, C.B.; Theoret, C. Short-term treatment of equine wounds with orf virus IL-10 and VEGF-E dampens inflammation and promotes repair processes without accelerating closure. Wound Repair Regen. 2016, 24, 966–980. [Google Scholar] [CrossRef] [PubMed]


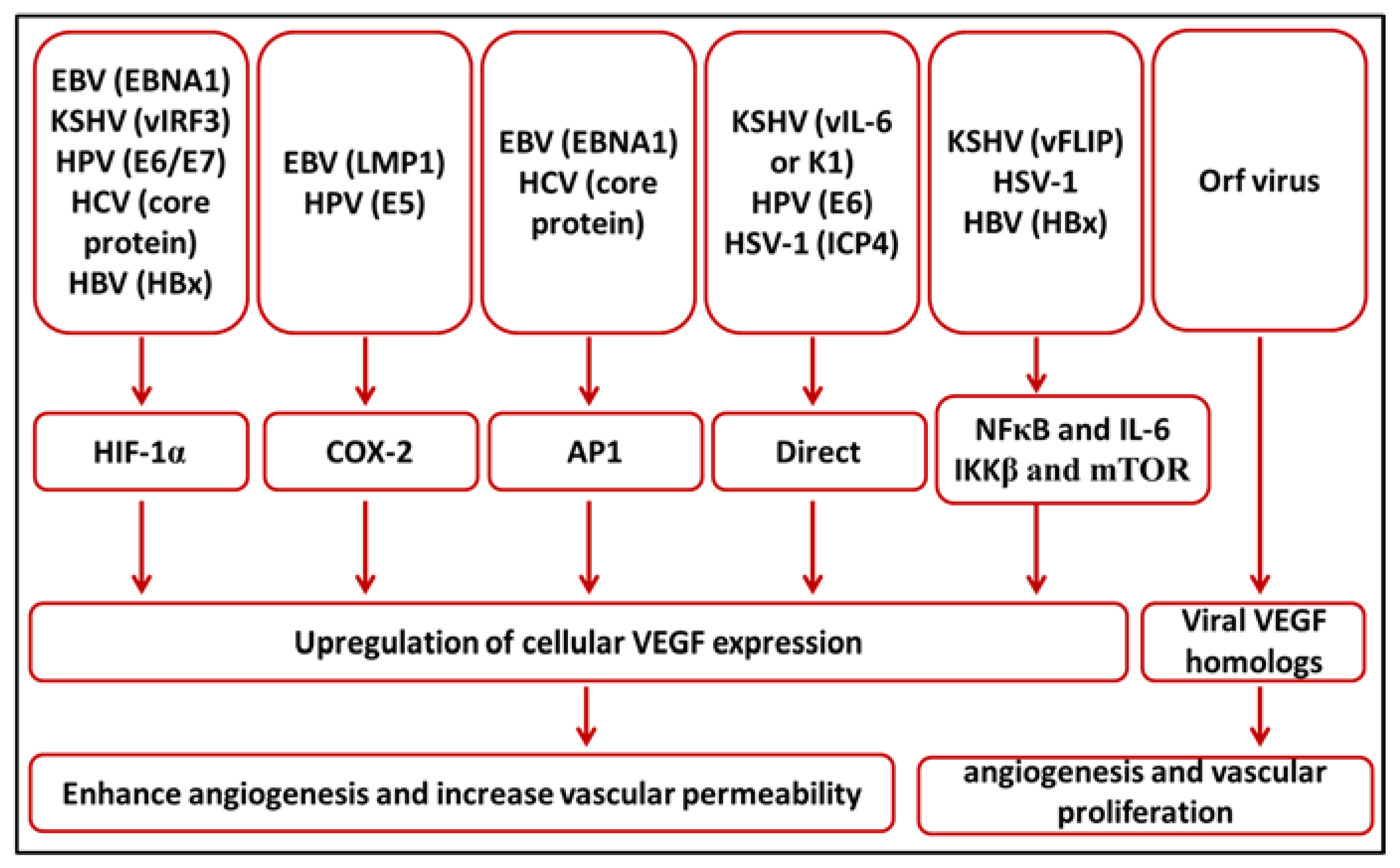{kind=link}
| Virus | Disease | Mechanism of VEGF Upregulation | References |
|---|---|---|---|
| EBV | Nasopharyngeal carcinoma | LMP1 upregulates the expression of VEGF through the phosphorylation of JNKs/c-Jun signaling pathways | [20,21] |
| LMP1 upregulates COX-2 expression which leads to the upregulation of VEGF expression | [22] | ||
| EBNA1 activates the AP-1 transcription factor which enhances the transcription of VEGF | [24] | ||
| Gastric carcinoma | Overexpression of HIF-1α leads to the upregulation of VEGF expression | [26] | |
| KSHV | Kaposi’s sarcoma | vFLIP activates the transcription factor NF-κB which induce inflammatory response leads to upregulation of VEGF | [38,39,40] |
| Viral K1 and vIL-6 directly implicated in expression of VEGF | [41,42] | ||
| vGPCR enhances the upregulation of VEGF | [43] | ||
| vIRF3 stabilizes HIF-1α to enhance VEGF expression | [44] | ||
| HPV | Cervical cancer, head and neck carcinoma | E6 binds to a responsive region consisting of four SP-1 sites in the VEGF promoter region | [58] |
| E7 upregulates VEGF expression through the telomerase reverse-transcriptase (hTERT) and telomerase activity | [59] | ||
| E5 activates the EGFR which in turn leads to the phosphorylation of PI3K and Akt, enhancement of the transcription of COX-2 leading to increase in VEGF expression | [56,60] | ||
| HCV | Hepatocellular carcinoma | Core protein stabilizes HIF-1α which automatically upregulates the expression of VEGF | [67,68,69] |
