Structural Biology of STAT3 and Its Implications for Anticancer Therapies Development

 , and
, and
Abstract
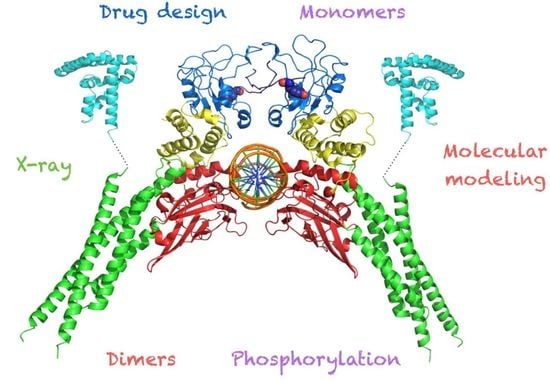
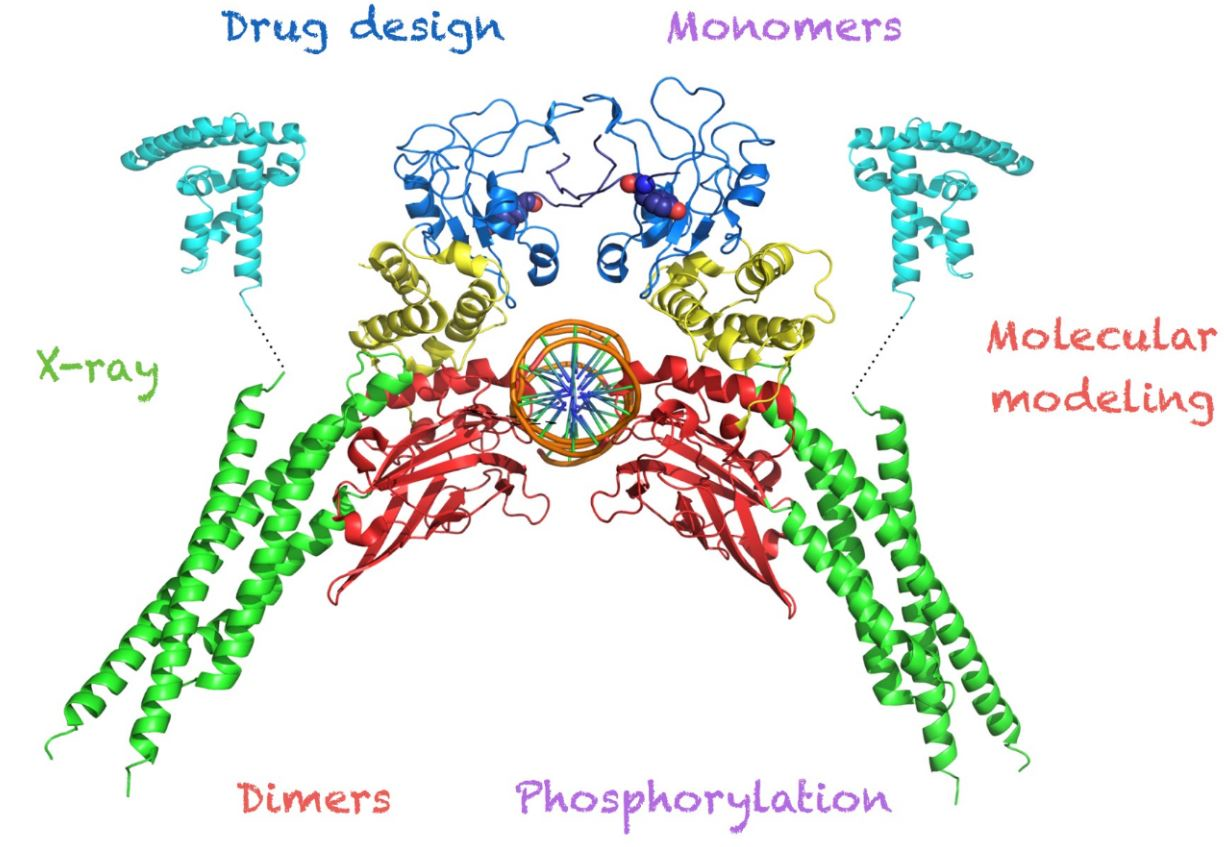
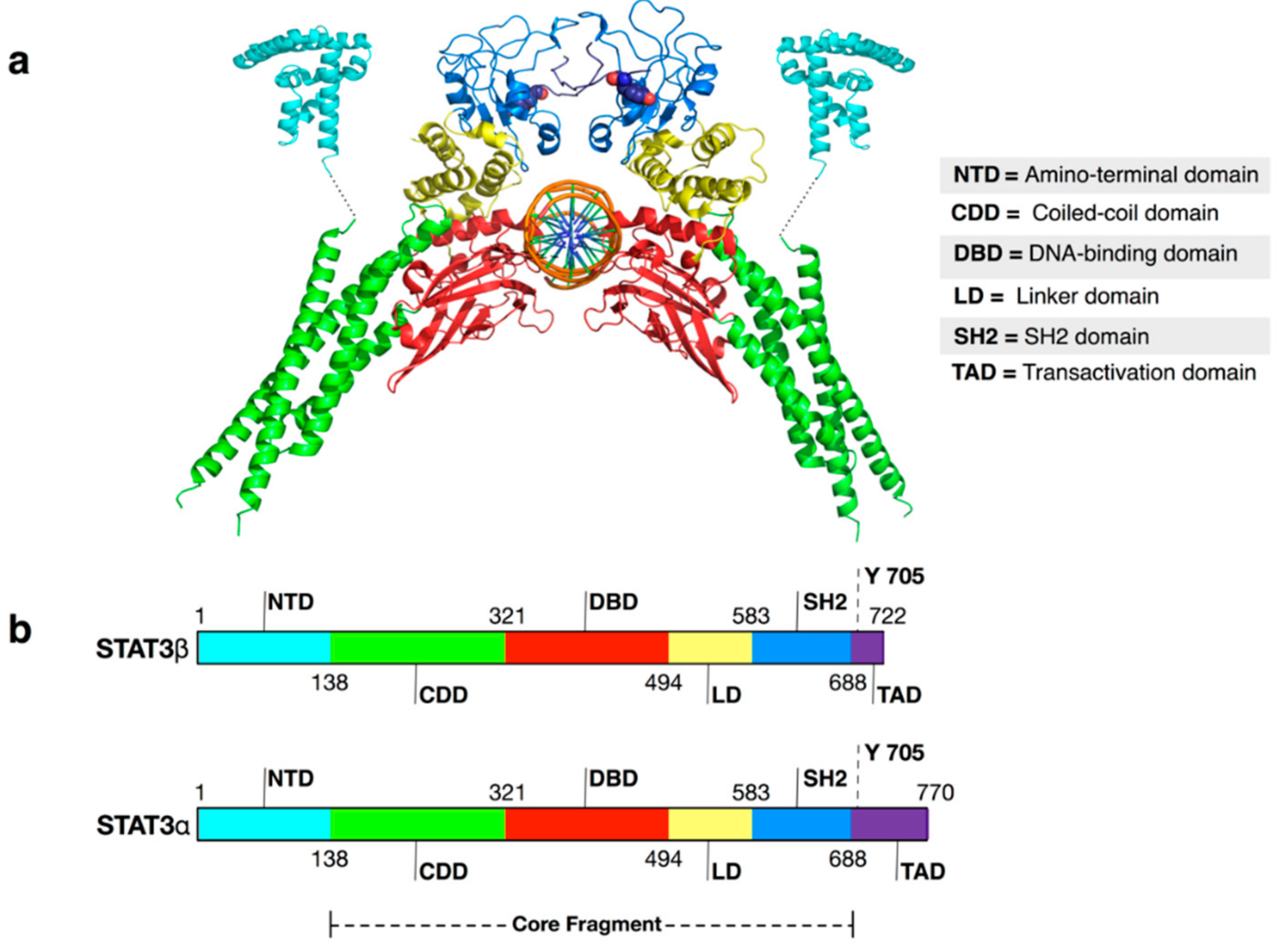
1. Introduction
2. Functional Mechanism
3. Post-Transcriptional Modifications and Their Role in the STAT3 Function
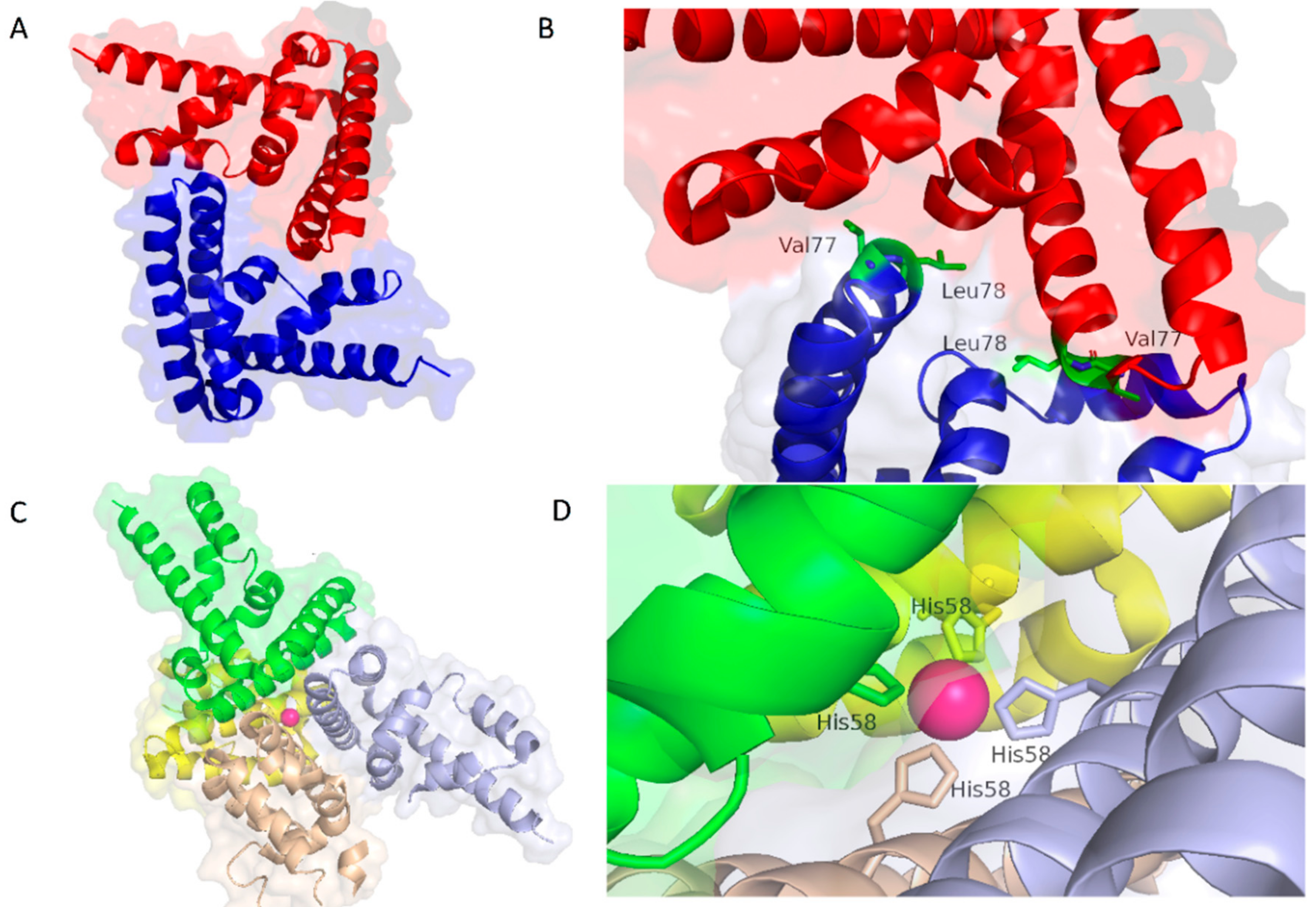
4. Structural and Biophysical Investigations
5. Drug Design
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| NTD | N-terminal domain |
| STAT3 | Signal transducer and activator of transcription 3 |
| USTAT3 | Unphosphorylated STAT3 |
| TAD | Trans activation domain |
| LD | Linker domain |
| DBD | DNA binding domain |
| CDD | Coiled coil domain |
| NTD | N-terminal domain |
| SH2 | SH2 domain |
| p-Tyr705 | Phosphorylated tyrosine 705 |
| PTMs | Post-transcriptional modifications |
References
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Koppikar, P.; Bhagwat, N.; Kilpivaara, O.; Manshouri, T.; Adli, M.; Hricik, T. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 2012, 489, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E., Jr. STATs: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Genini, D.; Brambilla, L.; Laurini, E.; Merulla, J.; Civenni, G.; Pandit, S.; D’Antuono, R.; Perez, L.; Levy, D.E.; Pricl, S.; et al. Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E4924–E4933. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.J.; Settleman, J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Masciocchi, D.; Gelain, A.; Villa, S.; Meneghetti, F.; Barlocco, D. Signal transducer and activator of transcription 3 (STAT3): A promising target for anticancer therapy. Future Med. Chem. 2011, 3, 567–597. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C.; Cerchia, C. Novel inhibitors of signal transducer and activator of transcription 3 signaling pathway: An update on the recent patent literature. Expert Opin. Ther. Pat. 2014, 24, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C.; Novellino, E. STAT-3 inhibitors: State of the art and new horizons for cancer treatment. Curr. Med. Chem. 2011, 18, 2359–2375. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Turkson, J. Targeting STAT3 in cancer: How successful are we? Exp. Opin. Investig. Drugs 2009, 18, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mule, J.; Kerr, W.G.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Swiderski, P.; Herrmann, A.; Wang, L.; Kowolik, C.; Kujawski, M.; Lee, H.; Scuto, A.; Liu, Y.; Yang, C.; et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat. Biotechnol. 2009, 27, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Mao, X.; Mertens, C.; Krishnaraj, R.; Qin, J.; Mandal, P.K.; Romanowski, M.J.; McMurray, J.S.; Chen, X. Crystal structure of unphosphorylated STAT3 core fragment. Biochem. Biophys. Res. Commun. 2008, 374, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Nkansah, E.; Shah, R.; Collie, G.W.; Parkinson, G.N.; Palmer, J.; Rahman, K.M.; Bui, T.T.; Drake, A.F.; Husby, J.; Neidle, S.; et al. Observation of unphosphorylated STAT3 core protein binding to target dsDNA by PEMSA and X-ray crystallography. FEBS Lett. 2013, 587, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Groner, B.; Müller, C.W. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature 1998, 394, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Wenta, N.; Strauss, H.; Meyer, S.; Vinkemeier, U. Tyrosine phosphorylation regulates the partitioning of STAT1 between different dimer conformations. Proc. Natl. Acad. Sci. USA 2008, 105, 9238–9243. [Google Scholar] [CrossRef] [PubMed]
- Droescher, M.; Vinkemeier, U. Self-association of STAT Proteins from Monomers to Paracrystals. In Jak-Stat Signaling: From Basics to Disease; Decker, T., Müller, M., Eds.; Springer: Vienna, Austria, 2012; pp. 47–63. [Google Scholar]
- Lim, C.P.; Cao, X. Structure, function, and regulation of STAT proteins. Mol. Biosyst. 2006, 2, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Sgrignani, J.; Olsson, S.; Ekonomiuk, D.; Genini, D.; Krause, R.; Catapano, C.V.; Cavalli, A. Molecular Determinants for Unphosphorylated STAT3 Dimerization Determined by Integrative Modeling. Biochemistry 2015, 54, 5489–5501. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Vinkemeier, U.; Zhao, Y.; Jeruzalmi, D.; Darnell, J.E.; Kuriyan, J., Jr. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell 1998, 93, 827–839. [Google Scholar] [CrossRef]
- Hu, T.; Yeh, J.E.; Pinello, L.; Jacob, J.; Chakravarthy, S.; Yuan, G.-C.; Chopra, R.; Frank, D.A. Impact of the N-Terminal Domain of STAT3 in STAT3-Dependent Transcriptional Activity. Mol. Cell. Biol. 2015, 35, 3284–3300. [Google Scholar] [CrossRef] [PubMed]
- Daria, D.; Ewa, B. Review: The JAK/STAT Protein Activation—Role in Cancer Development and Targeted Therapy. Curr. Signal Transduct. Ther. 2012, 7, 187–201. [Google Scholar]
- Shuai, K.; Horvath, C.M.; Huang, L.H.; Qureshi, S.A.; Cowburn, D.; Darnell, J.E., Jr. Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell 1994, 76, 821–828. [Google Scholar] [CrossRef]
- Yang, J.; Stark, G.R. Roles of unphosphorylated STATs in signaling. Cell Res. 2008, 18, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Timofeeva, O.A.; Chasovskikh, S.; Lonskaya, I.; Tarasova, N.I.; Khavrutskii, L.; Tarasov, S.G.; Zhang, X.; Korostyshevskiy, V.R.; Cheema, A.; Zhang, L.; et al. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J. Biol. Chem. 2012, 287, 14192–14200. [Google Scholar] [CrossRef] [PubMed]
- Haan, S.; Kortylewski, M.; Behrmann, I.; Müller-Esterl, W.; Heinrich, P.C.; Schaper, F. Cytoplasmic STAT proteins associate prior to activation. Biochem. J. 2000, 345, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, A.K.; Dinger, M.C.; Henze, C.; Brocke-Heidrich, K.; Horn, F. Analysis of Stat3 (signal transducer and activator of transcription 3) dimerization by fluorescence resonance energy transfer in living cells. Biochem. J. 2004, 377, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, J.; Brutsaert, S.; Olson, R.; Schindler, C. STATs dimerize in the absence of phosphorylation. J. Biol. Chem. 2003, 278, 34133–34140. [Google Scholar] [CrossRef] [PubMed]
- Domoszlai, T.; Martincuks, A.; Fahrenkamp, D.; Schmitz-Van de Leur, H.; Kuster, A.; Muller-Newen, G. Consequences of the disease-related L78R mutation for dimerization and activity of STAT3. J. Cell Sci. 2014, 127, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bhandari, R.; Vinkemeier, U.; van den Akker, F.; Darnell, J.E.; Kuriyan, J. A reinterpretation of the dimerization interface of the N-terminal Domains of STATs. Protein Sci. 2003, 12, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Pranada, A.L.; Metz, S.; Herrmann, A.; Heinrich, P.C.; Muller-Newen, G. Real time analysis of STAT3 nucleocytoplasmic shuttling. J. Biol. Chem. 2004, 279, 15114–15123. [Google Scholar] [CrossRef] [PubMed]