| Core protein activates the AP-1 transcription factor which potentiates VEGF expression through direct binding to its promoter | [71] | ||
| HBV | Hepatocellular carcinoma | HBx protein stabilizes the HIF-1α and enhances VEGF expression | [79,80,81] |
| HBx induces mTOR and IKKβ which in turn induces VEGF expression | [82] | ||
| Pre-S protein potentiates the expression of VEGF | [83] | ||
| HSV-1 | Herpetic stromal keratitis | ICP4 binds directly to the proximal promoter of VEGF gene and drives its transcription | [90] |
| HSV-1 disturbs the balance between VEGF and its neutralizing receptor sVEGFR1 | [91] | ||
| DENV | Dengue hemorrhagic fever, dengue shock syndrome | Upregulation of Th1 and Th2 cytokines and VEGF | [102,103] |
| Hantaviruses | Hantavirus Pulmonary Syndrome, Hemorrhagic Fever with Renal Syndrome | Mechanism jet to be elucidated | |
| Orf virus | Pustular skin disease | Encodes VEGF homolog called VEGF-E | [113,115] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkharsah, K.R. VEGF Upregulation in Viral Infections and Its Possible Therapeutic Implications. Int. J. Mol. Sci. 2018, 19, 1642. https://doi.org/10.3390/ijms19061642
Alkharsah KR. VEGF Upregulation in Viral Infections and Its Possible Therapeutic Implications. International Journal of Molecular Sciences. 2018; 19(6):1642. https://doi.org/10.3390/ijms19061642
Chicago/Turabian StyleAlkharsah, Khaled R. 2018. "VEGF Upregulation in Viral Infections and Its Possible Therapeutic Implications" International Journal of Molecular Sciences 19, no. 6: 1642. https://doi.org/10.3390/ijms19061642
APA StyleAlkharsah, K. R. (2018). VEGF Upregulation in Viral Infections and Its Possible Therapeutic Implications. International Journal of Molecular Sciences, 19(6), 1642. https://doi.org/10.3390/ijms19061642