- Timofeeva, O.A.; Tarasova, N.I.; Zhang, X.; Chasovskikh, S.; Cheema, A.K.; Wang, H.; Brown, M.L.; Dritschilo, A. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc. Natl. Acad. Sci. USA 2013, 110, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Timofeeva, O.A.; Tarasova, N.I. Alternative ways of modulating JAK-STAT pathway: Looking beyond phosphorylation. Jakstat 2012, 1, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Timofeeva, O.A.; Gaponenko, V.; Lockett, S.J.; Tarasov, S.G.; Jiang, S.; Michejda, C.J.; Perantoni, A.O.; Tarasova, N.I. Rationally designed inhibitors identify STAT3 N-domain as a promising anticancer drug target. ACS Chem. Biol. 2007, 2, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhou, X.; Zhou, M.; Xu, D.; Ma, Q.; Zhang, P.; Huang, X.; Li, Q.; Ma, D.; Zhou, J. Inhibition of STAT3 activity re-activates anti-tumor immunity but fails to restore the immunogenicity of tumor cells in a B-cell lymphoma model. Cancer Biol. Ther. 2014, 15, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Xiong, A.; Yang, Z.; Shen, Y.; Zhou, J.; Shen, Q. Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention. Cancers 2014, 6, 926–957. [Google Scholar] [CrossRef] [PubMed]
- Wakahara, R.; Kunimoto, H.; Tanino, K.; Kojima, H.; Inoue, A.; Shintaku, H.; Nakajima, K. Phospho-Ser727 of STAT3 regulates STAT3 activity by enhancing dephosphorylation of phospho-Tyr705 largely through TC45. Genes Cells 2012, 17, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S. Regulation of STAT signaling by acetylation. Cell. Signal. 2013, 25, 1924–1931. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, M.; Dermawan, J.K.; Willard, B.; Stark, G.R. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc. Natl. Acad. Sci. USA 2015, 112, 3985–3990. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.S.; Liang, J.J.; Wang, Y.; Zhao, X.J.; Xu, L.; Xu, Y.Y.; Zou, Q.C.; Zhang, J.M.; Tu, C.E.; Cui, Y.G.; et al. STAT3 Undergoes Acetylation-dependent Mitochondrial Translocation to Regulate Pyruvate Metabolism. Sci. Rep. 2016, 6, 39517. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Ramachandra, C.J.A.; Chitre, A.; Singh, P.; Lua, C.H.; Shim, W. Acetylated Signal Transducer and Activator of Transcription 3 Functions as Molecular Adaptor Independent of Transcriptional Activity During Human Cardiogenesis. Stem Cells 2017, 35, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Darra, E.; Chiavegato, G.; Cellini, B.; Cozzolino, F.; Monti, M.; Pucci, P.; Dell’Orco, D.; Mariotto, S. S-Glutathionylation at Cys328 and Cys542 Impairs STAT3 Phosphorylation. ACS Chem. Biol. 2014, 9, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ren, Z.; Parker, G.N.; Sondermann, H.; Pastorello, M.A.; Wang, W.; McMurray, J.S.; Demeler, B.; Darnell, J.E., Jr.; Chen, X. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol. Cell 2005, 17, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Vinkemeier, U.; Moarefi, I.; Darnell, J.E., Jr.; Kuriyan, J. Structure of the amino-terminal protein interaction domain of STAT-4. Science 1998, 279, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Droescher, M.; Begitt, A.; Vinkemeier, U. Paracrystals of STAT proteins and their dissolution by SUMO: How reduced transcription factor solubility increases cytokine signaling. Oncotarget 2011, 2, 527–528. [Google Scholar] [PubMed]
- Novak, U.; Ji, H.; Kanagasundaram, V.; Simpson, R.; Paradiso, L. STAT3 forms stable homodimers in the presence of divalent cations prior to activation. Biochem. Biophys. Res. Commun. 1998, 247, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Husby, J.; Todd, A.K.; Haider, S.M.; Zinzalla, G.; Thurston, D.E.; Neidle, S. Molecular Dynamics Studies of the STAT3 Homodimer:DNA Complex: Relationships between STAT3 Mutations and Protein–DNA Recognition. J. Chem. Inf. Model. 2012, 52, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Porta, F.; Facchetti, G.; Ferri, N.; Gelain, A.; Meneghetti, F.; Villa, S.; Barlocco, D.; Masciocchi, D.; Asai, A.; Miyoshi, N.; et al. An in vivo active 1,2,5-oxadiazole Pt(II) complex: A promising anticancer agent endowed with STAT3 inhibitory properties. Eur. J. Med. Chem. 2017, 131, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Masciocchi, D.; Gelain, A.; Porta, F.; Meneghetti, F.; Pedretti, A.; Celentano, G.; Barlocco, D.; Legnani, L.; Toma, L.; Kwon, B.-M.; et al. Synthesis, structure-activity relationships and stereochemical investigations of new tricyclic pyridazinone derivatives as potential STAT3 inhibitors. Med. Chem. Comm. 2013, 4, 1181–1188. [Google Scholar] [CrossRef]
- Porta, F.; Gelain, A.; Barlocco, D.; Ferri, N.; Marchiano, S.; Cappello, V.; Basile, L.; Guccione, S.; Meneghetti, F.; Villa, S. A field-based disparity analysis of new 1,2,5-oxadiazole derivatives endowed with antiproliferative activity. Chem. Biol. Drug Des. 2017, 90, 820–839. [Google Scholar] [CrossRef] [PubMed]
- Meneghetti, F.; Villa, S.; Masciocchi, D.; Barlocco, D.; Toma, L.; Han, D.; Kwon, B.; Ogo, N.; Asai, A.; Legnani, L.; et al. Ureido-Pyridazinone Derivatives: Insights into the Structural and Conformational Properties for STAT3 Inhibition. Eur. J. Org. Chem. 2015, 2015, 4907–4912. [Google Scholar] [CrossRef]
- Fagard, R.; Metelev, V.; Souissi, I.; Baran-Marszak, F. STAT3 inhibitors for cancer therapy: Have all roads been explored? Jakstat 2013, 2, e22882. [Google Scholar] [CrossRef] [PubMed]
- Lai, P.S.; Rosa, D.A.; Magdy Ali, A.; Gomez-Biagi, R.F.; Ball, D.P.; Shouksmith, A.E.; Gunning, P.T. A STAT inhibitor patent review: Progress since 2011. Expert Opin. Ther. Pat. 2015, 25, 1397–1421. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.S.; Klostergaard, J. Chapter 7—STAT3 Signaling in Cancer: Small Molecule Intervention as Therapy? In Anti-Angiogenesis Drug Discovery and Development; Elsevier: Amsterdam, The Netherlands, 2014; pp. 216–267. [Google Scholar]
- Szelag, M.; Wesoly, J.; Bluyssen, H.A. Advances in peptidic and peptidomimetic-based approaches to inhibit STAT signaling in human diseases. Curr. Protein Pept. Sci. 2016, 17, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Masciocchi, D.; Villa, S.; Meneghetti, F.; Pedretti, A.; Barlocco, D.; Legnani, L.; Toma, L.; Kwon, B.-M.; Nakano, S.; Asai, A.; et al. Biological and computational evaluation of an oxadiazole derivative (MD77) as a new lead for direct STAT3 inhibitors. Med. Chem. Comm. 2012, 3, 592–599. [Google Scholar] [CrossRef]
- Turkson, J.; Ryan, D.; Kim, J.S.; Zhang, Y.; Chen, Z.; Haura, E.; Laudano, A.; Sebti, S.; Hamilton, A.D.; Jove, R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001, 276, 45443–45455. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Cabell, L.A.; Schaefer, T.S.; McMurray, J.S. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorg. Med. Chem. Lett. 2003, 13, 633–636. [Google Scholar] [CrossRef]
- Coleman, D.R.; Ren, Z.; Mandal, P.K.; Cameron, A.G.; Dyer, G.A.; Muranjan, S.; Campbell, M.; Chen, X.; McMurray, J.S. Investigation of the binding determinants of phosphopeptides targeted to the SRC homology 2 domain of the signal transducer and activator of transcription 3. Development of a high-affinity peptide inhibitor. J. Med. Chem. 2005, 48, 6661–6670. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Limbrick, D.; Coleman, D.R.; Dyer, G.A.; Ren, Z.; Birtwistle, J.S.; Xiong, C.; Chen, X.; Briggs, J.M.; McMurray, J.S. Conformationally Constrained Peptidomimetic Inhibitors of Signal Transducer and Activator of Transcription 3: Evaluation and Molecular Modeling. J. Med. Chem. 2009, 52, 2429–2442. [Google Scholar] [CrossRef] [PubMed]
- Siddiquee, K.A.; Gunning, P.T.; Glenn, M.; Katt, W.P.; Zhang, S.; Schrock, C.; Sebti, S.M.; Jove, R.; Hamilton, A.D.; Turkson, J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem. Biol. 2007, 2, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wang, R.; Wang, S.; Lin, J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4700–4705. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Masuda, Y.; Uehara, Y.; Sato, H.; Muroya, A.; Takahashi, O.; Yokotagawa, T.; Furuya, T.; Okawara, T.; Otsuka, M.; et al. Identification of a New Series of STAT3 Inhibitors by Virtual Screening. ACS Med. Chem. Lett. 2010, 1, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Kasembeli, M.M.; Jiang, X.; Tweardy, B.J.; Tweardy, D.J. Chemical probes that competitively and selectively inhibit Stat3 activation. PLoS ONE 2009, 4, e4783. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, L.; Genini, D.; Laurini, E.; Merulla, J.; Perez, L.; Fermeglia, M.; Carbone, G.M.; Pricl, S.; Catapano, C.V. Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3). Mol. Oncol. 2015, 9, 1194–1206. [Google Scholar] [CrossRef] [PubMed]
- Dupont, E.; Prochiantz, A.; Joliot, A. Penetratin story: An overview. Methods Mol. Biol. 2011, 683, 21–29. [Google Scholar] [PubMed]
- Feng, T.; Cao, W.; Shen, W.; Zhang, L.; Gu, X.; Guo, Y.; Tsai, H.I.; Liu, X.; Li, J.; Zhang, J.; et al. Arctigenin inhibits STAT3 and exhibits anticancer potential in human triple-negative breast cancer therapy. Oncotarget 2017, 8, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Li, C.; Zhang, W.; Xia, Y.; Li, S.; Lin, J.Y.; Yu, K.; Liu, M.; Yang, L.; Luo, J.; et al. Discovery of an Orally Selective Inhibitor of Signal Transducer and Activator of Transcription 3 Using Advanced Multiple Ligand Simultaneous Docking. J. Med. Chem. 2017, 60, 2718–2731. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.Y.; Fu, J.Y.; Yang, M.K.; Han, H.W.; Wang, P.F.; Zhang, Y.H.; Lin, H.Y.; Tang, C.Y.; Qi, J.L.; Yang, R.W.; et al. Identification of new shikonin derivatives as STAT3 inhibitors. Biochem. Pharmacol. 2017, 146, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.Y.; Zhu, X.; Luo, Y.L.; Lin, H.Y.; Tang, C.Y.; Qi, J.L.; Pang, Y.J.; Yang, R.W.; Lu, G.H.; Wang, X.M.; et al. Identification of New Shikonin Derivatives as Antitumor Agents Targeting STAT3 SH2 Domain. Sci. Rep. 2017, 7, 2863. [Google Scholar] [CrossRef] [PubMed]
- Son, D.J.; Zheng, J.; Jung, Y.Y.; Hwang, C.J.; Lee, H.P.; Woo, J.R.; Baek, S.Y.; Ham, Y.W.; Kang, M.W.; Shong, M.; et al. MMPP Attenuates Non-Small Cell Lung Cancer Growth by Inhibiting the STAT3 DNA-Binding Activity via Direct Binding to the STAT3 DNA-Binding Domain. Theranostics 2017, 7, 4632–4642. [Google Scholar] [CrossRef] [PubMed]
- Kolosenko, I.; Yu, Y.; Busker, S.; Dyczynski, M.; Liu, J.; Haraldsson, M.; Palm Apergi, C.; Helleday, T.; Tamm, K.P.; Page, B.D.G.; et al. Identification of novel small molecules that inhibit STAT3-dependent transcription and function. PLoS ONE 2017, 12, e0178844. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Springer, M.Z.; Xu, D.; Liu, D.; Hudmon, A.; Macleod, K.F.; Meroueh, S.O. Small molecules inhibit STAT3 activation, autophagy, and cancer cell anchorage-independent growth. Bioorg. Med. Chem. 2017, 25, 2995–3005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Guo, J.; Li, S.; Ma, T.; Xu, D.; Han, C.; Liu, F.; Yu, W.; Kong, L. Discovery of monocarbonyl curcumin-BTP hybrids as STAT3 inhibitors for drug-sensitive and drug-resistant breast cancer therapy. Sci. Rep. 2017, 7, 46352. [Google Scholar] [CrossRef] [PubMed]
- Arpin, C.C.; Mac, S.; Jiang, Y.; Cheng, H.; Grimard, M.; Page, B.D.; Kamocka, M.M.; Haftchenary, S.; Su, H.; Ball, D.P.; et al. Applying Small Molecule Signal Transducer and Activator of Transcription-3 (STAT3) Protein Inhibitors as Pancreatic Cancer Therapeutics. Mol. Cancer Ther. 2016, 15, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Yuan, C.; Ma, S.; Fan, J.; Fu, W.; Qiao, C. 4-Carbonyl-2,6-dibenzylidenecyclohexanone derivatives as small molecule inhibitors of STAT3 signaling pathway. Bioorg. Med. Chem. 2016, 24, 6174–6182. [Google Scholar] [CrossRef] [PubMed]
- Daka, P.; Liu, A.; Karunaratne, C.; Csatary, E.; Williams, C.; Xiao, H.; Lin, J.; Xu, Z.; Page, R.C.; Wang, H. Design, synthesis and evaluation of XZH-5 analogues as STAT3 inhibitors. Bioorg. Med. Chem. 2015, 23, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Xiao, Q.; Zhang, M.; Li, Y. Design, synthesis and biological evaluation of benzyloxyphenyl-methylaminophenol derivatives as STAT3 signaling pathway inhibitors. Bioorg. Med. Chem. 2016, 11, 2549–2558. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, W.; Yu, W.; Jou, D.; Wang, Y.; Ma, H.; Xiao, H.; Qin, H.; Zhang, C.; Lu, J.; et al. A novel small molecule STAT3 inhibitor, LY5, inhibits cell viability, colony formation, and migration of colon and liver cancer cells. Oncotarget 2016, 7, 12917–12926. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Chen, Z.; Zhang, L.; Huang, Z.; Chen, Y.; Xu, J.; Zhang, J.; Shu, X. Screening and biological evaluation of a novel STAT3 signaling pathway inhibitor against cancer. Bioorg. Med. Chem. Lett. 2016, 26, 5172–5176. [Google Scholar] [CrossRef] [PubMed]
- Yesylevskyy, S.O.; Ramseyer, C.; Pudlo, M.; Pallandre, J.R.; Borg, C. Selective Inhibition of STAT3 with Respect to STAT1: Insights from Molecular Dynamics and Ensemble Docking Simulations. J. Chem. Inf. Model. 2016, 56, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ma, T.; Li, S.; Yang, Y.; Guo, J.; Yu, W.; Kong, L. Antagonizing STAT3 activation with benzo[b]thiophene 1,1-dioxide based small molecules. Eur. J. Med. Chem. 2017, 125, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Li, R.J.; Cheng, M.S.; Kim, Y.S. Alantolactone selectively suppresses STAT3 activation and exhibits potent anticancer activity in MDA-MB-231 cells. Cancer Lett. 2015, 357, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Botta, A.; Sirignano, E.; Popolo, A.; Saturnino, C.; Terracciano, S.; Foglia, A.; Sinicropi, M.S.; Longo, P.; Di Micco, S. Identification of Lead Compounds as Inhibitors of STAT3: Design, Synthesis and Bioactivity. Mol. Inform. 2015, 34, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; He, L.; Cao, P.; Yu, Q. Eriocalyxin B Inhibits STAT3 Signaling by Covalently Targeting STAT3 and Blocking Phosphorylation and Activation of STAT3. PLoS ONE 2015, 10, e0128406. [Google Scholar] [CrossRef] [PubMed]
- Szelag, M.; Czerwoniec, A.; Wesoly, J.; Bluyssen, H.A. Identification of STAT1 and STAT3 specific inhibitors using comparative virtual screening and docking validation. PLoS ONE 2015, 10, e0116688. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.H.; Liu, L.J.; Lin, S.; Lu, L.; Zhong, H.J.; Susanti, D.; Rao, W.; Wang, M.; Che, W.I.; Chan, D.S.; et al. Discovery of a small-molecule inhibitor of STAT3 by ligand-based pharmacophore screening. Methods 2015, 71, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, Y.; Pireddu, R.; Yang, H.; Urlam, M.K.; Lawrence, H.R.; Guida, W.C.; Lawrence, N.J.; Sebti, S.M. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013, 73, 1922–1933. [Google Scholar] [CrossRef] [PubMed]
- Dhanik, A.; McMurray, J.S.; Kavraki, L.E. Binding modes of peptidomimetics designed to inhibit STAT3. PLoS ONE 2012, 7, e51603. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
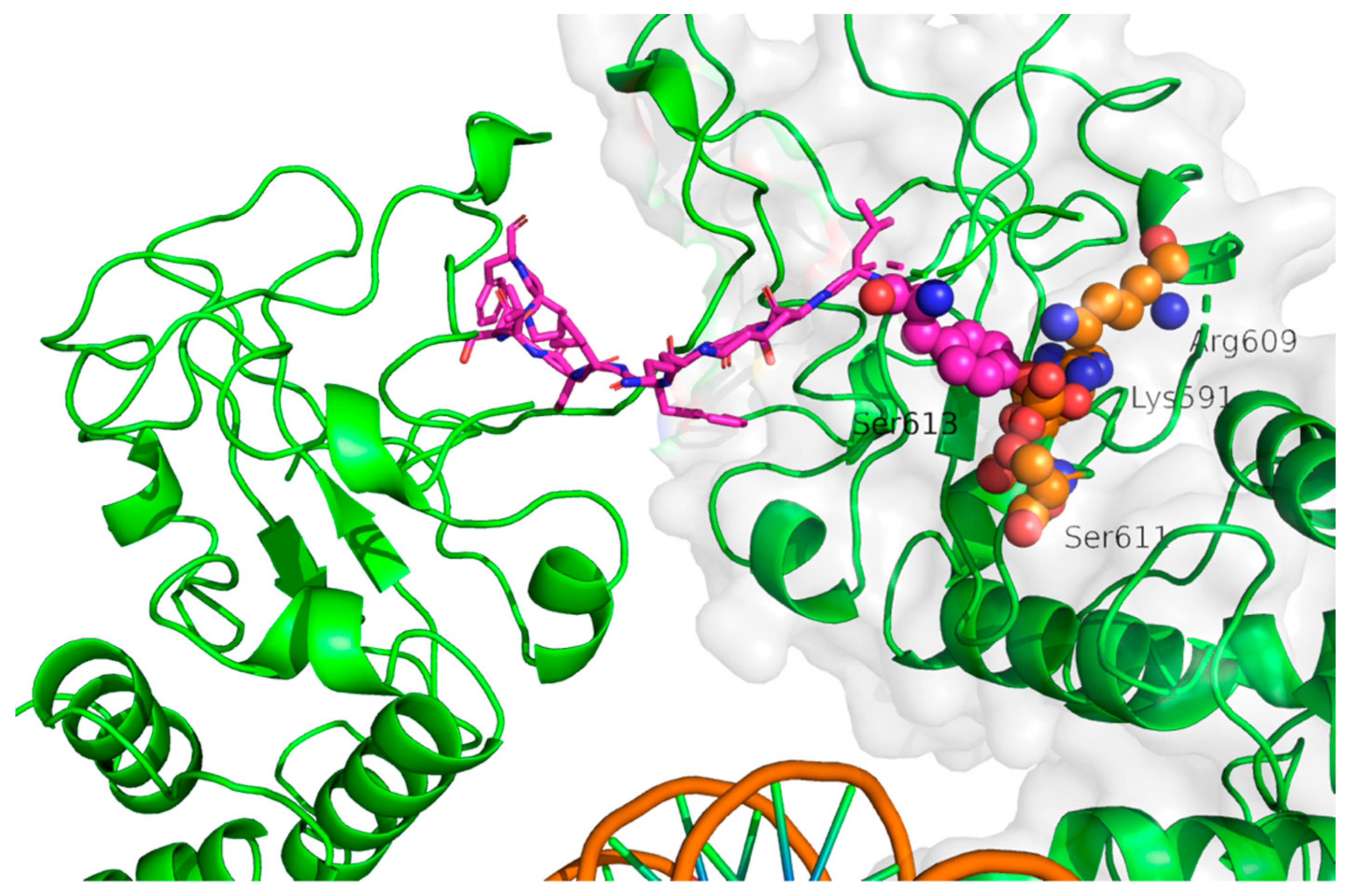{kind=link}
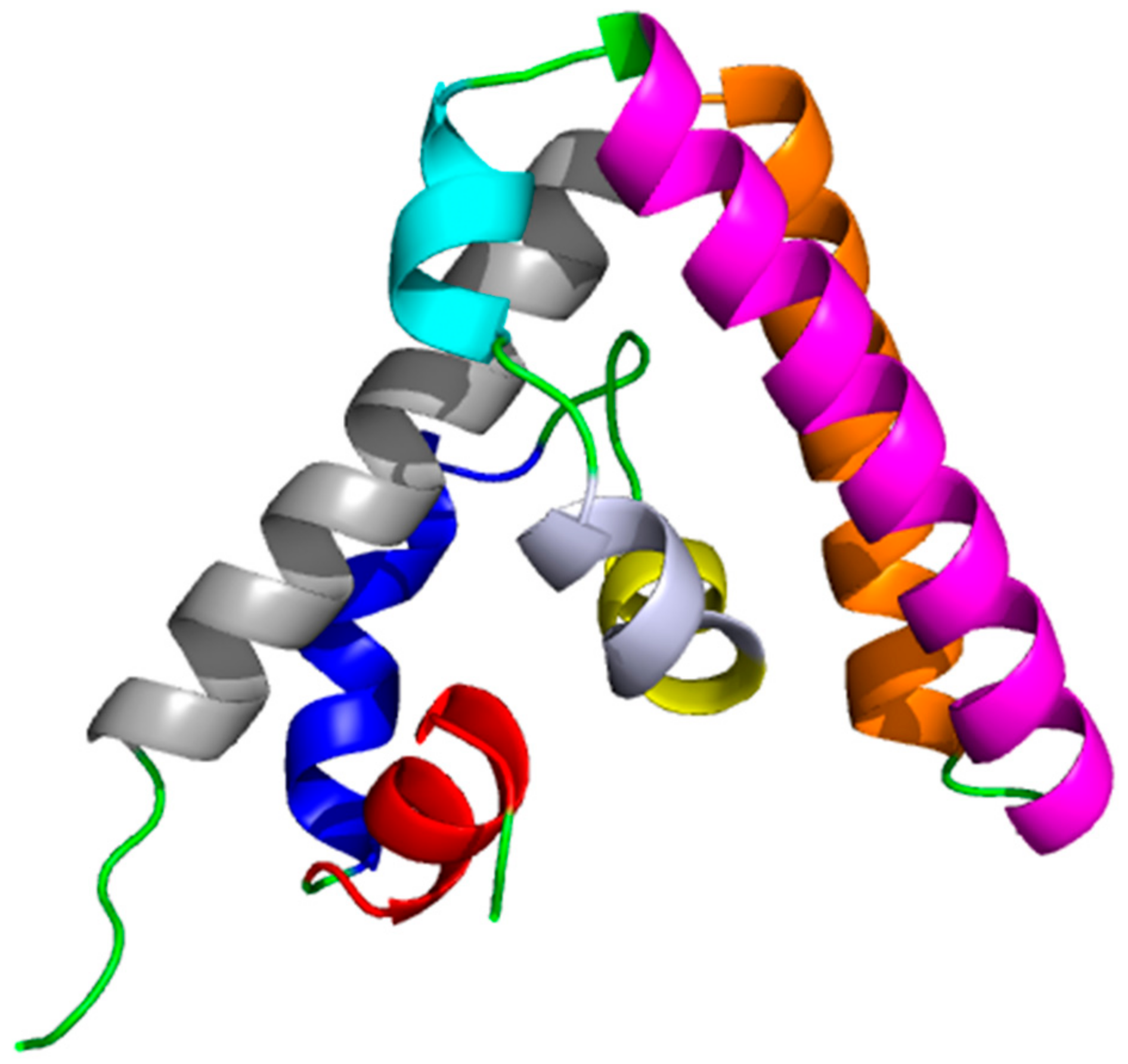{kind=link}
| PDB Code | Description | Reference |
|---|---|---|
| 3CWG | Unphosphorylated mouse STAT3 core fragment (full length without amino-terminal domain (NTD)) | [19] |
| 1BG1 | STAT3B/DNA complex (no N-terminal domain) | [21] |
| 4E68 | Unphosphorylated STAT3B (no N-terminal domain) core protein binding to dsDNA | [20] |
| 4ZIA | X-ray structure of STAT3 N-terminal domain | [27] |
| Title | Structural Experimental and/or Computational Structural Biology Contribution | Year | Reference |
|---|---|---|---|
| Arctigenin Inhibits STAT3 and exhibits anticancer potential in human triple-negative breast cancer therapy. | Use of docking and molecular dynamics simulations to understand the binding mode of Arctigenin (a STAT3 inhibitor). | 2017 | [78] |
| Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells | Use of docking and molecular dynamics simulations to characterize the binding mode of OPB-51602, a small molecule currently in clinical trials, to STAT3. | 2017 | [5] |
| Discovery of an Orally Selective Inhibitor of Signal Transducer and Activator of Transcription 3 Using Advanced Multiple Ligand Simultaneous Docking | Use of Advanced Multiple Ligand Simultaneous Docking (AMLSD) to design compounds able to directly inhibit both phosphorylation and dimerization of STAT3 protein. | 2017 | [79] |
| Identification of New Shikonin Derivatives as STAT3 Inhibitors | Discovery of to discover PMMB-187, a new STAT3 inhibitor by modification of shikonin scaffold guided by computational modeling. | 2017 | [80] |
| Identification of New Shikonin Derivatives as Antitumor Agents Targeting STAT3 SH2 Domain. | Discovery of PMM-172, a new STAT3 inhibitor, via molecular docking and scaffold modification of shikonin. | 2017 | [81] |
| MMPP Attenuates Non-Small Cell Lung Cancer Growth by Inhibiting the STAT3 DNA-Binding Activity via Direct Binding to the STAT3 DNA-Binding Domain | Use of molecular docking for the rational design of 16BHPB, a STAT3 inhibitor. | 2017 | [82] |
| Identification of novel small molecules that inhibit STAT3-dependent transcription and function | Use of docking studies to understand the binding mode of new STAT3 inhibitors identified by high-throughput screening. | 2017 | [83] |
| Small molecules inhibit STAT3 activation, autophagy, and cancer cell anchorage-independent growth | Molecular docking of STAT3 inhibitors to predict their binding mode. | 2017 | [84] |
| Discovery of monocarbonyl curcumin-BTP hybrids as STAT3 inhibitors for drug-sensitive and drug-resistant breast cancer therapy | Docking studies to predict the binding mode of new STAT3 inhibitors. | 2017 | [85] |
| Applying Small Molecule Signal Transducer and Activator of Trancription-3 (STAT3) Protein Inhibitors as Pancreatic Cancer Therapeutics | Docking of STAT3 inhibitors and visualization of the computed receptor-ligand complexes. | 2016 | [86] |
| 4-Carbonyl-2,6-dibenzylidenecyclohexanone derivatives as small molecule inhibitors of STAT3 signaling pathway | Docking studies of a new STAT3 inhibitor to confirm its binding mode. | 2016 | [87] |
| Design, synthesis and evaluation of XZH-5 analogues as STAT3 inhibitors | Structure activity relationship (SAR) to design new STAT3 inhibitors using docking studies to predict their binding mode. | 2016 | [88] |
| Design, synthesis and biological evaluation of benzyloxyphenyl- methylaminophenol derivatives as STAT3 signaling pathway inhibitors | SAR studies of benzyloxyphenyl-methylaminophenol scaffold and docking studies to confirm inhibitors binding mode | 2016 | [89] |
| A novel small molecule STAT3 inhibitor, LY5, inhibits cell viability, colony formation, and migration of colon and liver cancer cells | Evaluation of LY5 inhibition and docking studies to understand its binding mode | 2016 | [90] |
| Screening and biological evaluation of a novel STAT3 signaling pathway inhibitor against cancer | Identification of new STAT3 inhibitors (directed to SH2 domain) by docking studies using SPECS libraries (202,490 compounds). | 2016 | [91] |
| Selective Inhibition of STAT3 with Respect to STAT1: Insights from Molecular Dynamics and Ensemble Docking Simulations | Molecular mechanisms studies about the selectivity of 13 experimentally tested STAT3 inhibitors by molecular dynamics and ensemble docking simulations. | 2016 | [92] |
| Antagonizing STAT3 activation with benzo[b]thiophene 1, 1-dioxide based small molecules | Discovery of new STAT3 inhibitors via structure-based drug design methods, using benzo[b]thiophene 1, 1-dioxide (BTP) as lead compound. | 2016 | [93] |
| Alantolactone selectively suppresses STAT3 activation and exhibits potent anticancer activity in MDA-MB-231 cells | Structure-based molecular docking study used to investigate the binding mode of alantolactone, found to be a STAT3 inhibitor, and STAT3 | 2015 | [94] |
| Identification of Lead Compounds as Inhibitors of STAT3: Design, Synthesis and Bioactivity | Virtual screening to find new STAT3 inhibitors. | 2015 | [95] |
| Eriocalyxin B Inhibits STAT3 Signaling by Covalently Targeting STAT3 and Blocking Phosphorylation and Activation of STAT3 | Computational modeling analysis of the mechanism of action EB, a natural compound able to act as a STAT3 inhibitor. | 2015 | [96] |
| Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3) | In silico studies to understand the binding mode of a previously known STAT3 inhibitor. | 2015 | [76] |
| Identification of STAT1 and STAT3 Specific Inhibitors Using Comparative Virtual Screening and Docking Validation | Virtual screening to find new STAT3 inhibitors. | 2015 | [97] |
| Discovery of a small-molecule inhibitor of STAT3 by ligand-based pharmacophore screening | Pharmacophore based virtual screening. | 2014 | [98] |
| A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation | Structure activity relationship (SAR) studies using a previously published S3I-201 compound as a lead and docking studies to understand the binding mode of the new inhibitors | 2013 | [99] |
| Binding Modes of Peptidomimetics Designed to Inhibit STAT3 | Molecular docking combined with molecular dynamic simulations to model to model the structures of 12 peptidomimetic inhibitors bound to the SH2 domain of STAT3. | 2012 | [100] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sgrignani, J.; Garofalo, M.; Matkovic, M.; Merulla, J.; Catapano, C.V.; Cavalli, A. Structural Biology of STAT3 and Its Implications for Anticancer Therapies Development. Int. J. Mol. Sci. 2018, 19, 1591. https://doi.org/10.3390/ijms19061591
Sgrignani J, Garofalo M, Matkovic M, Merulla J, Catapano CV, Cavalli A. Structural Biology of STAT3 and Its Implications for Anticancer Therapies Development. International Journal of Molecular Sciences. 2018; 19(6):1591. https://doi.org/10.3390/ijms19061591
Chicago/Turabian StyleSgrignani, Jacopo, Maura Garofalo, Milos Matkovic, Jessica Merulla, Carlo V. Catapano, and Andrea Cavalli. 2018. "Structural Biology of STAT3 and Its Implications for Anticancer Therapies Development" International Journal of Molecular Sciences 19, no. 6: 1591. https://doi.org/10.3390/ijms19061591
APA StyleSgrignani, J., Garofalo, M., Matkovic, M., Merulla, J., Catapano, C. V., & Cavalli, A. (2018). Structural Biology of STAT3 and Its Implications for Anticancer Therapies Development. International Journal of Molecular Sciences, 19(6), 1591. https://doi.org/10.3390/ijms19061591